2. Antimicrobial Spectrum of Coumarins
Dhawan and co-workers [
17] reported the synthesis of hybrid derivatives, i.e., coumarin-clubbed−β-lactam triazole derivatives (
10a–o), and tested them for their antimicrobial potential against Gram-positive methicillin-resistant
Staphylococcus aureus, as well as four Gram-negative bacteria, such as
Pseudomonas aeruginosa,
Klebsiella pneumonia,
Escherichia coli, and
Acinetobacter baumannii, and two fungal strains, like
Candida albicans and
Cryptococcus neoforman. An outline for the synthesis of these novel analogues is depicted in
Scheme 1. The construction of the target hybrids (
10a–o) began by the reaction of 7-(ethynyloxy)-4,5-dimethyl-2H-chromen-2-one (
9a) and 7-(ethynyloxy)-4-methyl-2H-chromen-2-one (
9b) with various synthesized substituted azido lactams (
6a–h) in the presence of copper sulphate pentahydrate and sodium ascorbate in a ratio of dichloromethane:water (8:2) at room temperature. The starting materials
8a and
8b were synthesized by a reaction of orcinol
7a and phloroglucinol
7b with ethyl acetoacetate in sulphuric acid via the Pechmann rection. Compound
9a was obtained by the base-promoted reaction of
8a with propargyl bromide in dimethylformamide (DMF). Substitute azido lactams were synthesized by a reaction with acetic acid and imine at 0–5 °C. Substituted key intermediate azidoketene (
6a–h) were prepare in situ in the presence of p-toluene sulfonyl chloride and triethylamine to furnish the desired azido lactams. The antibacterial potency of the furnished compounds was detected by the disc diffusion method at a conc. Of 32 µg/mL with ≤ 1% DMSO. From the displayed results, most of compounds were moderately active against all bacterial excerpts and fungal stains. Further studying revealed compounds
10c,
10f, and
10g possess significant activity against
Staphylococcus aureus, with maximum percentage inhibitions of 7.75, 7.92, and 8.94%, respectively, while the other analogues were inactive with methicillin-resistant
Staphylococcus aureus (MRSA), whereas compounds
10i,
10j,
10k, and
10d emerged as active hybrids against
C. albicans with 21.65, 9.42, 6.24, and 14.96 percentage inhibition, respectively. The SAR study revealed analogues bearing methyl and iodo groups showing significant antimicrobial activity.
Dharavath and co-workers [
18] reported a set of coumarin-based 1,2,3-triazole hybrids
16(
a–l) and tested for their antibacterial activity towards Gram-positive, such as
S. aureus and
Bacillus subtilis, and Gram-negative bacteria like
E. coli, as well as
K. pneumonia. The route for the synthesis is outlined in
Scheme 2 [
14]. The substituted phenyl acetic acid
11 was activated by 1,1-carbonyldiimidazole and potassium carbonate as a base in acetone for 1 h to furnish intermediate
12. Later, compound
14 was prepared by the selective propargylation of
13 using K
2CO
3 in DMF as a solvent. Prepared alkyne-substituted hydroxy acetophenone
14 was refluxed with intermediate
12 using K
2CO
3 and acetone to obtain the key intermediate (
15a–d). In a last step, the title analogues (
16a–l) were afforded a copper (I)-catalyzed Huisgen cycloaddition reaction of intermediate
15a–d with different aryl azides using copper iodide and DMF/H
2O (1:1). The antibacterial and antifungal potentials of the furnished analogues detected by the disc diffusion method and percentage zone inhibition were compared to standard drugs Gatifloxacin and Clotrimazole, respectively. Among compounds
16a,
16d,
16g, and
16j possess potent antibacterial action due to methoxy group in the triazole moiety was shown, while compounds,
16b,
16c,
16e,
16f,
16h, and
16i showed significant potential against the tested organisms because of electron-withdrawing groups on the coumarin ring when compared to the standard drug.
Antifungal potential of the tested compound measured against
Aspergillus niger,
Aspergillus flavus, and
Fusarium sporum at a concentration of 50 mg mL
−1. Among the screened hybrids
16a–c displayed significant potential against all tested fungal strains due to the existence of fluorine on the coumarin ring, while compounds
16j–l showed good potential due to the appearance of -OCH
3 in the triazole ring. The docking study clearly showed the interaction profile between the coumarin-based 1,4-disubstituted 1,2,3-triazole moiety with amino acids. Active analogues
16d and
16h formed H-bond interactions with His-88, Val-191, and amino acids with water in the cavity of the selected proteins, i.e., DNA-gyrase B and topoisomerase IV (PDB ID: 4GEE) and cytochrome P450 EryK (PDB ID: 2XFH) (
Figure 2). When the results were compared to the well-reported antifungal agent ketoconazole (0.5 μg/mL against
A. niger), the above coumarin-based 1,2,3-triazole hybrids were not potentially active [
18].
S.M. Sutar et al. [
19] performed the synthesis of coumarin and 1-aza-coumarin derivatives of miconazole (
21a–e), (
22a–e), (
23a–e), and (
24a–e) and evaluate there
in vitro antimicrobial potential. The route for their synthesis is outlined in
Scheme 3. Initially,2-bromo-1-(2,4-dichlorophenyl) ethenone
18 was obtained via the bromination of
17. Further, corresponding with
18 in the reaction with imidazole and benzimidazole in THF under reflux furnished N-alkyl analogues
19a and
19b. Key intermediates
20a and
20b were afforded via a reduction of
19a and
19b using NaBH
4. Finally, the O-alkylation of
20a and
20b with various substituted 4-bromomethyl coumarins and 4-bromomethyl carbostyrils using K
2CO
3 and DMF yielded target compounds
21a–e,
22a–e,
23a–e, and
24a–e, respectively.
Among the series, majority of the analogues were found to be moderate-to-significantly active against Gram-positive strains, such as
S. aureus and
Bacillus cereus, whereas derivatives of miconazole (
21a–c) displayed reflective inhibitory activity against both Gram-positive strains, with MIC ranging from 1 to 4 μg/mL. Likewise, the replacement of imidazole with benzimidazole analogues (
23a–c) revealed the same potency as compared to
21a–c. The MIC value of the tested analogues explored that presence of electron-donating species like methyl
21a,
21b,
23a, and
23b and methoxy
21c and
23c on coumarin employed excellent potency as compared to an electron-withdrawing group (ex. Chloro and aryl ring). Therefore, analogues
21a and
23c against
B. Cereus and analogue
23c against
S. aureus displayed 100% potency (1 μg/mL) compared to the control ciprofloxacin (1 μg/mL); however, compounds
21a,
21c, and
23b against
S. aureus and compounds
21b,
21c,
23a, and
23b against
B. Cereus recorded 50% potency (2 μg/mL) when compared to ciprofloxacin. Two groups of fungal strains, i.e., Yeast (included
C. albicans,
Candida utilis, and
Candida krusei) and Filamentous (including
Aspergillus fumigatus,
A. niger, and
Rhizoctonia bataticola) fungi, were screened against the synthesized analogues and their potential compared with the standard drugs Itraconazole and Miconazole. None of analogues proved better than that of the standard drug against
A. niger and
R. bataticola, excluding
23e against
A. niger and
23c against
R. bataticola. However, the compounds bearing benzimidazole (
23a–24e) exhibited good-to-moderate potential (4–31.2 μg/mL) against
A. fumigatus compared to that of the compounds bearing an imidazole framework (
21a–22e). Candidates having an aza-coumarin framework with methyl and chloro substituents (
24b–e) showed better potency (4 μg/mL). In conclusion, the majority of candidates were found effective against the inhibition of all fungi species; in particular,
21e–24e were remarkable, while analogue
21e of the 7,8-benzo substitution on coumarin recorded 4 μg/mL of MIC, and the replacement of coumarin with 1-aza-coumarin (
22a–e) displayed 200−400% efficiency against all three strains. The molecular docking study revealed that compound
24e displayed the highest binding affinity of 11.2 kcal/mol with protein human lanosterol 14α-demethylase (PDB ID: 3LD6) by forming hydrogen-bonding interactions with amino acids MET 378 and ILE 379 (
Figure 3).
Rawan Alnufaie and co-workers [
20] reported the synthesis of some coumarin-substituted pyrazole hybrids for antimicrobial evaluation against methicillin-resistant
S. aureus. 4-hydrazino benzoic acid
25 refluxed with fluoro
26a, and hydroxy
26b bearing 3-acetylcoumarin furnishes corresponding to hydrazones
27a and
27b, respectively, which was subjected to reflux using POCl
3/DMF to yield formyl-substituted pyrazole derivatives
28a–b. Intermediate fluoro-substituted coumarin
28a and hydroxy-substituted coumarin derivative
28b reacted with substituted phenyl hydrazine under reflux to obtain coumarin-substituted pyrazole-derived hydrazones
29a–r and
30a–l outlined in
Scheme 4. Antibacterial potency of the synthesized analogues was detected against eight types of Gram-positive and three Gram-negative bacterial strains. Compound
29c with
N,
N-diphenyl substitution has a proven potential against all Gram-positive bacterial strains. Compound
29c inhibited the growth of methicillin-sensitive
S. aureus with the lowest MIC of 3.125 μg/mL, as well as four methicillin-resistant strains also inhibited by this candidate (MIC = 1.56 μg/mL). A compound bearing chloro (
29h) and bromo (
29i) substitutions displayed moderate activity against Gram-positive strains. This candidate was found to be active against all three
A. baumannii strains (MIC = 6.25 μg/mL). The candidate having 3,4-difluro substitution (
29k) was found moderately potent against both Gram-positive and Gram-negative bacterial strains with a MIC of 6.25 μg/mL. Candidates with very strong electron-withdrawing groups like trifluoromethyl (
29o) were found significantly active against Gram-positive strains with a MIC value of 3.125 μg/mL, while the other two analogues
29p and
29q with very strong electron-withdrawing frameworks were inactive.
Hydroxy-substituted coumarin analogues 30a–l failed to show antibacterial potential against all tested strains. Based on the observed results, the SAR study revealed that compounds bearing a fluoro substitution on coumarin produced potent antimicrobial action, while the hydroxy-substituted coumarin analogues almost eliminated the antimicrobial potential. This may happen due to the different natures of hydroxy and fluoro substituents. Candidates with fluoro and N,N-diphenyl substitutions possessed significant antimicrobial action, but one of the phenyls from the diphenyl substitution when replaced with methyl (29b) or benzyl (29d) species eliminated the potential of the compounds. Hydrophobic mono-substituted analogues were found to possess better potential than that of the disubstituted analogues. A compound with a trifluoromethyl framework (29o) was found the most potent among all the synthesized compounds against the Gram-positive bacterial strains.
Hanan M. Alshibl and colleagues [
21] stated the antimicrobial potential of the pyranocoumarin and coumarin−sulphonamide hybrids (
Scheme 5a,b). Initially, new pyranocoumarins (
32a,b) were furnished via one-pot synthesis by the reaction of substituted aryl aldehyde with malononitrile and
31b in the presence of potassium hydrogen phthalate. The carboxamide analogues
33a and/or
33b were prepared by the acid hydrolysis of
32a and/or
32b, respectively, with sulphuric acid. Similarly, the reactions of
32a and
32b with excess N, N-dimethylformamide-dimethylacetal afforded the corresponding
34a and
34b. Target analogues
35a–f were yielded by the reaction of intermediate
32a,b with various substituted aromatic aldehydes in 1,4-dioxane. Coumarin sulfonyl chloride
37a,b was prepared by reacting 4-hydroxy-6-substituted coumarin
36a and/or
31b with chlorosulfonic acid in DCM. Coumarin−3-sulphonamides (
38a–f) were obtained by refluxing
37a or
37b with substituted sulpha analogues. Similarly,
37a or
37b in a reaction with the nucleophilic agent aniline and p-substituted aniline in EtOH under reflux furnished the corresponding coumarin−3-sulphonamides (
39a–d and
40e–f, respectively). Coumarin−sulphonamide chalcone analogues
41a–f were prepared via the Claisen–Schmidt condensation of
p-acetyl derivatives (
40e or
40f) with different
p-substituted aromatic aldehydes by the addition of NaOH in absolute ethanol (
Scheme 5b). Synthesized analogues were screened for their in vitro antimicrobial potential against Gram-positive bacterial strains, namely,
S. aureus,
B. subtilis,
Bacillus, and
Megaterium, and Gram-negative strains such as
E. coli and
P. aeruginosa. Candidates were also screened against yeast and yeast-like pathogenic fungal strains
Saccharomyces cerevisiae and
C. albicans, respectively. Screening was accomplished by using agar well diffusion and Sabouraud dextrose agar. Among the synthesized analogues,
32a,
b,
35a,
c,f,
38a,
c,
d–f,
39a,
c,
d, and
41b–f were found to be potent antimicrobial agents with Izs larger than 25 mm against at least one tested microbial strain, while compounds
35e and
39b displayed moderate potential with Izs between 20 and 24 mm. However,
33a,
b, 34a,
b,
35d,
39e–f, and
41a exhibited marginal activity with Izs 15–19 mm, whereas analogues
38c,
d 39c,d, and
41c,d displayed the maximum Izs and revealed potent antimicrobial action towards the tested microorganisms when compared to the reference with a MIC of 125 µg/mL. However,
38d established remarkable broad-spectrum potential towards all the tested microorganisms with a MIC of 125 µg/mL. Analogues bearing sulfadiazine or 4-hydroxyphenyl moieties,
38c–d and
39c–d, revealed fairly excessive antibacterial potential (30 mm) compared to the standard ciprofloxacin (28 mm) against
S. aureus with a MIC of 125 µg/mL, while analogues containing sulphanilamide or 4-chlorophenyl substitutions (
38a and
41c,d) exhibited equal antibacterial potential against
S. aureus than the standard analogue ciprofloxacin (28 mm).
Abrar Bayazeed and co-workers [
22] reported the antimicrobial potency of novel coumarin derivatives obtained from 3-(2-oxo-2H-chromen-3-yl)-pyrazole-1-carbothioic acid amide with two diverse hydrazonoyl chlorides and phenacyl bromides when screened against fungal and bacterial strains. The synthesis of the target analogues is outlined in
Scheme 6. The key intermediates of carbothioamide (
44 and
45) were furnished via condensation and Michel-type addition succeeded by eliminating water and Me
2NH by a reaction of enaminone (
43) and thiosemicarbazide (
42). Key intermediate carbothioamide (
44) afforded chromen-3-yl-pyrazole derivatives
47a–e and
49a–e via a substitution reaction with two different types of hydrazonoyl chlorides,
46a–e and
48a–e, respectively. Finally, thiazole derivatives
51a–f were prepared via the nucleophilic substitution reaction of the intermediate carbothioamide (
44) with substituted phenacyl bromide under reflux. The synthesized chromenylpyrazoles candidates were examined in vitro for their antimicrobial potential versus two fungal Gram-positive and Gram-negative bacterial strains by using the agar diffusion method.
All test compounds were examined separately against all microbial strains at a concentration of 30 μg/mL. Among the series, four analogues, namely,
49e,
51b,
49b, and
51c, showed an exceeded potential compared to that of the reference/standard ketoconazole against
A. fumigatus with percent zone inhibitions of 164%, 158.8%, and 147.1%. Moreover, two analogues,
49b and
51c, showed excellent activity against
C. albicans above the reference ketoconazole 1.5-fold, while compounds
47b,
49e, and
51b exhibited good potency as compared to ketoconazole against
C. albicans with inhibition zone percentages 75–85%. The SAR studies stated that the presence of two ester groups in the synthesized derivatives is responsible for the improved antifungal potential, as seen in
49e. Moreover, analogues bearing electron-donating groups CH
3 and OCH
3 at the
para position of the aromatic skeleton (
51b,
49b, and
51c) had improved potential, and it exceed that of the reference drug. The antibacterial potential compounds
49b and
51c were found highly potent against
S. aureus with inhibition percentages 104.2% and 104%, respectively. Two other analogues included in the study, namely,
49e and
51b, exhibited the same excellent potential as compared to the reference Gentamicin with 107.7% and 103.8% inhibition. The Gram-negative bacteria
K. pneumoniae was highly affected by analogues
47a and
49e with 71.4% inhibition, while most of the analogues were found less active against
E. coli. The SAR analysis also revealed that, for the inhibition of Gram-positive bacteria, the presence of ester and electron-donating substitution at the
para position of the aromatic framework showed antimicrobial potency, as observed for analogues
49e,
49b,
51c, and
51b [
22].
R K Sharma et al. [
23] synthesized a set of 3-amidocoumarins analogues and evaluated their in vitro antimicrobial action against four Gram-negative, two Gram-positive bacterial, and three fungal pathogens. The synthesis of the designed analogues is depicted in
Scheme 7. In this study, 3-nitrocoumarin (
53) was obtained via the condensation of salicylaldehyde (
52) and ethyl nitroacetate in the presence of L-proline as a catalyst. Later, the reduction of
53 to 3-aminocoumarin (
54) using SnCl
2 subsequently by acylation with different substituted carboxylic acids using PCl
3 in acetonitrile under reflux furnished the target analogues of 3-amido coumarins
55a–p. On the other hand, coumarin-3-carboxylic acid
56 synthesized via the Knoevenagel condensation of salicylaldehyde (
52) with malonic acid in the presence of L-proline followed by the condensation of synthesized coumarin−3-carboxylic acid (
56) with suitable amines using DCC and DMAP in dry DCM gave the corresponding 3-carboxamide coumarins
57a–g. The MIC of amido−coumarins
55a–p and
57a–g detected against Gram-negative
Salmonella typhi,
P. aeruginosa,
K. pneumoniae, and
E. coli; two Gram-positive (
S. aureus and
Bacillus pumilus); and three fungal strains (
Candida tropicalis,
C. albicans, and
Aspergillus fumigatus) were determined by the two-fold serial dilution method using novobiocin and chloramphenicol as the antibacterial and fluconazole and amphotericin B as the antifungal standards. Among the series compounds,
55e–f with hydroxyl, chloro, methyl, and methoxy group substitutions on the phenyl ring demonstrated high MIC in the range of 50 to >200 µg/mL. Some enhancement in the potency was noticed upon the addition of disubstituted phenyl rings in analogues like
55i,
55j, and
55k. Analogue
55i inhibited the growth of
S. typhi and
P. aeruginosa at concentrations of 12.5 and 25 µg/mL, respectively, while analogue
55j bearing amino and hydroxy groups as substitutions on the phenyl ring exhibited their inhibitory potential against
S. typhi and
S. aureus with a MIC value of 25 µg/mL. Enhancement in the potency was shown upon the insertion of an electron-withdrawing nitro group in the place of amino group
55k against three bacterial strains with MIC 12.5–25 µg/mL. However, analogue
55l with a diiodo and hydroxyl group at the phenyl ring explored a significant antibacterial potential against
S. typhi,
S. aureus,
E. coli, and
B. pumilus at lower MIC (6.25–25 µg/mL). Compounds bearing a long alkyl chain dodecyl group (
57b) and oleyl group showed good inhibitory potential against the
S. typhi,
E. coli, and
S. aureus bacterial strains, whereas compound
57f with a piperidinyl ring displayed excellent inhibitory potential against
P. aeruginosa,
S. typhi,
E. coli, and
S. aureus bacterial strains with MIC 6.25–25 µg/mL. If we look at the antifungal potential, analogues
55l,
57b,
57c, and
57f displayed significant potency against
C. albicans and
A. fumigatus, with MICs in the range of 6.25–25 μg/mL as compared to standard drugs, while other analogues from the series exhibited poor activity, with MIC in the range of 50 to >200 μg/mL against the fungal strains. The structure−activity relationship (SAR) has stated that compounds bearing a hydroxy group along with electron-donating methyl group
55i, amino group
55j, and, similarly, electron-withdrawing nitro
55k and diiodo
55l on the phenyl ring exhibited higher antibacterial potential, with MIC ranging from 6.25 to 25 μg/mL as compared to the analogues with a monosubstituted phenyl group (
55i–l vs.
55e–h). This may happen due to di- and tri-substituted phenyl frameworks, which possibly make additional intermolecular interactions leading to the improved penetration of the compounds through the bacterial membrane. Among the series compounds,
55l,
57b, and
57f were identified as the most promising analogues and exhibited broad-spectrum antimicrobial activity. For better understanding, molecular docking studies of the synthesized analogues was performed against
A. fumigatus chitinase (PDB ID: 1W9U). Compound
55l explored the highest binding affinity toward a protein with a binding energy −8.44 Kcal/mol. It showed van der Waals interactions with hydrophobic, as well as polar, residues in the binding pocket of the selected protein (
Figure 4).
Mohit Sanduja et al. [
24] designed and synthesized a series of uracil−coumarin-based bifunctional molecular hybrids roped by a 1,2,3-triazole moiety. All the synthesized derivatives were screened for their in vitro antibacterial potency against the
E. faecalis and
S. aureus (Gram-positive) and
P. aeruginosa and
E. coli (Gram-negative) bacterial strains using the agar well diffusion and broth microdilution methods. Target hybrids were synthesized by starting with 4-hydroxy coumarin (
58) on a treatment with dibromoalkanes in the presence of K
2CO
3 in DMF to yield the desired alkylated coumarins (
59). Obtained intermediate
59 on further treatment with NaN
3 in DMF gave azide-substituted coumarins (
60). Additionally, 5-substituted uracils (
61) on a treatment with propargyl bromide in the presence of K
2CO
3 furnished propargylated uracil analogue
62. Finally, the alkylated coumarins (
60) on a reaction with the propargylated uracil analogue (
62) in the presence of copper sulphate and sodium ascorbate afforded target triazole-linked uracil-coumarin hybrids (
63a,b), and it is outlined in
Scheme 8. Among this series of analogues,
63b and
63c were found to possess promising activity against
S. aureus with a zone of inhibition 26 mm and 28 mm. Moreover, both hybrids exhibited equipotent activity against
Enterococcus faecalis and
P. aeruginosa and were found less active against
E. coli. The MIC value of both the potent hybrids was determined by the broth microdilution method against
S. aureus and found to be 7.23 lg/mL for
63b and 11.7 lg/mL for
63c, which was compared to the standard drug levofloxacin (3.12 lg/mL). The SAR was defined for the compounds with electronegative species on the uracil skeleton, and it favoured an antimicrobial potency. The analogues with -F (
63b) and -Cl (
63c) found similar and activity. Moreover, the chain length in between the triazole and coumarin moieties also affected the potency. The analogues with two carbon chain lengths were found potent compared to the candidates with chain lengths with three, four, and five carbons. The docking study demonstrated that compound
63c fit well in the cavity of dihydrofolate reductase (DHFR) (PDB ID: 3SRQ). One of the coumarin rings of
63c formed hydrophobic interactions with residues Leu55, Leu29, and Val32, while a second coumarin moiety formed interactions with residues Gly95, Ile15, and Leu21. The triazole ring nitrogen exhibited hydrogen-bonding interactions with the backbone nitrogen of Ala8, as well as strong
p−
p stacking interactions observed in between the triazole moiety and Phe93, while a second triazole skeleton showed
p−
p stacking interactions with Phe99 (
Figure 5).
Mohsen K.A. Regal et al. [
25] synthesized some new coumarins and dicoumarol derivatives to screen for their in vitro antibacterial potency against Gram-negative
E. coli, Gram-positive
S. aureus, and antifungal activity detected against
C. albicans using the disc diffusion method at a 1 mg/mL concentration. Ampicillin was used as the standard drug for antibacterial and clotrimazole for antifungal action. The reported derivatives were obtained by reacting coumarin
31b with different alkyl halides under phase transfer catalysis conditions using K
2CO
3 as the base and TBAC as the catalyst. This oxygen alkylation through nucleophilic displacement furnished analogues
64a–d and a smaller amount of compound
65 in the case of benzyl chloride. On the other hand, coumarin
31b on the treatment with phenylisothiocyanate via the C
3 addition on the carbon nitrogen double bond of the isothiocyanate yielded 3-(N-phenyl) thiocarbamide coumarin derivative
66. Similarly, coumarin
31b via a one-pot three-component phase transfer catalysis reaction converted into intermediate
67 by a reaction with carbon disulphide and different alkyl halides and/or aroyl halides under K
2CO
3 as the base and TBAC as the catalyst (
Scheme 9a). Coumarin
31b on reaction with aromatic aldehydes such as 3,4-methylene dioxybenzaldehyde and/or 4-methoxy benzaldehyde in an equimolar ratio via condensation gave 3-arylidene-6-methyl-4-oxocomarin
69a,
b under base catalyst piperidine. Repeated reactions using an excess of
31b could yielded the dicoumarol analogues
70a,b in good yields (
Scheme 9b). Compound
69a underwent Michael-type cycloaddition reactions and afforded cycloaddition products of pyranochromene (
71) and pyrano pyridone (
72) when reacted with ethyl acetoacetate in the presence of methoxide and/or ammonium acetate as the base under fusion conditions (
Scheme 9c). The reaction of 3-arylidenes (
69a,
b) and/or dicoumarols (
70a,
b) with hydrazine hydrate in boiling ethanol furnished the corresponding 1,2-dibenzyldene hydrazine derivatives (
73a,
b), whereas
70b converted to pyridine derivatives
75a–c via a cyclocondensation reaction of two carbonyl groups with nitrogen nucleophiles by treatment with ammonium acetate, methyl amine, and/or
p-toluidine under reflux (
Scheme 9d). From the set of synthesized analogues, 81% were found to be active against all tested microorganisms. Compounds
64a,
64b,
66,
70a,
70b,
71, and
72 were proven to be promising results against antibacterial and antifungal potential with activity-indexed values ranging between 50% and 87%. The minimum potential was shown by analogues
64c,d with an activity-indexed value of 40%.
Kavita Bhagat et al. [
26] synthesized the novel indolinedione−coumarin hybrids (
Scheme 10) and screened for their antimicrobial potential against Gram-negative (
E. coli and
Salmonella enterica); Gram-positive (
S. aureus and
Mycobacterium smegmatis) bacterial strains; and four fungal strains (
C. albicans,
Alternaria mali,
Penicillium sp., and
Fusarium oxysporum) by using the agar gel diffusion method by Different substituted indolinedione (
76) on treatment with 1,2-dibromoalkanes by using K
2CO
3 as a base in dimethylformamide to give
77 which on reaction with NaN
3 in DMF at RT furnish 1-(4-azidoalkyl)indoline-2,3-diones (
78). Intermediate 4(prop-2-ynyloxy)-2H-chromen-2-one (
79) was obtained by reacting 4- hydroxycoumarin (
58) and propargyl bromide under basic condition using K
2CO
3. Finally, the obtained intermediate on reaction with different substituted 1-(4-azidoalkyl) indoline-2,3-diones (
78) in the presence of catalytic amount of pentahydrate CuSO
4 and sodium ascorbate yields desired indolinedione−coumarin hybrids
80a–u. From this series, analogue
80b exhibited potent antibacterial activity against
S. aureus and
S. enterica with zone of inhibition 2.5 and 1.3 cm, respectively. Along with most of the hybrids have shown good potential against fungi
Penicillium sp. analogue
80a was found to exhibit excellent antifungal potential with zone of inhibition 2.5 cm. Similarly,
80b was found to be the second promising hybrid with zone of inhibition1.3 cm against
Penicillium sp. SAR analysis revealed that electron density at the fifth position of indolinedione framework greatly impacted antibacterial potential. The screening clarifies that chain length of two carbon atom which links the triazole moiety to indolinedione framework was the most tolerable linker while the unsubstituted indolindione is the highest crucial for antifungal potential. The docking study of the most active analogues from the series, i.e.,
80b, was accomplished on
S. aureus DHFR (PDB ID: 3SRQ). Study demonstrates that compound
80b fits well in the cavity of enzyme and gets stabilized by different electrostatic interactions including major van der Waal’s, π−π stacking, and H-bond interactions (
Figure 6).
Mohd. Shahnawaz Khan et al. [
27] reported an eco-friendly itinerary series of newly substituted chromene-3-carboxamide derivatives. The green synthesis approach involves condensation of substituted salicylaldehyde (
82) with N-(substituted) phenyl malonic acid (
81) in the presence of a base catalyst, piperidine obtained chromene-3-carboxamide derivatives
83a–j (
Scheme 11). The newly furnished derivatives were evaluated for their antimicrobial potential against two bacterial strains,
E. coli (Gram-negative) and
B. cereus (Gram-positive) and seven fungal strains, namely,
A. niger,
A. fumigatus,
A. flavus,
Rhizopus,
Mucor,
Penicillium, and
C. albicans, by the agar well diffusion method using fluconazole as a reference drug. Compounds
83c,
83d,
83e,
83f, and
83j exhibited broad-spectrum antibacterial potential due to the presence of bromo, chloro or nitro substitutions at C6 and C8 position of chromene ring l. The inhibitory potential against
E. coli and
B. cereus at 1000 μg/mL concentration with a zone of inhibition of 10–16 mm was reported. All the analogues from the series were also evaluated for their antifungal potential against seven fungal strains. Among them
83a has shown moderate antifungal activity with MIC 500 μg/mL against
A. flavus,
A. niger, and penicillium, while
83b was shown against
A. niger and
C. albicans with MIC of 500 and 250 μg/mL, respectively. Moreover, compounds
83c and
83f displayed lower MIC value against
A. fumigatus,
A. flavus, Mucor and Penicillium. Results conclude that compounds
83c–g has displayed good to moderate antifungal potency against all tested fungal strains. Some analogues
83d and
83e exhibited excellent MIC of 125 μg/mL.
Milenko N. Ristić and co-workers [
28] carried out synthesis of some novel asymmetric azines containing coumarin analogues. The title hybrids were prepared using the synthetic strategy depicted in (
Scheme 12). The coumarin ketone
84 obtained via acylation of
58 with add full form (GAA) in the presence of POCl
3. Hydrazone of 4-hydroxy-3-acteyl coumarin
85 can be afforded by reaction of
84 with hydrazine hydrate in a 1:1 ratio using ethanol. The title compounds asymmetric azines (
86a–i) were synthesized by reacting
85 with different substituted aromatic aldehydes in absolute ethanol. All the title compounds tested for antimicrobial potential against four Gram-positive, three Gram-negative bacterial strains and two fungal strains. Found results shows that all the compounds were effective inhibitors of microbial growth against all the tested strains except
86i which is active against
C. albicans. Compounds
86a and
86f displayed highest antimicrobial potency with MIC value of 1.32 and 1.4 µmol mL
−1, respectively. Among the series compound
86c found to be potent antifungal candidate.
Megharaja Holiyachi et al. [
29] reported one-pot multi-component synthesis of tri and tetra-substituted coumarin-imidazole hybrids. Synthesized derivatives were tested for their antimicrobial potential against
Bacillus flexus (Gram-positive)
Pseudomonas spp. (Gram-negative) bacterial and
Scopulariopsis spp. and
Aspergillus terreus fungi strains using via the well diffusion method using ciprofloxacin and nystatin as reference drugs. Compounds were prepared as described in (
Scheme 13). Reaction of mixture of 4-formylcoumarins, substituted anilines and 1, 2-diketone (
87) with ammonium citrate under reflux condition in the presence of acid catalyst has yielded title analogues
88a–m. Likewise analogues
90a–f were obtained using the similar above-mentioned reaction conditions. Further corresponding ester
91a–f were furnished by treatment of
90a–f to sulphuric acid in methanol under reflux. Derived results have shown that most of the analogous display promising activity against both bacterial strains. Analogues having
N-1-phenyl substation on imidazole and substitutions on coumarin ring
88a–g possess excellent antibacterial potency with MIC values of 0.1 to 0.4 µg/mL except compound
88b, while analogues bearing nitrobenzene substitution on N-1 imidazoles
88h–m displayed higher activity. Compounds
91a–f were also found to be promising inhibitors against both tested strains of bacteria at low concentrations. All synthesized candidates were screened for antifungal potency and found excellent inhibitors of
Scopulariopsis spp. and
Aspergillus terreus fungi strains. These antifungal results are almost like that of the antibacterial potential. SAR studies revealed from the series
88a–g that compound
88b with (R1-CH
3 and R-H) at
N-1-phenyl substitution was inactive, however introduction of electron-withdrawing species in
88i (NO
2),
90b (COOH), and
91b (COOCH
3) on the
N-1-phenyl ring exhibited potent antibacterial and antifungal activity. It indicates that for antimicrobial potential C7 substitution at coumarin nucleus and electron-withdrawing groups on
N-1-phenyl ring at
p-position is necessary.
Farzanabi Shaikh et al. [
30] carried out the synthesis and evaluation of substituted coumarin and phenyl-1,2,4-triazolidine-3-thiones for their antitubercular and antimicrobial activity. Title analogues were prepared by two component rection of coumarin, phenyl and thiosemicarbazide using PEG-400. Initially intermediate thiosemicarbazone were obtained via nucleophilic addition of thiosemicarbazide (
43) to the carbonyl carbon of substituted 4-formylcoumarine/benzaldehyde, followed by intramolecular nucleophilic attack of –NH
2 of thiosemicarbazone to azomethine carbon to furnish title analogues coumarin and phenyl 1,2,4-triazolidine-3-thion
92a–i and
93a–e (
Scheme 14). The title compounds were tested for their antibacterial potential against Gram-positive (
S. aureus and
B. subtilis) and Gram-negative (
E. coli and
P. aeruginase) and four fungal strains, namely,
C. albicans,
A. niger,
A. flavus, and
A. fumigate by using broth dilution method. From the obtained results it clears that all compounds were active against both Gram-positive and Gram-negative bacterial strains, as well as all tested fungi. Amongst the coumarin-fused triazolothiones (
92a–i) series compounds
92a,
92b,
92c,
92d, and
92i found to be highly potent against
S. aureus and
Bacillus spp., as well as
E. coli, with MIC values ranging from 0.8 to 1.6 μg/mL while, standard ciprofloxacin exhibited MIC 2 μg/mL, whereas the in vitro antifungal evaluation concludes that all the hybrids displayed outstanding potential against fungal strains with MIC values ranging from 0.4 to 6.25 μg/mL. Molecular docking was performed against the
E. coli FabH target protein (PDB ID: 1HNJ) to support given results. Compound
92d makes three hydrogen bonding interactions with amino acids residues THR28, ASP27, ARG151, and compound
92i makes interaction with amino acids THR28, ASP27, and ARG151, while compound
92a against active site of enzyme cytochrome P450 14 alpha-sterol demethylase (CYP51; PDB ID: 1EA1) makes hydrogen bonding interaction with residues ARG96 and HEM460, as shown in (
Figure 7).
Mohd Imran [
31] synthesized benzimidazole-based coumarins derivative for their antimicrobial and antioxidant activity. A mixture of substituted 2-(propylthio)-1H-benzo[d]imidazole (
94a,b) and substituted 3-(2-bromoacetyl)-2H-chromen-2-one (
95a–e) was stirred in acetone furnished title analogues 2-butylthio-1H-benzimidazole-based coumarin derivatives (
96a–j) outlined in
Scheme 15. All the synthesized compounds were screened for their antibacterial potential against
S. aureus (ATCC-25923),
E. coli (ATCC-25922),
E. faecalis (ATCC-29212),
K. pneumoniae (ATCC700603),
C. albicans (ATCC-2091), and
P. citrinum (NCIM-768). Compounds having –F, -Cl, and –Br groups at C-6 of the coumarin moiety together with –OCH
3 at C-5 of the benzimidazole found to be potent against tested microorganism.
Chirag G. Naik et al. [
32] worked on the synthesis of some novel coumarin fused pyrazolones and methyl thiazole derivatives for their antimicrobial potential. 3-(2-bromoethyl)-4-hydroxy-2
H-chromen-2-one (
97a,b) were obtained from 4-hydroxy coumarin and dibromo ethane in the presence of base. Synthesis of
99a,b was done by reacting corresponding
97a,b with ethyl-4,5-dihydro-2-(4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (
98) in acetone. Analogues
100a and
100b were prepared by reaction of 4-hydroxy coumarin with 3-methyl-1- phenyl-1H-pyrazol-5(4H)-one and 3-methyl-1H-pyrazol-5(4H)-one in the presence of K
2CO
3 (
Scheme 16). Synthesized derivatives were evaluated for antimicrobial potential against
S. aureus,
Bacillus subtilis (Gram-positive) and
E. coli,
P. aeruginosa (Gram-negative) by using agar diffusion method and fungi
C. albicans (MTCC 227) using Sabouraud dextrose agar medium. Compounds
99a and
99b were found to possess significant inhibitory potential against
P. aeruginosa and
E. coli, while compounds
99b and
100a displayed good to moderate activity against
Bacillus subtilis. All the compounds revealed moderate potency against
Candida albicans (fungi).
Asha V. Chate and co-workers [
33] reported a series of novel coumarin-linked pyrazoline inhibitors of D-alanine-D-alanine ligase via one-pot four component synthesis and screened for their antimicrobial potential against
E. coli DdlB ligase. The outline for synthesis of title analogues is depicted in (
Scheme 17). Title hybrids
101a–l and
101s–w and
101m–r were obtained via one-pot four component synthesis between salicylaldehyde, ethyl acetoacetate, hydrazine hydrate, and benzaldehyde in water in the presence of cyclodextrin. Prepared analogues were subjected to in vitro evaluation for antibacterial and antifungal potential against human pathogenic bacterial strains (
E. coli,
B. subtilis,
S. aureus) and fungal strains (
C. albicans,
Candida glabrata,
A. fumigates,
A. flavus,
A. niger and
C. neoformans) using Miconazole as a standard drug. In vitro antibacterial analysis revealed that compound
101f is potent and has displayed excellent potential with MIC values of 14 µg mL
−1, 14 µg mL
−1 and 32 µg mL
−1 against
E. coli 1411,
E. coli SM1411 and
S. aureus NCIM-2901 respectively. The results are comparable to the standard drug D-cycloserine. Compound
101g has proven to be the second best from the series with MIC of 16 µg mL
−1, 18 µg mL
−1 and 40 µg mL
−1 against
E. coli 1411,
E. coli SM1411 and
S. aureus NCIM-2901, respectively. Moreover, compounds
101i and
101h also displayed good potential among the series. Similarly, compound
101j having thiophene moiety displayed excellent antifungal potential than the standard drug miconazole against
C. albicans,
C. glabrata,
F. oxysporum, and
A. fumigates with MIC values 20 µg mL
−1, 20 µg mL
−1, 22 µg mL
−1, 34 µg mL
−1, 12 µg mL
−1, 14 µg mL
−1 and 12 µg mL
−1 respectively. The SAR analysis revealed that candidates
101m–r with substitution on ‘NH’ group of pyrazole moiety are inactive or displayed less antibacterial potential as compared to the compounds having no substitution on ‘NH’ group of pyrazole moiety (
101a–l). Candidates having di-chloro substitution on phenyl ring (
101i) was found more active than compound
101h with
p-chloro substitution on the phenyl ring. Analogue
101c bearing 3,4,5-tri-methoxy substitution on the phenyl ring exhibited more antifungal potency than compound
101b containing 3,4-dimethoxy substitution on the phenyl ring. Compound 3-(5-(3-methoxynaphthalen-2-yl)-4,5-dihydro-1Hpyrazol-3-yl)-2H-chromen-2-one) (
101e) also found to possess good potential against tested fungi.
Coumarin bearing dithiocarbamate derivatives were synthesized and evaluated by their in vitro antimicrobial potential against both Gram-positive, Gram-negative bacterial strains by S.N. Mangasuli et al. [
34]. The study includes evaluation against bacterial strains, namely,
S. aureus,
B. subtilis,
E. coli, and
P. aeruginosa, and fungal strains, namely,
A. flavus,
Trichoderma harzianum, and
Penicillium chrysogenum, and yeast
C. albicans using the macro-dilution broth method. Bromoalkoxy-chromen-2-ones (
102) were obtained by treatment of aliphatic dibromo species with
58 in the presence of K
2CO
3 in DMF. Condensation of 4-(2-bromoethoxy)-2Hchromen-2-one with dithiocarbamate salt in ethanol furnished desired 2-(2-oxo-2H-chromen-4-yloxy) ethyl pyrrolidine1-carbodithioate analogues
103a–l (
Scheme 18). Compound
103i was found to be the most potent from the series with MIC of 0.5 mg/mL against
S. aureus, 1 mg/mL against
B. subtilis, 2 mg/mL against
E. coli and
P. aeruginosa. Likewise, compound
103h displayed good potential against
S. aureus,
B. subtilis and
P. aeruginosa with MIC of 1 mg/mL and against
E. coli with MIC of 1 mg/mL. Further, most of the compounds from the series have shown good to moderate antibacterial activity as compared to the standard. Moreover, prepared candidates were screened for their antifungal potential. Among them compound
103i displayed excellent potential against
A. flavus,
T. harzianum and
C. albicans with MIC 1 mg/mL and
C. albicans with 0.5 mg/mL which is very less MIC value as compared to the standard. Similarly, compound
103h exhibited activity against
A. flavus,
T. harzianum and
C. albicans with MIC of 1 mg/mL and 0.5 mg/mL against
P. chrysogenum. SAR studies explored that candidate
103i with electron-donating species (-CH
3) at
p-position of piperidine framework of dithiocarbamate linked via alkyl ether linkage -O(CH
2) nS- (
n = 4) to coumarin scaffold displayed lower MIC 0.5 mg/mL as compared to the standard analogue. Likewise, the analogue
103i has shown a C-score value of 7.57 which is superior in comparison to the standard fluconazole. SAR also revealed variation in alkyl chain of the respective derivatives (
n = 4) with different substituents found excellent in activity in comparison with alkyl chain of
n = 2. Docking results have shown that all the docked analogues exhibited good docking score against
C. albicans DHFR (PDB ID: 1Al9). Active compounds from series
103h makes hydrogen bonding interactions with amino acid residues ARG56, SER78, ARG79, LYS57 and
103i with ARG79, LYS57 (
Figure 8). The study has proposed mechanism of action for coumarin bearing dithiocarbamate analogues by inhibiting the dihydrofolate reductase enzyme.
In another study S. N. Mangasuli and co-workers [
35] contributed to synthesis of novel coumarin-theophylline hybrids and screened their antitubercular and antimicrobial potential. The synthesis methodology is outlined in (
Scheme 19) were starting from substituted 4-bromomethyl coumarin were obtained via Pechmann cyclization of phenol with 4-bromoethylacetoacetate in sulphuric acid. The furnished substituted 4-bromomethyl coumarins upon condensation with theophylline (
104) in the presence of anhydrous K
2CO
3 in acetone yield 1,3-dimethyl-9-[(substituted-2-oxo-2Hchromen-4-yl) methyl)-1H-purine-2,6(3H,9H]-dione derivatives (
105a–j). The prepared derivatives were screened for their in vitro antimicrobial potency against
S. aureus (Gram-positive) and
E. coli, and
S. typhi (Gram-negative) bacteria, as well as fungi
C. albicans. Among the series, compound
105a with 6-CH
3 substitution was found active against
S. aureus,
E. coli with MIC of 3.9 µg/mL and against
S. typhi with MIC of 7.8 µg/mL. Additionally, compound
105c with 5,6-Benzo,
105f with 6-OCH
3, and
105j with 6-tert-butyl substitutions displayed good antibacterial potency against
S. aureus and for
E. Coli with MIC of 7.8 µg/mL, 7.8 µg/mL, and 3.9 µg/mL, respectively. The antiviral potency was compared with Amphotericin B as the standard drug. From the obtained results, compounds
105a (6-CH
3),
105b (7-CH
3), and
105f (6-OCH
3) were found most active against
C. albicans with a MIC value of 31.3 µg/mL. Additionally,
105c,
105d,
105i, and
105j displayed sensible potential against
C. albicans with MIC value of 62.5 µg/mL. The SAR studies have shown that presence of electron-donating species substituted to C-6 of coumarin moiety contributes towards the antibacterial potential.
Nilesh B. Chauhan et al. [
36] reported a new series of 4-methyl-6-nitro-2-oxo-2
H-chroman-7yl-2-(4-(4-fluorophenyl)-6-phenyl-2
H-1,3-thiazin-2-yl-amino) acetates (
Scheme 20). Compound
8b was synthesized via Pechmann condensation, which, on nitration with nitric acid, obtains
106, followed by a reaction with chloroacetyl chloride, gives 4-methyl-3-nitro-2-oxo2H-chromen-7-yl 2-chloroacetate (
107). Title derivatives
111a–j were obtained by condensation of
107 with amino thiazine derivatives (
110a–j) via cycloaddition reaction in between substituted chalcones (
109a–j) and thiourea. The test compounds were screened for their antimicrobial potential against four bacterial and three fungal strains using broth microdilution method and compared with chloramphenicol, ciprofloxacin, and griseofulvin as a standard. Analogue
111c attached to -Cl and
111h attached to –CH
2CH
2CH
3 at C-4 of benzaldehyde were found to be more active against
E. coli with MIC value of 50 µg/mL and the results were compared to standard drugs chloramphenicol and ciprofloxacin, whereas compounds
111a (-H) and
111b (2-Cl) displayed significant antifungal potential against
S. pyogenes with MIC value of 250 µg/mL compared with griseofulvin. Other test analogues were found to be less to moderately active.
Synthesis of some coumarin-piperazine derivatives was carried out by Shrinivas Koparde et al. [
37]. Outline for synthesis of title compounds is depicted in (
Scheme 21). Substituted 4-bromomethyl coumarins were obtained via Pechmann cyclization of phenols with 4-bromoethylacetoacetate. Title coumarin-piperazine derivatives (
113a–h) were prepared by condensation of obtained substituted 4-bromomethyl coumarins with 1-(4-(4-hydroxyphenyl) piperazin-1-yl) ethanone (
112) in the presence of K
2CO
3 using DMF as solvent. Label hybrids evaluated for their in vitro antimicrobial activity against both Gram-negative and Gram-negative bacterial strains, as well as four fungi. Among the series compound
113a was exhibited excellent potential against
S. aureus and
E. faecal (MIC = 0.5 µg/mL) and with
E. coli and
P. aeruginosa with MIC value 1 µg/mL, whereas
113d and
113f were found as good in action against
S. aureus and
E. faecal with MIC value 2 µg/mL. Antifungal potential of the test compounds was evaluated against
A. flavus,
P. chrysogenum,
Trichoderma harzianum, and
C. albicans. Obtained results have concluded that displayed compound
113a has excellent activity against
A. flavus and
T. harzianum with MIC of 0.5 µg/mL and against
P. chrysogenum and
C. albicans with MIC value of 1 µg/mL, which is equivalent to the standard fluconazole. Moreover, compounds
113d,
113f, and
113h exhibited good potency with MIC 2 µg/mL against
A. flavus,
T. harzianum and
P. chrysogenum when compared to the standard. Further, molecular docking studies were encouraged and supported the results of in vitro antimicrobial activities, the compounds
113a,
113e,
113f, and
113h have higher C score values than that of the standard drug ciprofloxacin.
V.K.A. Kalalbandi et al. [
38] have illustrated in vitro antimicrobial potency of some dihydropyran-bis coumarins. 6-formylcoumarins (
114a) were synthesized via the Reimer−Tiemann reaction in the presence of chloroform and KOH. 3-formyl-4-cholorocoumarin (
114b) furnished through the Vilsmeier−Haack reaction of 4-hydroxy coumarin in POCl
3-DMF and 8-formyl-7-hydroxy-4-methylcoumarin (
114c) prepared via Duff reaction of 7-hydroxy-4-methylcoumarin. Title compounds dihydropyran-bis coumarins (
115a–i) were synthesized via one pot multicomponent reaction of different substituted hydroxycoumarins, formyl coumarins (
114a–c) and malononitrile in the presence of catalytic amount of triethylamine in methanol (
Scheme 22). The in vitro antimicrobial activity was carried out against bacterial strains, namely,
E. faecalis,
S. aureus,
P. aeruginosa, and
E. coli, and fungi
C. albicans and
A. Niger using broth micro dilution method. Biological study exposes most of the derivative active against Gram-positive bacterial strains and compounds
115a and
115b have emerged as a most potent form the synthesized hybrids by exhibiting MIC of 0.4 and 0.8 mg mL
−1, respectively, against
S. aureus as compared to the standard ciprofloxacin. Additionally,
115a was found active against Gram-negative strain
P. Aeruginosa, while the rest of the test compounds were moderately active against remaining microorganisms. On the other hand, compounds
115a and
115b also found significantly active against
C. albicans with MIC values 0.8 and 0.4 mg mL
−1, respectively. The SAR study concluded that core biscoumarin attached dihydropyran moiety with no substitution found better in potency against Gram-positive strains as compared to the substitution on core biscoumarin attached dihydropyran framework.
Jyoti M. Madar and co-workers [
39] developed a new series of structurally identical coumarin triazoles and evaluated for their cytotoxic and antimicrobial action. Initially, substituted 4-bromomethyl coumarins obtained via Pechmann cyclization which is converted into substituted dipolar coumarin azides by reaction with sodium azide in acetone. 1,4,5-trisubstituted-1,2,3- triazoles (
116a–f) were prepared via base catalysed cycloaddition reaction of substituted dipolar coumarin azides with malononitrile (active methylene compounds). In the same way, coumarin triazole carboxylate (
117a–f) were obtained by the reaction of substituted coumarin azide with ethylacetoacetate in the presence of NaH in THF via reflux. The yielded compounds
117a–f were converted to coumarin triazole carboxylic acid (
118a–f) using NaOH under reflux (
Scheme 23). The newly furnished analogues
116a–f,
117a–f and
118a–f were screened for their in vitro antimicrobial activity against different bacterial and fungal strains. Out of the tested hybrids from the series of coumarin triazole amino carbonitriles (
116a–f), compounds
116a,
116b,
116d, and
116e were proved as excellent inhibitors against
S. aureus with MIC values 0.4, 0.8, 0.4, and 0.4 μg/mL, respectively, when compared to the standard Ciprofloxacin with MIC value 2 μg/mL. In addition,
116a,
116c and
116e displayed good potential against
E. coli with MIC value 3.12, 12.5 and 1.6μg/mL, respectively. From the series of coumarin triazole carboxylate (
117a–f), compounds
117d,
117e, and
117f exhibited excellent potency against
S. aureus with MIC values 0.8, 0.8 and 0.4 μg/mL, respectively, when compound
117b and
117e displayed good potential against
E. coli with MIC 3.12 and 12.5 μg/mL when compared to the standard. Furthermore, from the series
118a–f, three analogues
118d,
118e, and
118f found excellent activity against
S. aureus with MIC values 0.2, 0.8 and 0.4μg/mL, respectively. Wherein candidate
118d has proven to be good in activity against
E. coli. The antifungal screening has revealed that among the 18 furnished candidates,
116a,
116b,
116d,
116e,
117f, and
118e emerging as excellent candidate with MIC values 0.4, 12.5, 3.12, 1.6, 6.25, and 6.25 μg/mL, respectively, against
A. niger compared to the standard fluconazole. SAR study revealed that electron-releasing groups (CH
3 and OCH
3) at C5, C6 and C7 of coumarin moiety contributes to the antibacterial potency of the title compounds.
Priscila López-Rojas et al. [
40] analysed in vitro antimicrobial activity of novel series of 4-substituted 1,2,3-triazole-coumarin derivatives. The synthesis of title analogues is depicted in (
Scheme 24). When 4-hydroxy-coumarin (
58) reacted with 3-bromoprop-1-yne in the presence K
2CO
3 in anhydrous acetone it gives O-propargylated coumarin (
79). Similarly, 4-bromo-coumarin (
120) on reaction with prop-2-yn-1-amine via nucleophilic substitution reaction in DMF afforded N-propargylated coumarin (
121). The title 4-substituted 1,2,3-triazole-coumarin derivatives (
119a–m and
122a–m) prepared via copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction of corresponding O-propargylated coumarin (
79) and N-propargylated coumarin (
121) with different substituted alkyl or aryl azides, respectively. The synthesized analogous were tested against
S. aureus (ATCC 6538),
E. faecalis (PCM 2673),
E. coli (ATCC 8739),
K. pneumoniae and yeast
C. albicans (ATCC 10231) and were compared with the standard chloramphenicol and ketoconazole. Among the screened series, compounds
119a,
119b,
119f,
122h and
122k have exhibited promising activity with MIC ranging from 12.5 to 50.0 µg/mL against
E. faecalis. Compound
119b with 2-OMe–Ph group attached at the triazole moiety and an –OCH
2– linker remained the finest candidate of the series. In the nitrogenated series candidate
122h with 3-NO
2–Ph and
122k with an undecyl chain has proven to possess the best potential.
Antibacterial, antitubercular, and antiviral activity of a new thiazolyl-coumarin hybrids was executed by Hasnah Osman and co-workers [
41]. Initially, the substituted 3-acetylcoumarins on bromination with ethanol free chloroform afford key intermediates substituted-3-(2-bromoacetyl)-2H-chrome-2-ones (
123a,b). Subsequently thiazolyl-coumarin analogues (
124a–o) were obtained via Hantzsch cyclization of the corresponding intermediate
123a,b with
N-substituted and
N,
N-disubstituted thiourea (
Scheme 25). Analogues were tested by in vitro antibacterial activity against two bacteria
S. pneumonia and
S. aureus (Gram-positive) and
E. coli,
E. aerogenes and
S. typhi (Gram-negative) by using broth microdilution method. The obtained results were compared with the standard drugs streptomycin, kanamycin, and vancomycin. Out of the tested hybrids, compounds
124d,
124h,
124k, and
124m have exhibited promising antibacterial action. The bromophenol analogues
124d (MIC 79 µM) and
124h (MIC 73 µM) displayed highly significant inhibition against all the tested microorganisms with MIC values fairly less than that of vancomycin (MIC 86–176 µM). The disubstituted
N,
N-diphenyl candidate
124l exhibited very low potency (MIC 158–316 µM). However, replacement with bulky alkyl groups led to compounds retaining their antibacterial potency such as
124k (MIC 115–230 µM) and
124m (MIC 49–98 µM). From the obtained results, the SAR analysis revealed that mono-substituted thiazoles remained more effective as compared to the di-substituted thiazoles. Candidates having lipophilic electron-withdrawing bromo group at the phenyl ring displayed promising potential while substitution of electron releasing (-OCH
3) group at coumarin moiety is contributing towards increased potency.
A new series of coumarin−carbonodithionate hybrids were contributed by Sumitra N. Mangasuli and co-workers [
42] to evaluate their in vitro antimicrobial potential. The preparation strategy is as depicted in (
Scheme 26). The mixture of 4-hydroxy (
58a) and 7-hydroxy coumarins (
58c) treated with various dibromoalkanes and anhydrous K
2CO
3 in DMF gives bromoalkoxy coumarins (
125a–d). The obtained intermediates
125a–d when condensed with potassium O-ethyl/methyl carbonodithioate in absolute ethanol furnished 4- and 7-substituted coumarin-carbonodithioate hybrids (
126a–d and
127a–d) via microwave irradiation technique. Prepared candidates were tested for their in vitro antimicrobial potential against
S. aureus,
B. subtilis,
E. coli and
P. aeruginosa and fungi
A. flavus,
T. harzianum, and
P. chrysogenum, as well as yeast C.
albicans.
Among the synthesized compounds, 126c and 126a have displayed excellent antibacterial potency against S. aureus and B. subtilis with MIC value 0.5 µg/mL and 2 µg/mL, respectively, and against E. coli and P. aeruginosa with MIC 1 µg/mL and 4 µg/mL, respectively. Moreover, analogues 126b and 126d have displayed good to moderate potential against tested pathogens. Compound 126c also demonstrated potential against A. flavus, T. harzianum, and P. chrysogenum (MIC 0.25 µg/mL) and against C. albicans with (MIC 1 µg/mL), while candidate 126b was found to possess significant activity against A. flavus, T. harzianum, and P. chrysogenum (MIC 0.5 µg/mL) and C. albicans with (MIC 1 µg/mL) as compared to the standard fluconazole, while the rest of the test compounds were found good to moderately active against tested fungal strains.
Megharaja Holiyachi and co-workers [
43] designed and synthesized by one pot multicomponent method some coumarin–imidazole analogues and phenyl imidazoloacrylates for both antimicrobial and anti-inflammatory potential. Initially, coumarin–imidazole hybrids (
128a–g) were prepared by reacting 4-formycoumarin and 1,2-diketone (
87) in the presence of the ammonium citrate as catalyst via conventional method. The next step obtained
128a–g on treatment with
p-toluenesulfonyl chloride under triethylamine and yields coumarin– imidazole sulphonamides (
129a–f). On the other hand, analogues
128a–g on treatment with sodium hydroxide via ring opening mechanism produced analogues
130a–f which is isolated in the form of sodium salts (
Scheme 27). The assay was performed against
Bacillus flexus (Gram-positive) and
Pseudomonas spp. (Gram-negative) bacterial strains, as well as fungi
Scopulariopsis spp. and
Aspergillus terreus via the agar well diffusion method using ciprofloxacin and nystatin as the reference candidates. The derived results conclude that all the screened analogues are active against both the tested bacterial strains compared to the standard drug. Compounds
128c with –OCH
3 and
128e having –Cl at C-6 of coumarin displayed excellent potential with MIC value 0.3 μg/cm
3. N-sulfonyl bearing imidazole derivatives (
129a–f) has shown slight increase in the potential compared to compounds
128a–g. Compound
129d with bromo substitution at C6 position of coumarin and
129f having 7,8-benzo substitution on the coumarin with MIC value 0.2 μg/cm
3. The newly formed modified coumarin–imidazole hybrids to sodium salt of phenyl acrylate of imidazole
130a–f, were found highly active against both tested bacterial strains compared to the standard and the other series of analogues
128a–g and
129a–f.
Among this series compound
130c having –OCH
3 and –OH on benzene,
130e with chloro substitution and –OH on benzene ring and
130f emerged as highly potent with MIC 0.1 μg/cm
3. From the above evidence, analogues
130a–f displayed excellent potential against both bacterial strains, and it might be the ionic nature of molecules which are completely soluble in water, while, against the tested fungal strains, analogues
128a,
128b,
128e, and
128g found active with MIC ranges from 1 to 4 μg/cm
3. In case of series
129a–f, all the complexes exhibited promising activity against both fungi except
129d having bromo substitution at C6 position of coumarin, whereas improved activity was observed in case of series
130a–f with MIC value ranging from 1 to 2 μg/cm
3. Further, to explore antimicrobial potential of the synthesized analogues molecular docking study was perform against
C. albicans dihydrofolate reductase (PDB IDs: 1AI9) and
A. fumigatus N-myristoyl transferase (PDB ID: 4CAW; A Chain). Among the series compounds
130a–f exhibited very good interaction with the selected enzyme target. Compound
130a shows six hydrogen bonding interactions with cavity of enzyme 1AI9 involved amino acids residues LYS57 and ARG79 and three others with ARG56, while compound
130c displayed hydrogen-bonding interactions with amino acids, namely, LYS57, ARG79, and GLU116, in the active pocket of the enzyme. Moreover, compound
130e has shown interaction at the active site of N-myristoyl transferase (PDB: 4CAW; A-Chain with amino acids GLY25 and GLY251) (
Figure 9). All the three analogues displayed better C score values against the enzyme (PDB ID: 1AI9).
Several dimers of coumarin-1,2,3-triazole hybrids bearing alkyl spacer were synthesized and evaluated for their antimycobacterial and antimicrobial potency by A. Dongamanti et al. [
44]. An outline for the synthesis of new dimers is presented in (
Scheme 28). 6-substituted-7-hydroxycoumarins (
131a,b) were prepared via Pechmann condensation of resorcinols with ethyl acetoacetate. 6-substituted-4-methyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-ones (
132a,b) were obtained by the reaction of the corresponding
131a,b with propargyl bromide in the presence of potassium carbonate in acetone under reflux. Coumarin–1,2,3-triazole hybrids (
133a–j) were afforded via nucleophilic substitution reaction of dibromoalkanes with sodium azides to produce in situ diazidoalkanes, which is coupled with
132a,b by copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition reaction. The in vitro antimicrobial screening was performed against
B. subtilis,
S. aureus,
E. coli and
Proteus vulgaris bacterial strains and
A. niger and
C. albicans fungal strains via Agar well-diffusion method using gentamicin and fluconazole as reference drugs.
Among the series compound 133j was found to be highly active antibacterial candidate against B. subtilis, S. aureus and E. coli (MIC 3.125 µg/mL) with zone of inhibition 19, 16 and 14 mm, respectively. Additionally, compounds 133e and 133i were found as promising inhibitors against tested organism with MIC value of 6.25 µg/mL, while analogues 133d and 133j displayed good antifungal potential (MIC 12.5 µg/mL) with a zone of inhibition 12 and 14 mm against A. niger and 14 and 10 mm in contrast to C. albicans, respectively. Along with that, hybrids 133e and 133i exhibited good to moderate potential (MIC 12.5–25 µg/mL) with inhibitory zone of 12 mm, 16 mm and 10 mm, 11 mm, respectively. The SAR study proposed that compounds having lipophilic n-octyl and n-hexyl spacer and chloro substitution on the coumarin moiety were found to be promising for antimicrobial potential.
LR Singh et al. [
45] analysed in vitro antibacterial activity of coumarin-benzimidazole conjugates. The reported compounds were synthesized by a procedure depicted in (
Scheme 29). The salicylaldehyde and ethyl acetoacetate in the presence of piperidine was converted into intermediate
134 via Knoevenagel condensation, which on bromination in dry chloroform yields compound
135. The obtained intermediate
135 was reacted with benzimidazole in acetonitrile gave
136 via nucleophilic substitution reaction. Later condensation of
136 with hydroxyl amine hydrogen chloride under refluxed furnish key intermediate
137. Finally, the title analogues
138a–k were achieved by the reaction of
137 with different substituted benzyl halides under potassium tert-butoxide in DMSO via etherification. The synthesized derivatives were screened against
B. subtilis,
S. aureus (Gram-positive) and
P. aeruginosa,
Proteus vulgaris,
E. coli (Gram-negative) bacterial strains. MIC values were established by using ampicillin, kanamycin, tetracycline, and ciprofloxacin as standard drugs. The derived biological data represents, compounds
138a and
138c have efficiently inhibited growth against the
B. subtilis with MIC values 0.95 and 6.25 μg ml
−1, respectively, while compounds
138c,
138f,
138j and
138k exhibited better potential against
P. vulgaris with MIC values 1.56, 3.12, 1.56 and 1.56 μg mL
−1, respectively, along with compound
138a active against
P. aeruginosa (MIC 3.12 μg mL
−1) as compared to the standard ampicillin. Compound
138a was found active against Gram-negative strains (
S. aureus, and
E. coli with MIC 1.56 and 3.12 μg mL
−1, respectively). The SAR of the synthesized compounds revealed molecules with halogen species could display antibacterial potential. Compound
138a having chlorine species at para position of coumarin–benzimidazole framework found to have highest broad spectrum antimicrobial potential.
Synthesis of novel 3-((dicyclohexylamino)-(substituted phenyl/heteryl)-methyl)-4-hydroxy-2H-chromen-2-onederivatives (
140a–o) was carried out under solvent-free condition and there in vitro antimicrobial potential was evaluated by Shailee V. Tiwari et al. [
46]. The title analogues were prepared via three component reactions of appropriate aldehydes, 4-hydroxy coumarin (
58) and dicyclohexylamino (
139) in the presences of [Et
3NH] [HSO
4] acting as a catalyst and a solvent (
Scheme 30). The synthesized coumarin analogues were assessed for their antimicrobial potential against bacterial strains, namely,
E. coli,
B. subtilis, and
S. aureus, and fungal strains
C. albicans,
C. glabrata,
F. oxysporum,
A. fumigates,
A. flavus,
A. niger, and
C. neoformans via standard agar method using ampicillin and miconazole as reference drugs. Out of the test compounds, analogues
140e having 2,4-difluoro group on the phenyl ring was found to be most active against
E. coli (MIC 48 µg/mL),
B. subtilis (MIC 50 µg/mL) and
S. aureus (MIC 52 µg/mL). Furthermore, compound
140c with 2,6-dichloro group on the phenyl ring has displayed excellent potency against
E. coli,
B. subtilis and
S. aureus with MIC values 50, 48 and 50 µg/mL, respectively. The archived results revealed that analogues bearing electron-withdrawing substitutions such as
140e (2,4-difluro),
140d (4-fluro),
140c (2,6-dichloro) and
140b with 4-chloro on the phenyl ring with coumarin, dicyclohexylamine, and β-aminocarbonyl framework build up the antibacterial potential. Similarly, the obtained hybrids
140b,
140c,
140d and
140e with electron-withdrawing groups displayed significant in vitro antifungal potential against
A. fumigates,
A. flavus and
A. niger. Interestingly, compound
140l having 4-hydroxy-3-ethoxy on the phenyl ring has proven to be the most active antifungal agent from the series against
C. albicans,
C. glabrata,
Fusarium oxysporum,
A. fumigates,
A. flavus,
A. niger, and
C. neoformans with MIC values 25, 28, 28, 36, 15, 12, and 12 µg/mL, respectively, while analogue
140e seemed to be equally active against
A. fumigates,
A. flavus,
A. niger, and
C. neoformans when compared with the standard miconazole.
Renuka et al. [
47] reported antimicrobial and antioxidant activity of coumarin appended pyrazolyl-1,3,4-oxadiazoles and pyrazolyl-1,3,4-thiadiazoles. Initially, 1-aryl-3-(7-hydroxy-4-methyl-2- oxo-2H-chromen-8-yl)-1H-pyrazole-4-carboxaldehydes (
141a–e) was treated with semicarbazide hydrochloride (
142) in the presence of sodium acetate in ethanol under reflux to furnish the corresponding semicarbazones (
143a–e), which is via oxidative cyclization using bromine in acetic acid gives 1,3,4-oxadiazoles (
144a–e) (
Scheme 31a). In the similar manner, precursors
141a–e when reacted with thiosemicarbazide hydrochloride (
145) in the presence of sodium acetate gives thiosemicarbazones (
146a–e), which was converted into 1,3,4-thiaoxadiazoles (
147a–e) on reaction with
146a–e via oxidative cyclization using bromine in acetic acid (
Scheme 31b). Among the series, chloro-substituted thiosemicarbazone was found to be excellent against all the tested pathogens, while methyl substituted thiosemicarbazone displayed good potential against
E. coli.
Samina Khan Yusufzai and co-workers [
48] investigated in vitro antibacterial and antitubercular potential of some new hydrazinyl thiazolyl coumarin hybrids. Various 3-acetyl coumarins (
149a–g) were prepared by the condensation of selective salicyladehydes (
148a–g) with ethylacetoacetate in catalytic amount of piperidine, which on treatment with thiosemicarbazide furnishes coumarin thiosemicarbazone analogues (
150a–g). The second precursor 3-(2-bromoacetyl)-2H-chromen-2-one (
151a,b) was produced via bromination of 3-acetyl coumarins (
149a and
f). The title analogues
152a–k were synthesized via Hantzsch cyclization of coumarin thiosemicarbazones (
150a–g) and α-bromo-3-acetyl coumarins (
151a,b) by cyclocondensation in CHCl
3/EtOH followed by treatment with NH
4OH (
Scheme 32). In vitro antibacterial evaluation was carried out against
S. pneumoniae,
S. aureus (Gram-positive) and
E. coli,
E. aerogenes,
S. typhi (Gram-negative) bacterial strains by employing colorimetric microdilution method. Among the newly furnished hybrids, Compound
152c has displayed highest antibacterial potential against
S. typhi,
S. pneumoniae, and
S. aureus with MIC values in the range of 31.25–62.5 μg/mL and are comparable to the standard kanamycin. Along with that compounds
152i,
152j, and
152k has also exhibited good to moderate potential against all the tested pathogens with MIC in the range of 62.5–125 μg/mL. The SAR analysis has revealed that introduction of hydroxyl and halogens especially bromine on coumarin ring (
152c and
152b) could improve the antibacterial potential against all the pathogens compared to other substitutions. It was concluded that lipophilicity or hydrophobicity might be concerned with their mechanism of action. Halogens are highly reactive species due to their electronegativity thus, sufficient halogens (Cl, Br, and I) can be fatal to microorganisms.
Mei-Hang Chen et al. [
49] synthesized and screened several novel coumarin substituted amide derivatives (
Scheme 33) as potential antibacterial agents against
Xanthomonas oryzaepv. Oryzae (Xoo) and
Xanthomonas citri subsp.
Citri (Xcc). 4-hydroxycoumarin (
58) was refluxed with POCl
3 using Et
3N reagent to yield 4-chlorocoumarin (
153) via chlorination. Esterification of
153 with 4-aminoophenol in the presence of K
2CO
3 in CH
3CN afforded 4-(4-aminophenoxy) coumarin (
154). Finally, the compound
154 was treated with aromatic acid and 4-dimethylaminopyridin via condensation in CH
2Cl
2 gives amide derivatives containing coumarin (
155a–q). The tested compounds
155f,
155h,
155k,
155l,
155m,
155n,
155o, and
155q found excellent antibacterial potency against
Xanthomonas oryzaepv. oryzae at 200 μg mL
−1 with percent zone of inhibition 92.7, 90.5, 97.3, 98.6, 94.1, 95.2, 92.4, and 96.0%, respectively, and were compared to the activity profile of the standard bactericide thiodiazole−copper (63.1%). Meanwhile, the same analogues displayed potential against Xoo at 100 μg mL
−1 with percent inhibition of 55.1, 52.3, 55.7, 57.8, 52.1, 55.0, 52.6, and 56.3%, respectively, which is more than that of thiodiazole−copper (30.2%). Likewise, analogues
155f,
155h,
155k,
155l,
155m,
155n,
155o, and
155q has displayed acceptable antibacterial action against Xcc, with the inhibition rates of 93.8, 93.7, 98.1, 99.2, 96.1, 97.3, 93.2, and 97.0%, respectively, which is closer to the standard thiodiazole−copper (86.2%) at 200 μg mL
−1. Based on the preliminary evaluation, the obtained EC
50 value indicated that compounds
155f,
155g,
155h,
155i,
155j,
155k,
155l,
155m,
155n,
155o, and
155q could display outstanding potency against Xoo (EC
50: 135.8, 156.0, 145.7, 178.5, 160.2, 103.6, 96.4, 126.8, 119.0, 127.3, and 117.5 in all μg mL
−1 respectively), while the same analogues exhibited significant potency against Xcc with EC
50 of 115.0, 138.1, 101.2, 142.4, 136.8, 79.4, 73.2, 119.0, 112.4, 107.4, and 83, respectively, which is better than that of thiodiazole−copper (138.3 μg mL
−1). The SAR study revealed that the electron-withdrawing groups F, Cl, Br, NO
2, CF
3, and OCF
3 could display excellent potency against both tested pathogens (Xoo and Xcc) when compared to the electron-donating substitutions (Me, OMe) at the R position. From the above evidence, most of the amide bearing compounds with coumarin have shown improved potency against Xoo and Xcc than that of imine with the same coumarin moiety.
Mohd. Imran Ansari et al. [
50] has reported a series of novel 3-(2-(5-(2-chloroquinolin-3-yl)-3-substituted phenyl-4,5-dihydro-1H-pyrazol-1-yl)-thiazol4-yl)-6-H/halo-2H chromen-2-ones and assessed for their in vitro antimicrobial activity. Compounds
156a–e were prepared by reacting suitable substituted hydroxy benzaldehyde with ethylacetoacetate in catalytic amount of piperidine, which is via bromination to get converted into key intermediate 3-(2-bromoacetyl)-6-H/halo-2H-chromen-2-ones (
157a–e). Meanwhile, 2-chloroquinoline-3-carbaldehyde on treatment with substituted acetophenone in ethanol obtained
159a–e, which on treatment with hydrazine carbothioamide under reflux yields 5-(2-chloroquinolin-3-yl)-3-substituted phenyl-4,5-dihydro-1Hpyrazole-1-carbothiamide (
160a–e). Finally, the title analogues
161a–y were prepared via condensation of
157a–e and
160a–e in ethanol (
Scheme 34). The prepared compounds screened for their in vitro antimicrobial action against
S. aureus,
E. faecalis,
Staphylococcus epidermidis,
B. subtilis, and
B. cereus (Gram-positive),
E. coli,
P. aeruginosa,
K. pneumoniae,
Bordetella bronchiseptica, and
P. vulgaris (Gram-negative) bacterial strain and five fungal strains, namely,
C. albicans,
A. niger,
A. flavus,
Monascus purpureous, and
Penicillium citrinum, by using the serial plate dilution method. Some analogues
161q,
161r, and
161s having fluoro-substituted coumarin on the phenyl ring has proven potential against tested bacteria as compared to the corresponding H/chloro/iodo/bromo-substituted compounds. Moreover, compounds
161q and
161r were found to possess excellent antifungal potential than that of the standard ofloxacin, and ketoconazole.
In another study Renuka Nagamallu et al. [
51] developed a series of coumarin appended 1,3-oxazines for their antimicrobial and antioxidant potential. The outline for the synthesis of title compounds is described in (
Scheme 35). The corresponding hydrazones (
164a–g) were synthesized via condensation of
162 with substituted phenylhydrazines (
163a–g) in catalytic amount of AcOH under reflux. The title coumarins appended 1,3-benzoxazine analogues (
165a–g) were prepared by treatment of corresponding hydrazones with triphosgene in the presence of Net
3 and CH
2Cl
2 via cyclization. The obtained oxazine derivatives
165a–g were evaluated for in vitro antimicrobial potential against
S. aureus,
S. pyogenes,
E. coli, and
P. aeruginosa bacterial strains and
C. neoformans,
A. niger,
A. flavus, and
C. albicans fungal strains. Compound
165b having methoxy group exhibited potent activity against all the tested bacterial strains. After that compounds
165d (chloro) and
165f (dimethyl) have displayed greater extent of inhibition against
Streptococcus pyogenes and
E. coli. Compounds
165c and
165g with CH
3 group have shown significant inhibitory potential against
S. pyogenes and
E. coli, whereas compounds
165b (methoxy) and
165d (chloro) had proved inhibitory potential against the tested fungi by inhibiting spore germination, while analogues
165c and
165f with a methyl group displayed good-to-moderate activity against
A. niger and
A. flavus.
Uzma Salar et al. [
52] synthesized and examined a series of coumarin-3-carboxamide derivatives for antimicrobial activity. The title analogues, coumarin-3-carboxamide (
166a–ab) were synthesized form coumarin-3-carboxylic acid (
56) with different substituted anilines in the presence of 1,1′-carbonyldiimidazole as a coupling agent (
Scheme 36). In vitro antimicrobial activity was assessed against
B. subtilis,
C. xerosis,
S. aureus,
S. faecalis, and
MRSA (Gram-positive) and
K. pneumoniae,
P. aeruginosa,
E. aerogene,
and S. dysenteria (Gram-negative) bacterial strains along with
A. niger,
A. terrus,
C. tropicalis,
C. sacromyces,
C. neoformen,
C. albicans,
Rhizopus,
Microsporumcanis, and
Absidia fungal strains by using disc diffusion method. Compound
166ab has exhibited highest antibacterial potential against
B. subtilis,
C. xerosis,
S. aureus,
S. faecalis,
MRSA,
K. pneumoniae,
P. aeruginosa,
E. aerogene, and
S. dysenteria, along with
166b and
166o, while none of the derivatives were found active against the tested fungal strains.
Jyotirmaya Sahoo et al. [
53] has produced new transitional metal complexes derived form 3-aryl-azo-4-hydroxy coumarin analogues for their antimicrobial action. The metal complexes
169a–h were yielded via condensation of different hydro alcoholic solutions of metal chlorides with 3-(4-chloro phenyl/4-methoxy phenyl)-azo-4-hydroxy coumarin derivatives (
168a,b) (
Scheme 37). The prepared complexes were assessed for antimicrobial potential against
E. coli,
K. pneumonia,
S. aureus,
C. albicans and
C. neoformans and compared the derived results with standard ampicillin and fluconazole. Among all the synthesized complexes, only
169a and
169e exhibited better antimicrobial potential against
E. coli,
K. pneumonia,
S. aureus,
C. albicans and
C. neoformans in comparison to the standard drugs (
p < 0.05).
A series of 2-(1-(2-oxo-2Hchromen-3-yl) ethylidene) hydrazinecarbothioamide derivatives were contributed by R H. Vekariya et al. [
54] in order to assess their antimicrobial activity (
Scheme 38). Salicylaldehyde on reaction with ethyl acetoacetate in the presence of starch sulfuric acid or cellulose sulfuric acid gives 3-acetyl coumarin under solvent free conditions. The obtained 3-acetyl coumarin was treated with isothiocyanatobenzene and hydrazine hydrate in the presence GAA under reflux to furnish title coumarin-clubbed thiosemicarbazone derivatives (
170a–s). The prepared hybrids were screened for their in vitro antimicrobial activity against bacterial strains (
S. aureus,
S. pyogenes,
E. coli,
P. aeruginosa) and fungal strains (
A. clavatus,
C. albicans, and
A. niger) by employing agar dilution method using ampicillin, ciprofloxacin, chloramphenicol, nystatin and griseofulvin as standard control drugs. Biological studies have revealed that compound
170o with electron deactivating fluoro group on phenyl (2 and 4 positions) has excellent potential against
S. aureus with MIC 50 lg/mL. Analogues
170b,
170e,
170j, and
170m bearing chloro group at phenyl ring were found excellent inhibitors next to
170o against
S. aureus with MIC 125 lg/mL. Moreover, candidates
170b,
170e,
170h,
170j,
170m,
170n, and
170s were found as potent inhibitors against
E. coli at MIC of 62.5 lg/mL. On the other hand, compound
170o bearing fluoro substitution at phenyl was found active against fungi
C. albicans with MIC value 100 lg/mL which is equal to the standard nystatin and greater than that of griseofulvin, while analogues
170b,
170e,
170j,
170m, and
170n having a chloro group at phenyl displayed potency against
C. albicans with MIC 250 lg/mL. Moreover, analogue
170i with an iodo group at the para position of the phenyl ring had proven better activity against
C. albicans (MIC 250 lg/mL) compared to the standard drugs.
In continuing efforts to find potent antimicrobial candidates R. Nagamallu and co-workers [
55] reported novel coumarin appended bis (formyl pyrazole) derivatives for their antimicrobial and antioxidant potential. The outline of synthesis is displayed in (
Scheme 39). Initially, 6-acetyl-7-hydroxy-4-methyl-2H-chromen-2-one (
172) was furnished via Pechmann reaction of 1-(2, 4-dihydroxyphenyl) ethanone (
171), and ethyl acetoacetate. The obtained
172 was treated with acetic anhydride to yield 6-acetyl-4-methyl-2-oxo2H-chromen-7-yl acetate (
173) which was subjected to Fries rearrangement using AlCl
3 to give 1,1′-(7-hydroxy-4-methyl-2-oxo-2H-chromene-6,8-diyl) diethanone (
174). The prepared
174 was reacted with substituted phenylhydrazines, semicarbazine, and thiosemicarbazine in ethanol and catalytic amount of glacial acetic acid (GAA) was refluxed to yield corresponding bis-hydrazones
175a–h followed by conversion to label coumarin appended bis (formyl pyrazole) (
176a–h) under Vilsmeier-Haack conditions. The biological assays were performed against
E. coli,
P. aeruginosa and
S. aureus bacterial strains, as well as
A. niger,
A. flavus, and
C. albicans via the broth dilution method using ciprofloxacin and fluconazole as the standard drugs. Most of the analogues from the series were found moderately active against tested bacterial strains with MIC values ranging from 6.25–75 µg/mL. Analogues
175g and
175h with CONH
2 and CSNH
2 exhibited excellent antibacterial activity against selected bacteria and compounds
176a and
176h with same group at pyrazole ring were found potent as compared to the standard. Compounds
175d and
175b bearings methoxy and methyl group have displayed good to moderate activity. All the test compounds were further tested for antifungal potential and compounds
175g and
175h with CONH
2 and CSNH
2 revealed excellent potential, while
176g with CONH
2 has shown potency similar to standards and the hybrid
176h having CSNH
2 was found potent against
A. niger.
Megharaja Holiyachi et al. [
56] in another study demonstrated the antibacterial and anticancer activity of coumarin benzimidazole hybrids. The basic skeleton of coumarin-benzimidazole (
177a–f) were obtained by rection of 4-formylcoumarins with ortho-phenylenediamine in the presence of p-toluene sulphonic acid in DMF under reflux. Further the resulting intermediate was converted to coumarin-benzimidazole sulphonamide analogues (
178a–f) by treatment of
p-toluenesulfonyl chloride with
177a–f via N-sulphonation. On the other hand, obtained
177a–f on reaction with methyl iodide in the presence of anhydrous potassium carbonate yield
179a–f (
Scheme 40). All the furnished derivatives were screened against
Bacillus flexus (Gram-positive) and
Pseudomonas spp. (Gram-negative) bacteria. Compounds substituted with
177c (OCH
3),
177d (Br) and
177e (Cl) at C-6 of coumarin moiety were found to be highly active against both the bacterial strains as compared to the standard ciprofloxacin. Among the series
178a–f and
179a–f, analogues
178c,
178d and
178e have displayed excellent potency against both the tested pathogens, whereas N-methylated analogues were found less active except
179e having chloro substitution. For antifungal potency, compound
117e with chloro substitution at C-6 of coumarin was found active against
Scopulariopsis spp., while different substitutions at C6 of the coumarin moiety in compounds
178c (OCH
3),
178d (Br), and
178e (Cl) had proven promising results against both
Scopulariopsis spp. and
Aspergillus tereus. Thus, the obtained results revealed that presence of larger group such as OCH
3, Cl and Br at C-6 of coumarin framework are responsible to show potent antibacterial and antifungal potential, whereas, for series
177a–f, N-sulphonation did not reveal any improvement in the potency; however, N-methylation reduced the potential.
Kinga Ostrowska et al. [
57] investigated antitumor and antimicrobial potential of 5-[4-(4-aryl-1- piperazinyl)butoxy]coumarins against
M. luteus,
B. subtilis,
B. cereus,
S. aureus, and
E. hirae (Gram-positive) and
E. coli and
P. aeruginosa (Gram-negative) bacterial strains, as well as
C. albicans and
C. parapsilosis fungal strains via the modified cylinder-plate method. The intermediate 1-(4,7-dimethylcoumarin-5-yloxy)-4-bromobutane (
181) and 1-(5-acetyl-4,7-dimethylcoumarin-5-yloxy)-4-bromobutane (
181b) were prepared by reaction of 5-hydroxy-4,7-dimethylcoumarin (
180a) or 6-acetyl-5-hydroxy-4,7-dimethylcoumarin (
180b) with sodium hydroxide in toluene by using 1,4-dibromobutane, tetrabutylammonium bromide alkylating agents via microwave. The obtained bromoalkyls (
181a and
181b) with N-substituted piperazine derivatives in the presence of potassium carbonate in acetonitrile yield title analogues (
182a–I and
183a–h, respectively) (
Scheme 41). The two analogues
182f and
183f displayed good antibacterial potency. It was observed that 4,7 dimethyl-5-[4-[4-(1-(4-pyridyl)) piperazin-1-yl] butoxy] coumarin (
182f) was active against
M. luteus with MIC 15 µg/cm
3. Another 5-acetyl-4,7- dimethyl-5-[4-[4-(1-(4-pyridyl)) piperazin-1-yl] butoxy] coumarin (
183f) with acetyl substitution was found less active, while other compounds were found inactive. Overall, all the analogues bearing methoxy substituents on aromatic ring or 4-pyridyl substituent were found active against tested organisms. Similarly, Compounds
182f and
183f were found active against tested fungi.
Tatjana Gazivoda Kraljević and co-workers [
58] reported anticancer and antibacterial activity of some 4-substituted 1,2,3-triazole–coumarin hybrids. In vitro antibacterial evaluation was done against bacterial strains, namely,
S. aureus,
E. faecalis, and
E. faecium (Gram-positive) and
P. aeurigonsa,
E. coli,
A. baumannii, and
K. pneumoniae (Gram-negative), and the obtained results were compared to the standard ceftazidime and ciprofloxacin. The title compounds 1,2,3-triazole–coumarin hybrids (
187–218) were furnished via Huisgen 1,3-dipolar cycloaddition reaction of a terminal alkyne with an azide using copper(I) as the catalyst (
Scheme 42). The synthesized analogues failed to display considerable antibacterial potential against both tested bacterial species, with the exclusion of some selective activity against
Enterococcus species. The obtained results have shown that coumarin–1,2,3-triazole analogues bearing substituted phenyl rings (
196–202), hybrids having p-alkyl phenyl (
199–201), and (p-alkylcyclohexyl) phenyl (
202) at 4-substituted 1,2,3-triazole nucleus exhibited promising potency against
Enterococcus faecalis with MICs values ranging from 8–64 µg/mL. Among the 4-(benzene sulphonamide) methyl-1,2,3-triazole–coumarin series, compounds having p-methyl (
203) and p-fluorobenzene sulphonamide subunits (
205 and
206) have proven their potential against
E. faecalis with MICs 64 and 32 µg/mL. In addition to that, compounds
208 (morpholine) and
210 (piperazine) displayed significant potential with MICs 32 µg/mL and 16 µg/mL, respectively.
A series of 2-piperidinomethylamino-4-(7-H/substituted coumarin-3-yl)-6- chloro substituted phenyl pyrimidines was prepared and screened for antimicrobial potential by Mohd Imran et al. [
59]. Title analogues
121–238 were furnished by reactions of 2-amino-4-(7-H/substitutedcoumarin-3-yl)-6-(chlorosubstitutedphenyl) pyrimidines (
220) with piperidine and formaldehyde (
Scheme 43). Among the synthesized series, compound
226 was found to be highly potent against
S. aureus,
E. faecalis,
S. epidermidis,
B. subtilis, and
B. cereus with MIC 50 μg/mL (
p < 0.05 or less). Compound
226 has exhibited significant activity against tested Gram-negative bacterial strains (
E. coli,
P. aeruginosa,
K. pneumonia,
B. bronchiseptica, and
P. vulgaris) with MIC value 25 μg/mL (
p < 0.05 or less). Moreover, compounds
226 and
237 have proven excellent inhibitors against tested fungi (
C. albicans,
A. niger,
A. flavus,
M. purpureous, and
P. citrinum) with MIC 25 μg/mL (
p < 0.0001) and 25 μg/mL (
p < 0.0001), respectively. The derived SAR concludes that the presence of bromo substitution at 7-position of coumarin ring along with 4-chlorophenyl at 6-position of the pyrimidine ring has played a critical role for inhibition of tested bacterial and fungal strains.
M.H. Shaikh et al. [
60] developed a series of coumarin incorporated triazoles analogues and assessed for their antitubercular, antioxidant and in vitro antimicrobial activity. The title analogues were synthesized by procedure depicted in (
Scheme 44). Initially the starting material required for synthesis of label analogues was benzyl azides and
239a–f were furnished by reaction of corresponding benzaldehydes with sodium azide via NaBH
4 reduction, bromination, and nucleophilic substitution reaction. The 7-hydroxy-4-methyl coumarin (
8b) obtained via Pechmann condensation, reaction of
8b and
58 with propargyl bromide in the presence of K
2CO
3 in DMF afforded 4-Methyl-7- (prop-2-yn-1-yloxy)-2H-chromen-2-one (
9b), and 4-(Prop-2-yn-1-yloxy)-2H-chromen-2-one (
79), respectively. In the final step, prepared benzyl azides (
239a–f) and coumarin-based alkynes (
9b and
79) via 1,3-dipolar cycloaddition reaction in the presence of t-BuOH-H2O (1:1) afforded corresponding 1,4-disubstituted-1,2,3-triazolebased coumarin derivatives (
240a–f and
240g–k, respectively). The test compounds were subjected to in vitro antimicrobial evaluation against
S. aureus,
M. luteus, and
B. cereus (Gram-positive)
E. coli,
P. fluorescens, and
F. devorans (Gram-negative) bacterial strains, as well as
A. niger,
P. chrysogenum, and
C. lunata fungal species via serial dilution method using kanamycin, ampicillin, chloramphenicol, miconazole, fluconazole, and amphotericin B as the standard drugs. Among the series, most the analogues were good inhibitors against all the tested bacterial and fungal strains. Among the series, compounds
240g and
240d revealed excellent antimicrobial potential against all the tested pathogens as compared to the standard analogues.
Mert Olgun Karatas et al. [
61] evaluated antimicrobial activity of novel silver (I) complexes with coumarin substituted N-heterocyclic carbene ligands. Novel eight coumarin substituted silver(I) N-heterocyclic carbene (NHC) complexes were produced via the interaction of the corresponding imidazolium or benzimidazolium chlorides and Ag
2O in dichloromethane (
Scheme 45). All the synthesized complexes were screened for their antimicrobial potency against
E. faecalis,
S. aureus,
E. coli, and
P. aeruginosa bacterial strains and
C. albicans and
C. tropicalis fungal stains. The obtained results demonstrated that about all the complexes had the potential to inhibit the growth of all the tested pathogens, while some of them displayed good action against different microorganism as compared to the standard analogues. From the tested complexes, the most lipophilic compound
245e (bis [1-(4-methylene-6,8-dimethyl-2H-chromen-2-one)-3-(naphthalene-2-ylmethyl) enzimidazole-2-ylidene] silver(I) dichloroargentate) was found to be most active as compared to the standard next to
245d.
Xin-Mei Peng and co-workers [
62] reported antimicrobial activity of some new coumarin-derived azolyl ethanols including imidazolyl, triazolyl, tetrazolyl, benzotriazolyl, thiol-imidazolyl, and thiol-triazolyl. The synthetic strategies to prepare title compounds coumarin-derived azolyl ethanols is depicted in (
Scheme 46a,b). Key intermediates
246 and
255 were synthesized via O-alkylation of 2-(chloromethyl) oxirane with hydroxyl coumarins (
8b and
254, respectively). The open ring compounds with suitable azoles using K
2CO
3 as base in ethanol gives corresponding mono-azolyl ethanols (
247–252) and bis-azolyl ethanols (
256–260). The resultant analogues evaluated for in vitro antimicrobial potential against Methicillin-resistant
S. aureus,
B. subtilis,
M. luteus (Gram-positive) and
E. coli,
P. aeruginosa, and
S. dysenteriae (Gram-negative) bacterial strains, as well as fungi (
C. albicans,
B. yeast,
C. utilis,
C. mycoderma, and
A. flavus) via the two-fold serial dilution method. Antibacterial screening has revealed that some of the analogues could display good to moderate potential against the selected bacterial strains as compared to the standard chloramphenicol and norfloxacin. Among the series, compound
257 bearing bis-triazolyl ethanol has displayed low MCI value of 8 mg/mL against MRSA, which was equivalent or better than that of standard drug norfloxacin MIC 8 mg/mL and chloramphenicol MIC 16 mg/mL. Compound
257 also found promising to inhibit growth of all tested fungi as compared to standard fluconazole.
R.R. Zhang et al. [
63] performed microwave assisted synthesis of some coumarin fused pyrano [3,2-c] chromene-2,5-diones (
Scheme 47) and the results of antifungal action against
Botrytis cinerea,
Colletotrichum copsica,
Alternaria solani,
Gibberella zeae, and
Rhizoctorzia solani were compared to a marketed broad-spectrum fungicide azoxystrobin. The prepared 3-(trifluoro) methyl
364a–e, 8-alkoxy
265a–d, 8-allyloxy
266a–c, pyrano[3,2-c] chromene-2,5-diones
267a–c and 3,4-cyclohexane pyrano[3,2-c] chromene-2,5-diones
268a–d evaluated for their fungicidal potential. Among the series, some of the analogues found to be potent inhibitors of
Botrytis cinerea and
Collecterichum capsica at conc < 50 ppm. Compounds
264d,
265c, and
266b were found to be highly active against
Botrytis cinerea with EC
50 values 0.141, 0.082, and 0.091 µM, respectively, which is much better than the standard drug azoxystrobin. Additionally, compound
264d (EC
50 = 0.115 µM) also found to be an excellent inhibitor against
Collecterichum capsica as compared to reference (EC
50 = 0.222 µM). Therefore, from above biological results it can be concluded that
264d is highly active and most promising compound for further studies.
In another study R.R. Zhang and co-workers [
64] investigated antifungal properties of some coumarin[8,7-e] [
1,
3] oxazine derivatives. Initially coumarins (
269a–e) were synthesized via Pechmann condensation using ZnCl
4 as the catalyst, which, on reactions with different and catalytic amounts of 4-N, N, dimethylaminopyridine under microwave, afforded target analogues
270a–t (
Scheme 48). The antifungal potency of these analogues was evaluated at concentration 50 ppm using reference drug osthole. Experimental studies revealed that compounds
270e,
270m, and
270s possess inhibitory potency against
Botrytis cinerea with zone of inhibition 88.2, 98.7, and 78.6, respectively, and are comparable to the control analogues and osthole (86.8%), while compounds
270g and
270m showed growth in their inhibition against
Colletotrichum capsica with 66.7 and 79.6%, respectively. However, analogues
270e,
270m, and
270o revealed 73.3, 88.2, and 81.5% inhibition, respectively, against
Rhizoctonia solani.


 ,
, 





































































{kind=link}
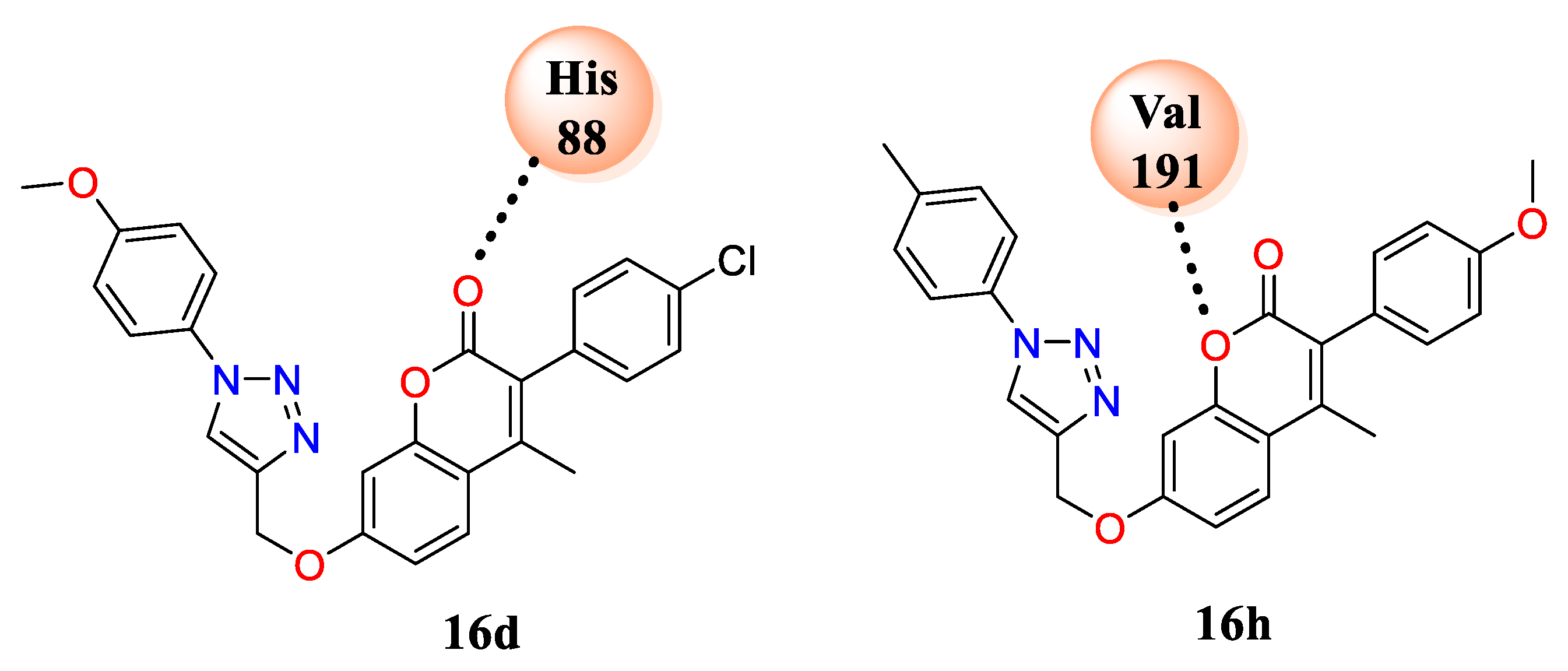{kind=link}
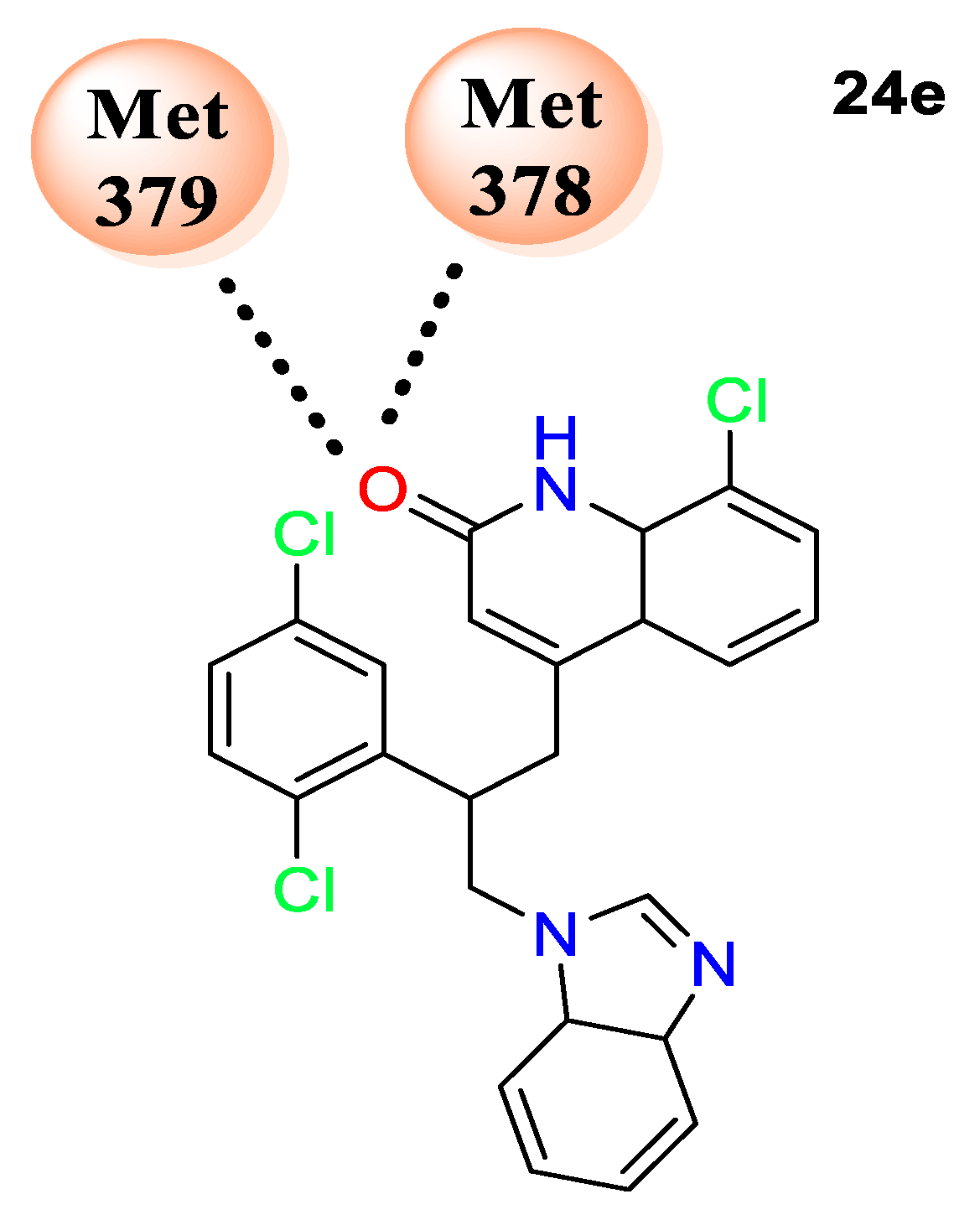{kind=link}
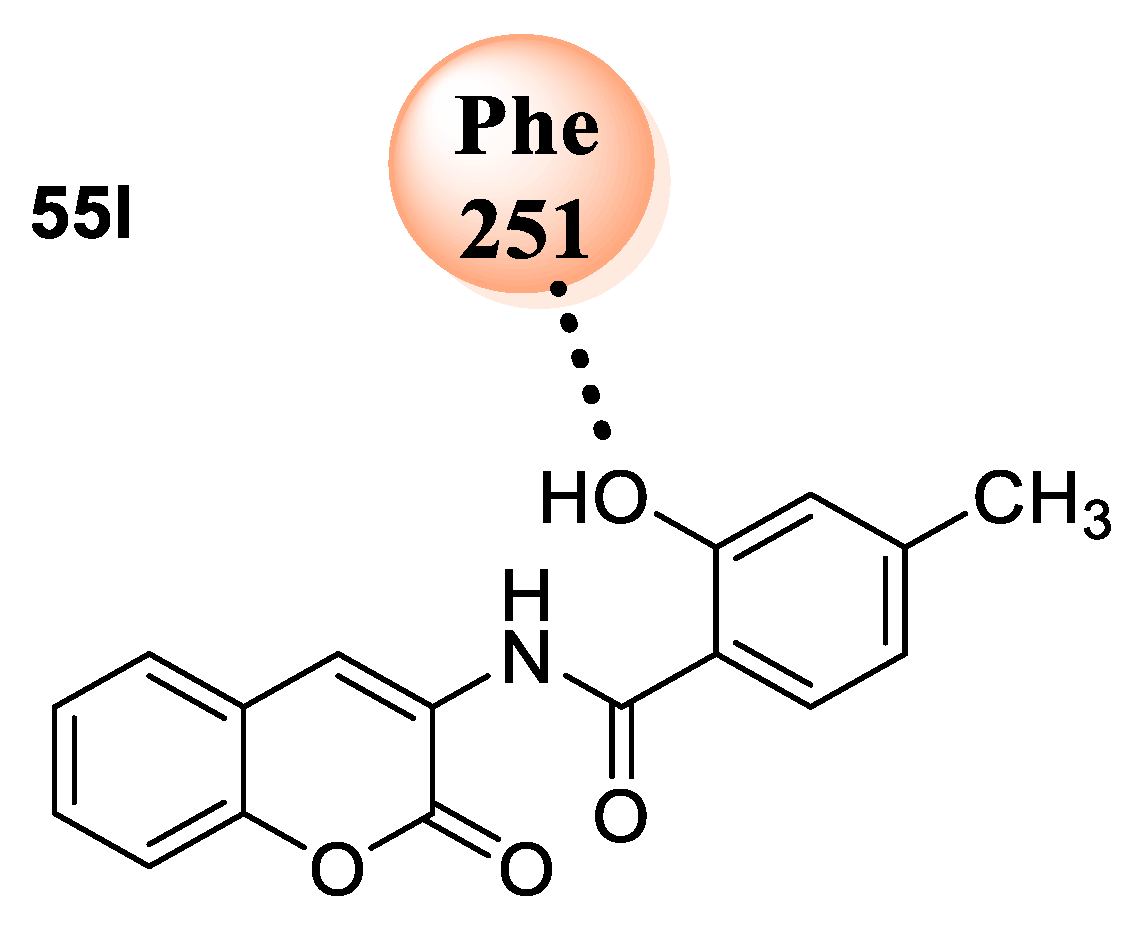{kind=link}
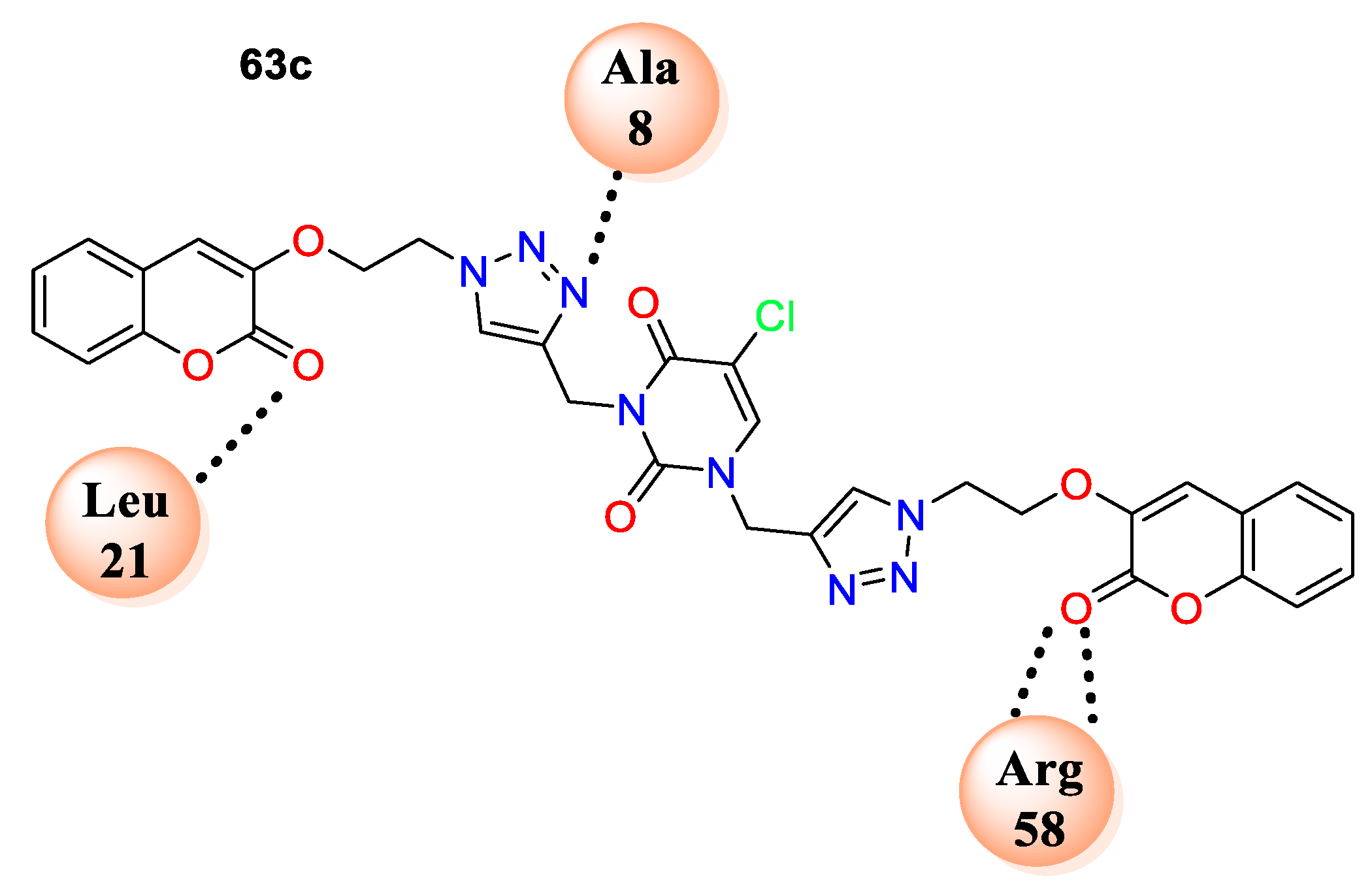{kind=link}
{kind=link}
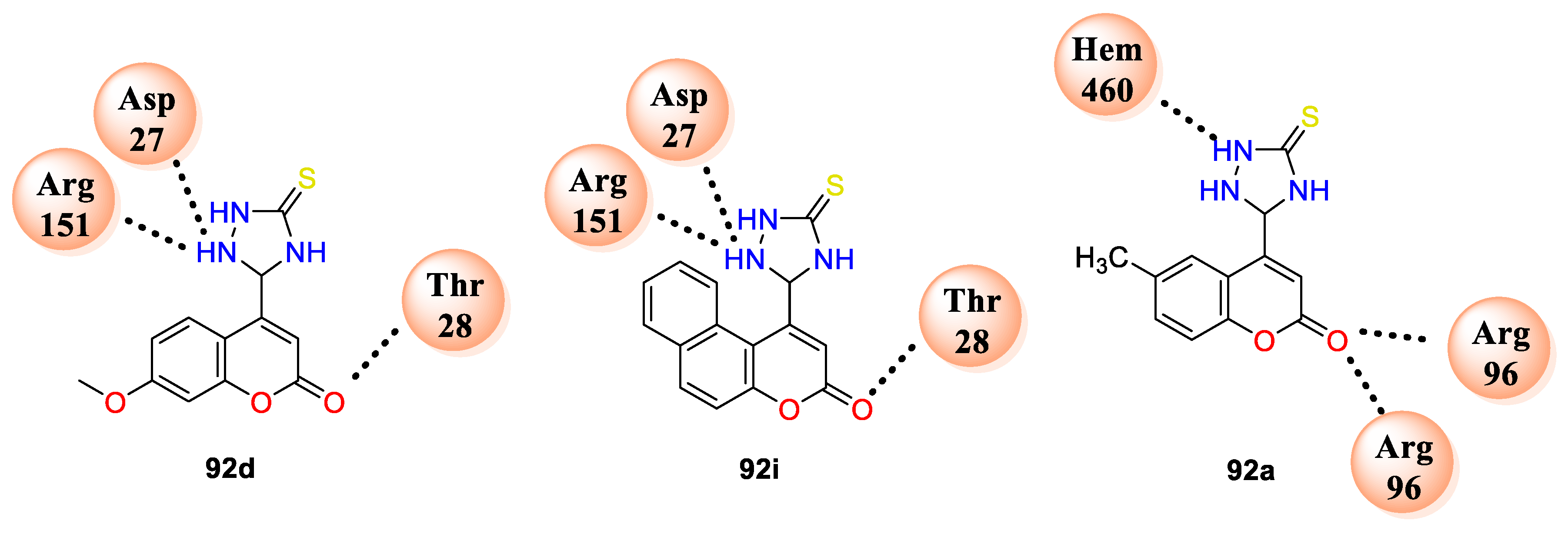{kind=link}
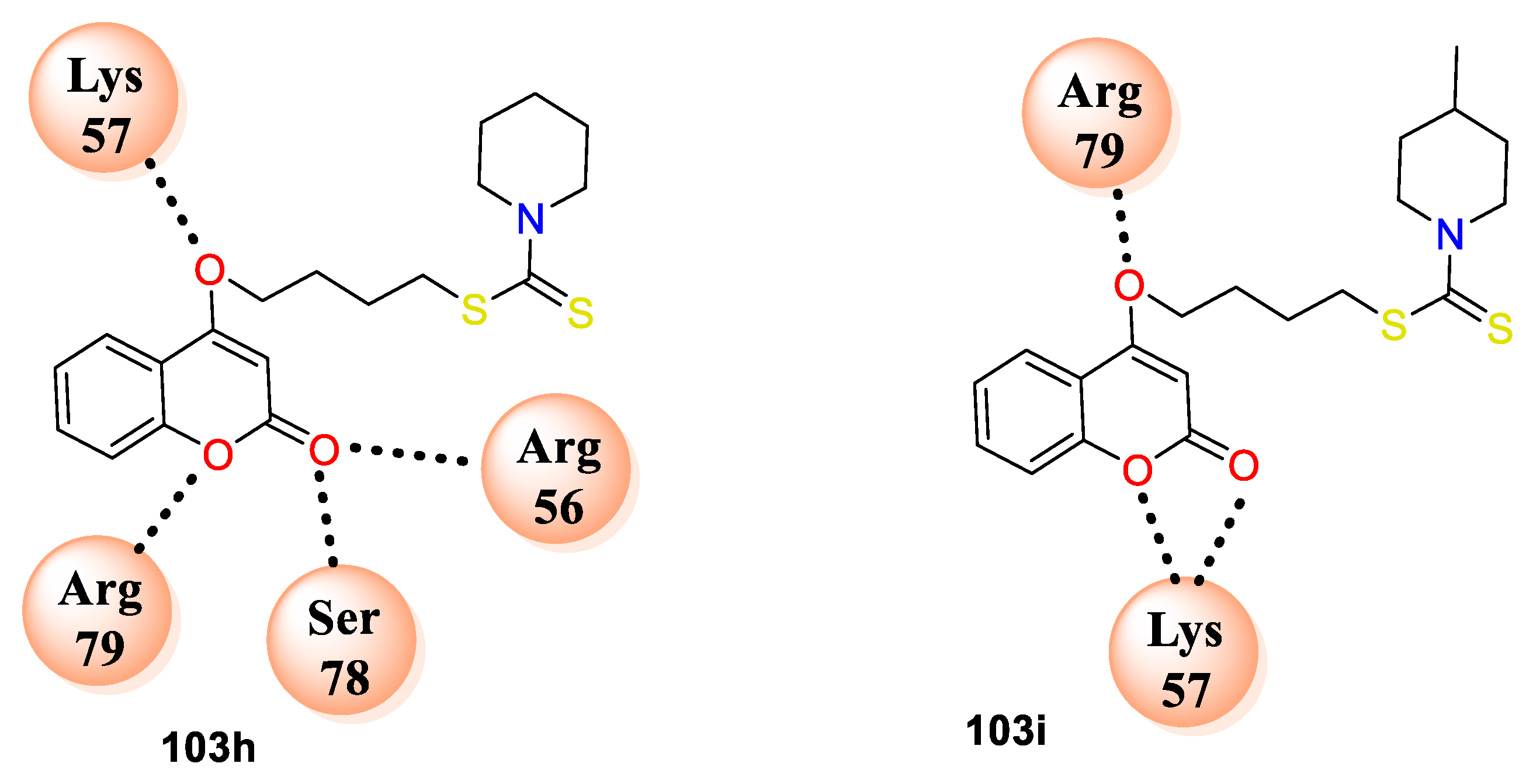{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
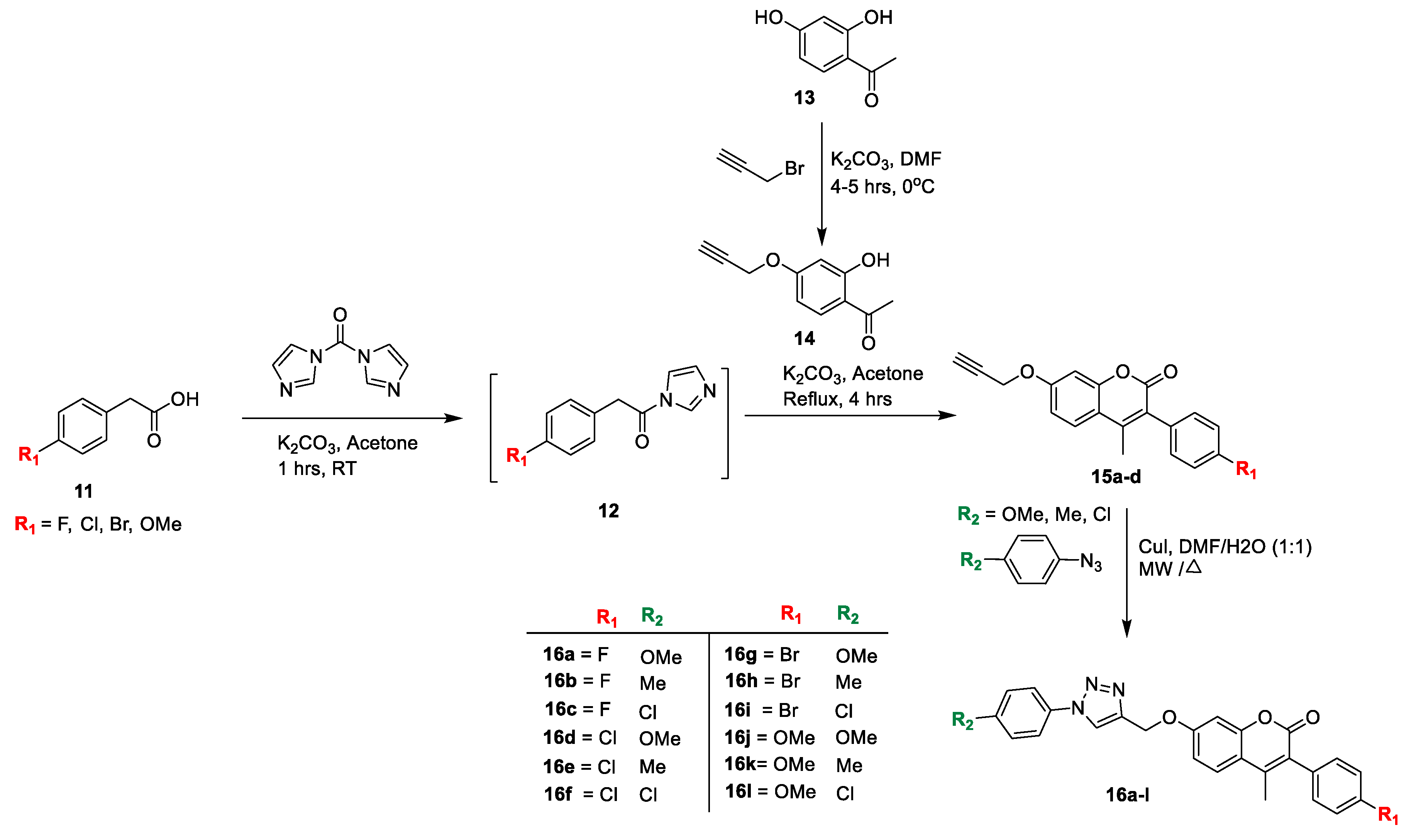{kind=link}
{kind=link}
{kind=link}
{kind=link}
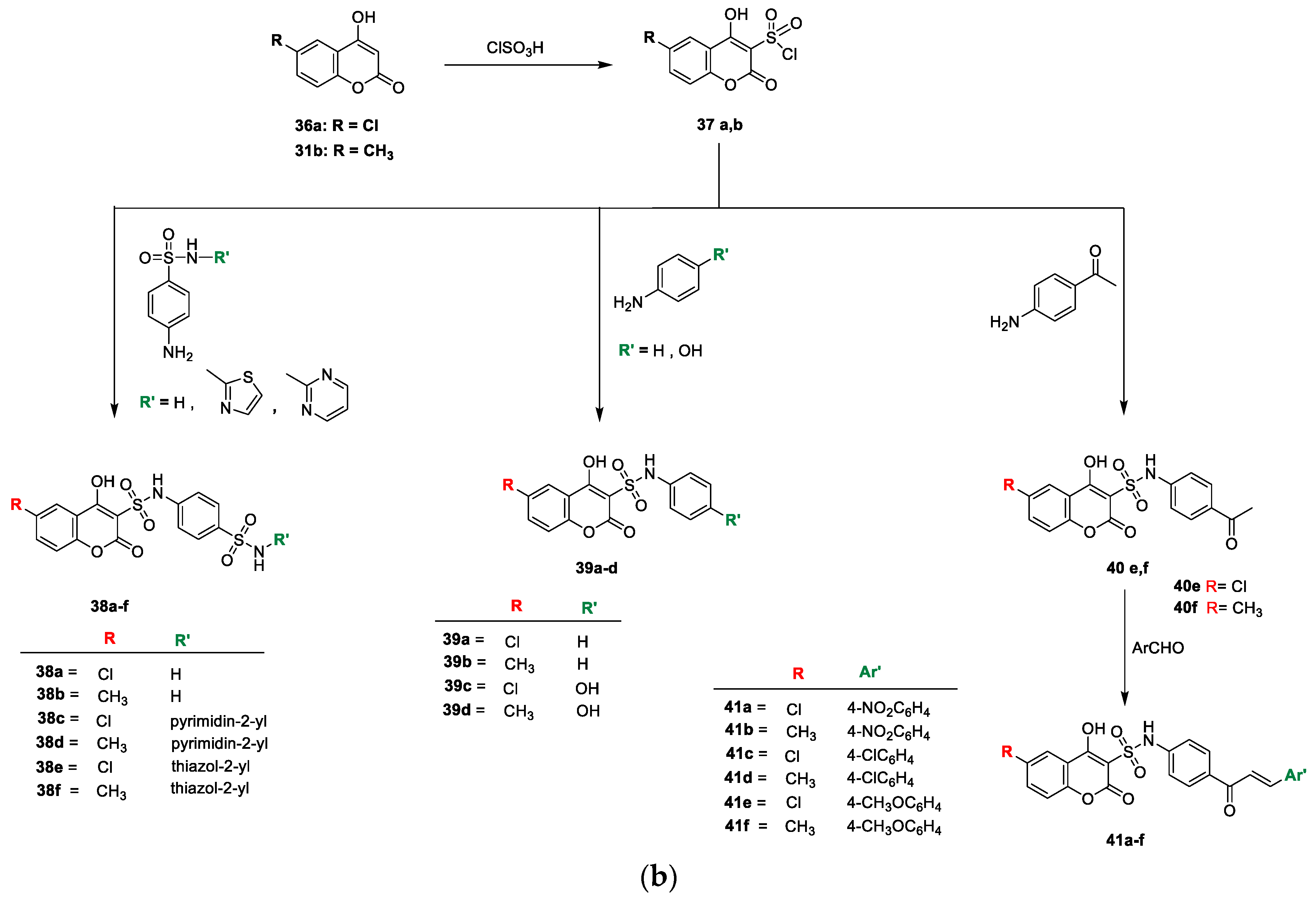{kind=link}
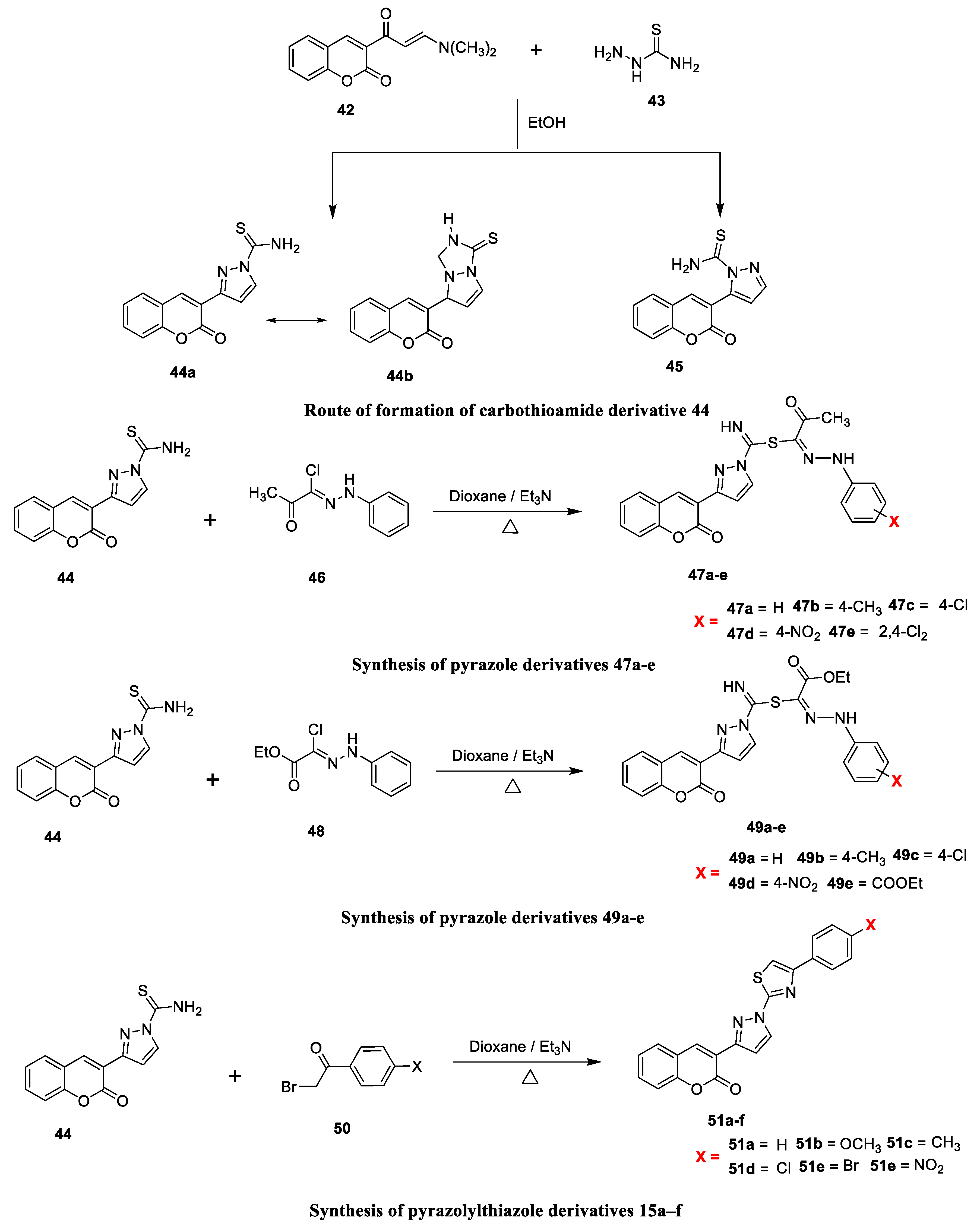{kind=link}
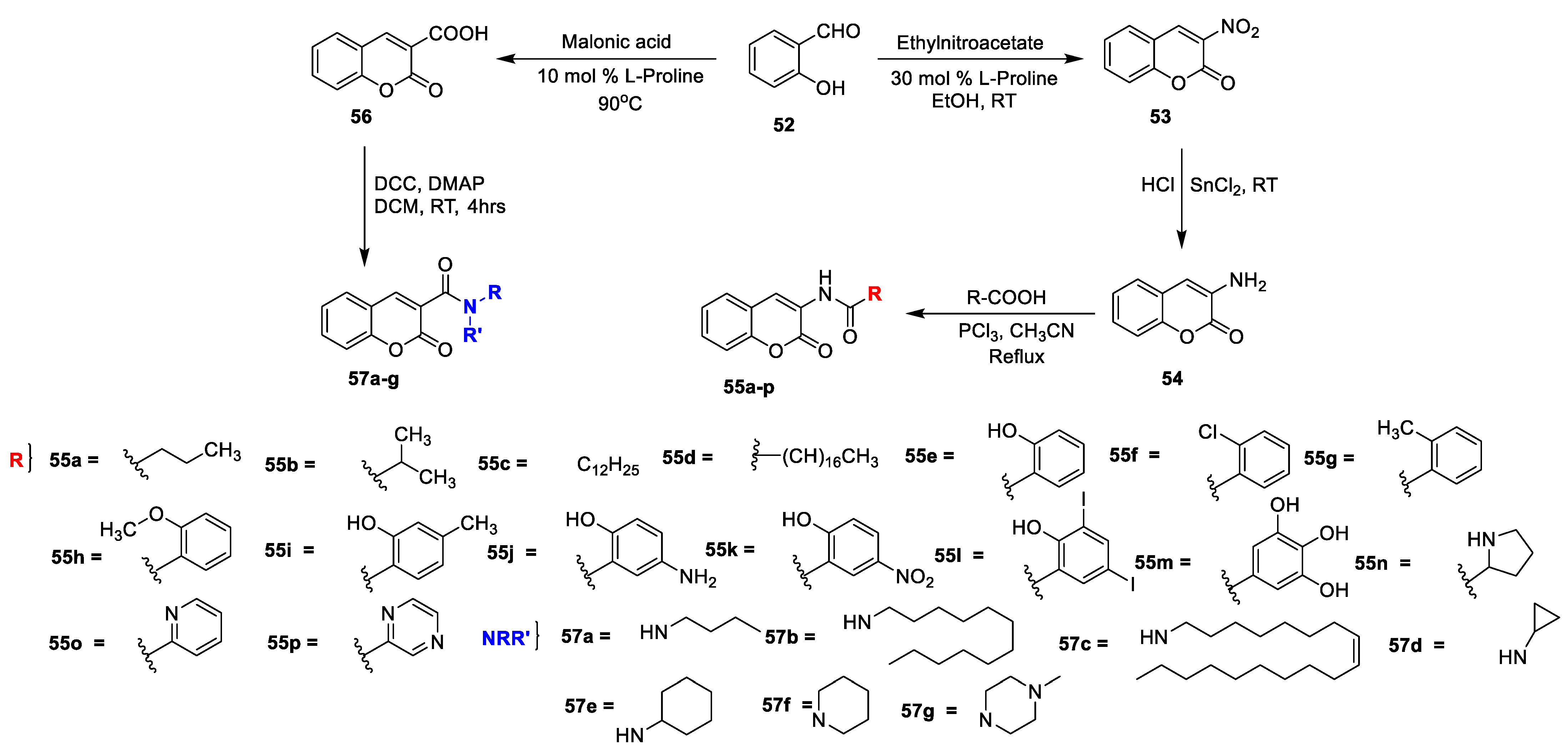{kind=link}
{kind=link}
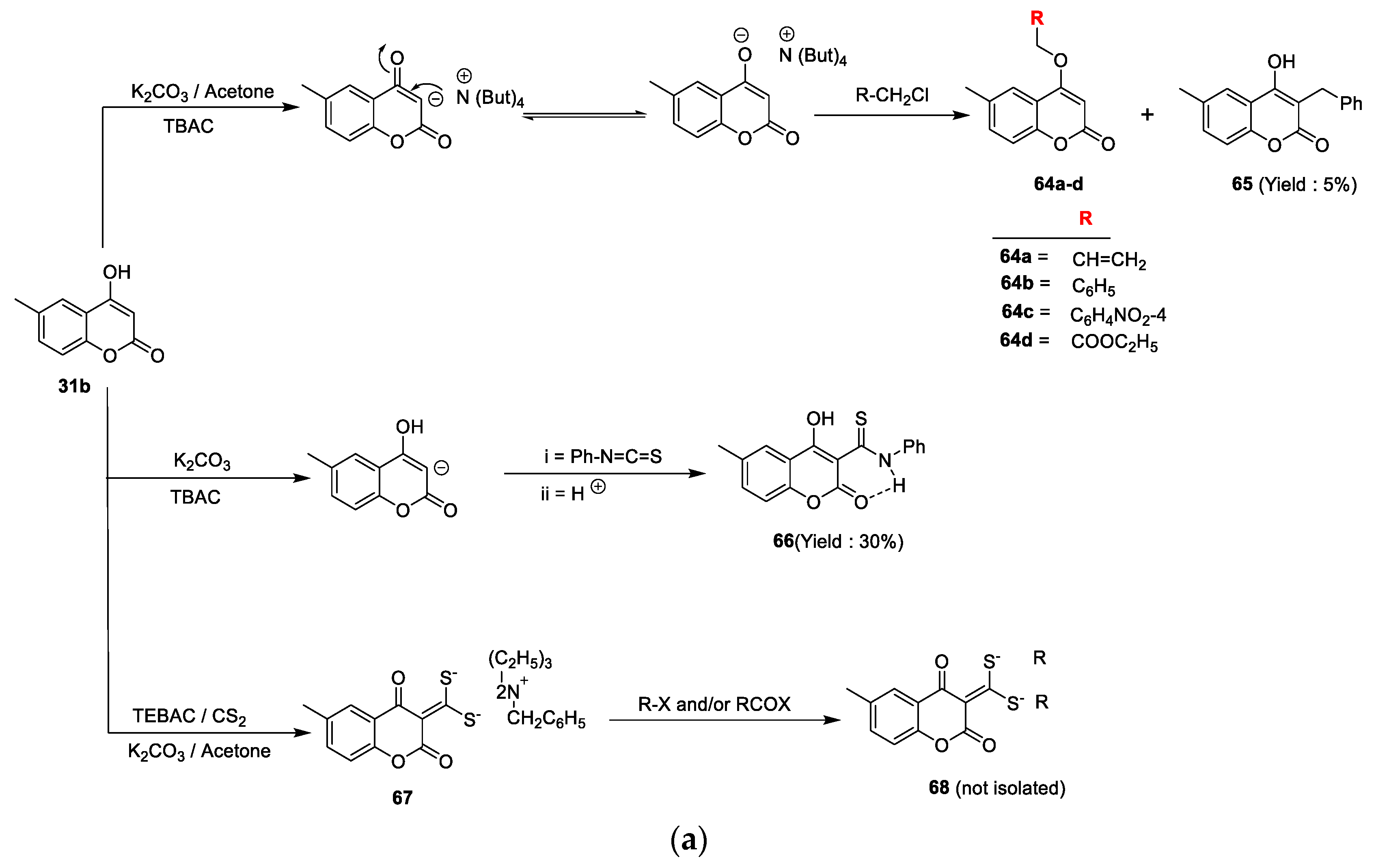{kind=link}
{kind=link}
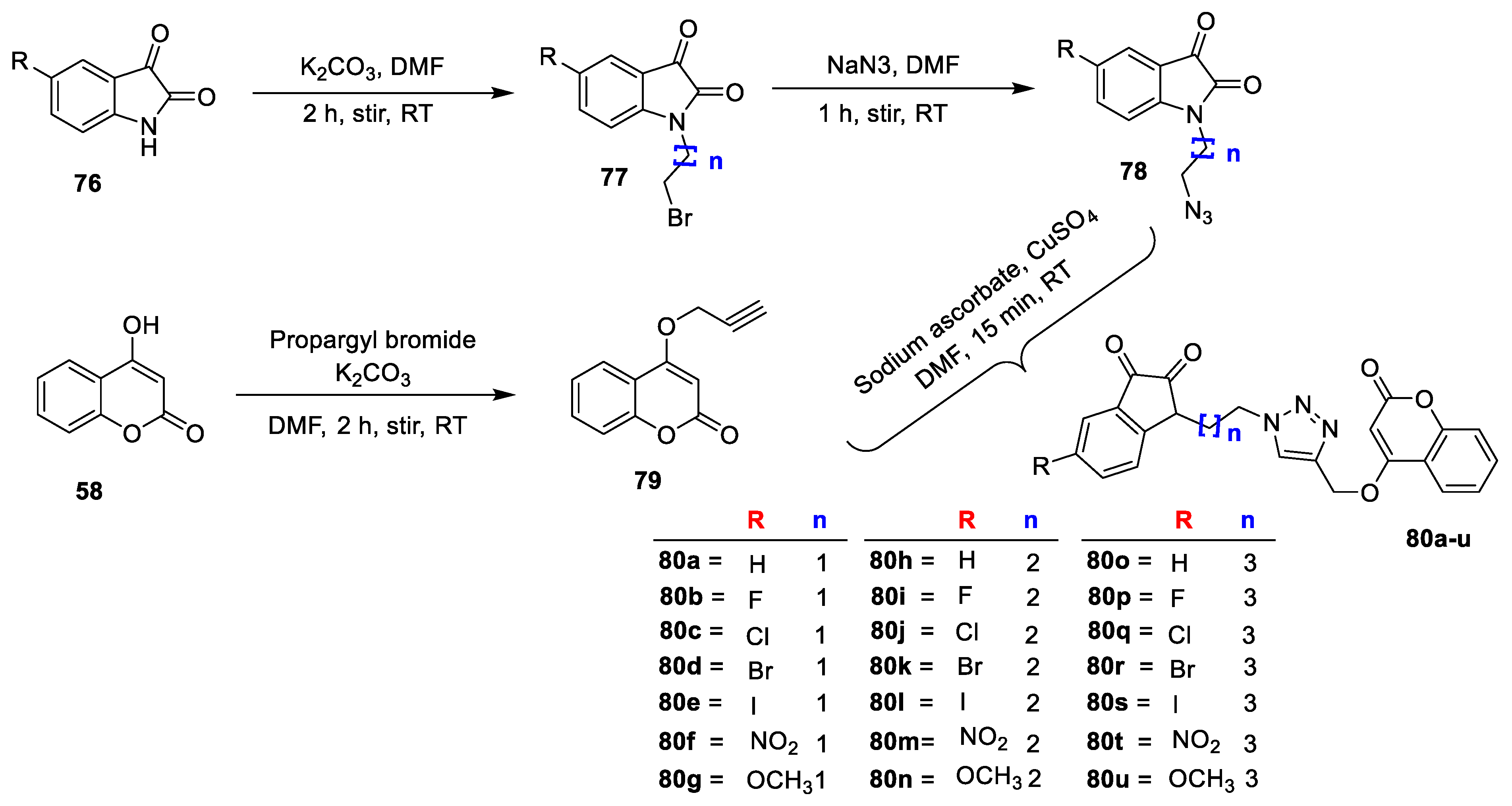{kind=link}
{kind=link}
{kind=link}
{kind=link}
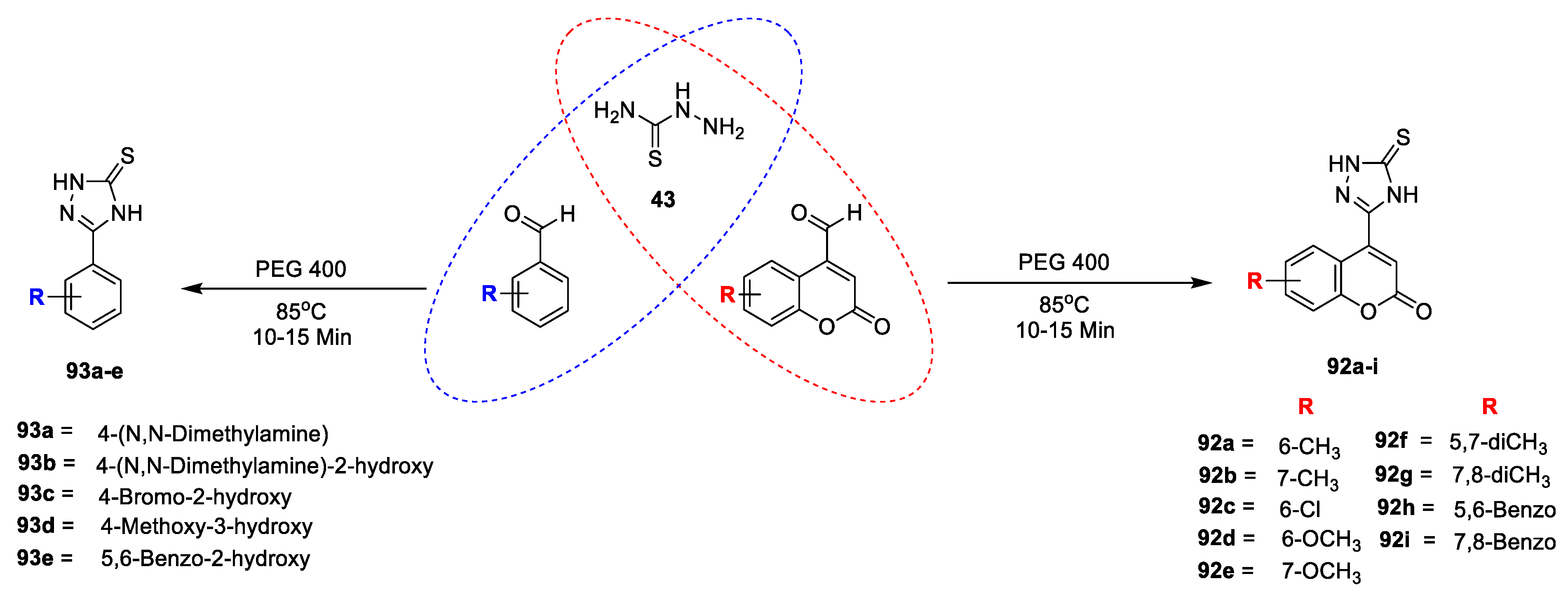{kind=link}
{kind=link}
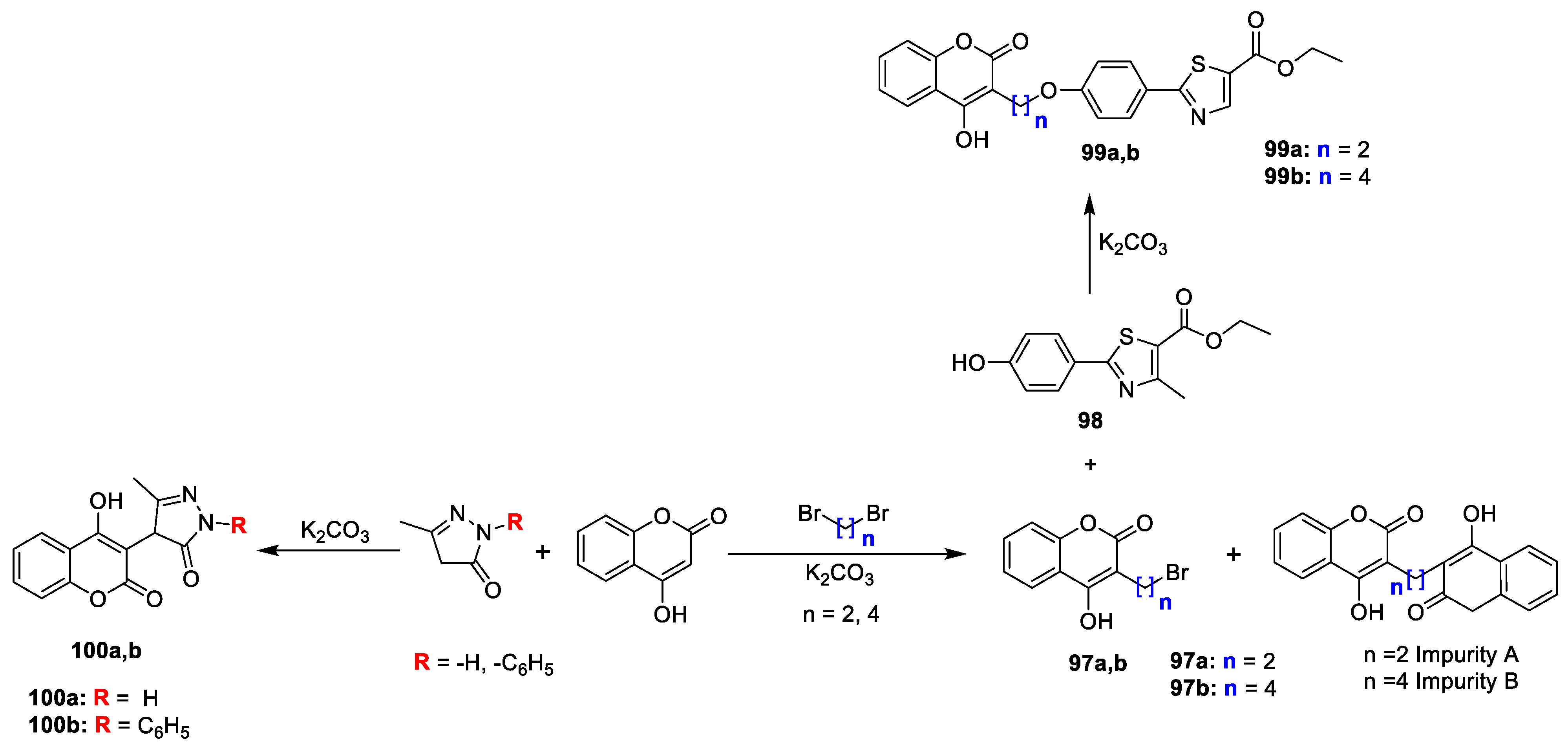{kind=link}
{kind=link}
{kind=link}
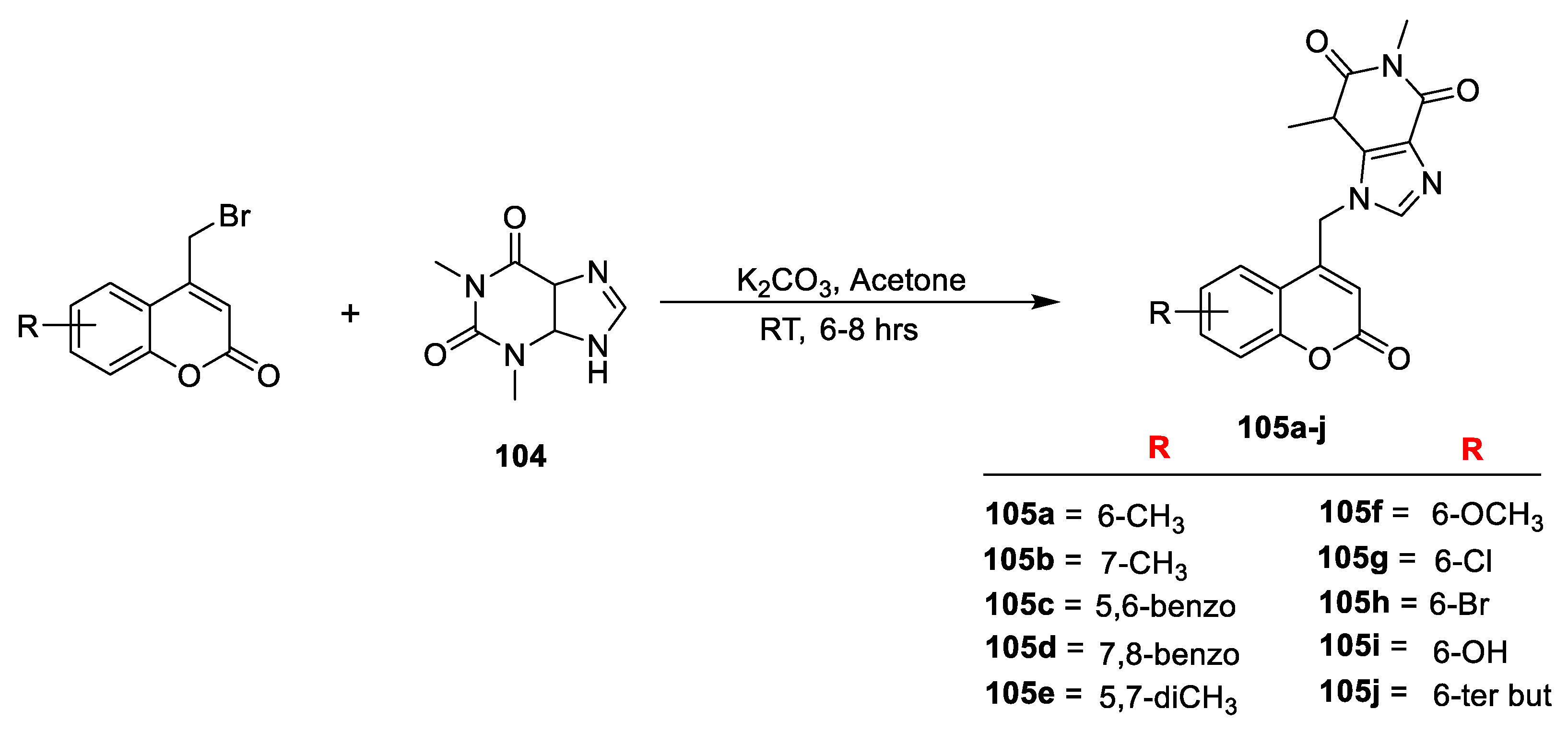{kind=link}
{kind=link}
{kind=link}
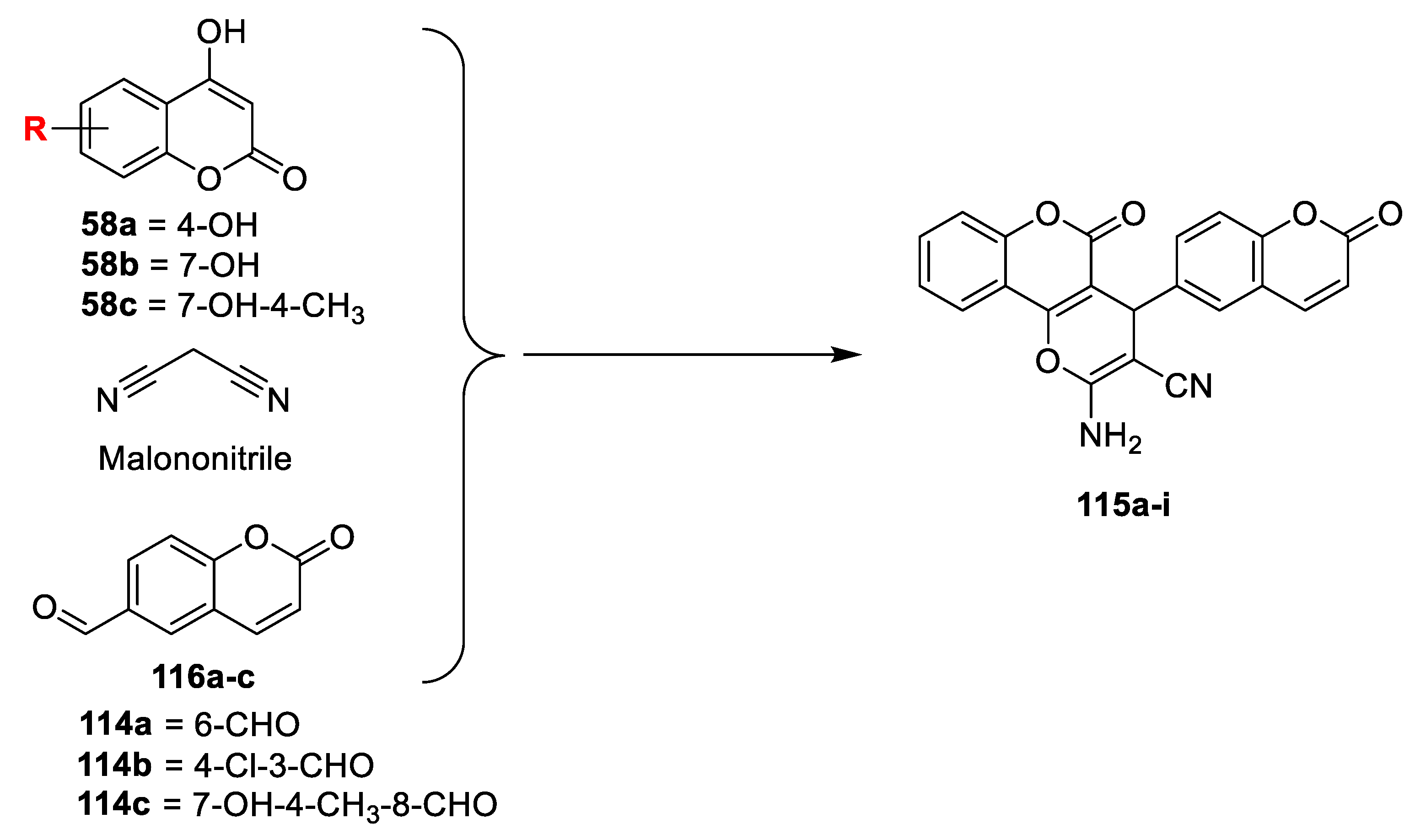{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
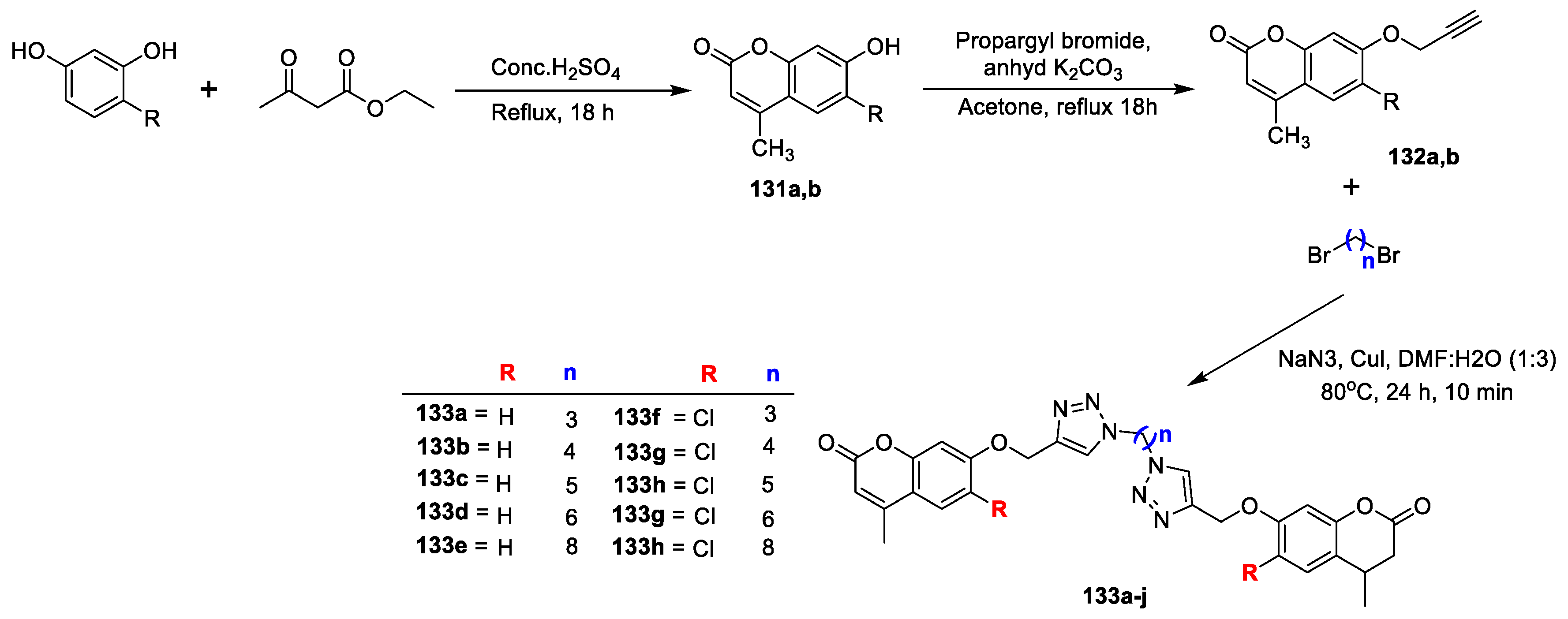{kind=link}
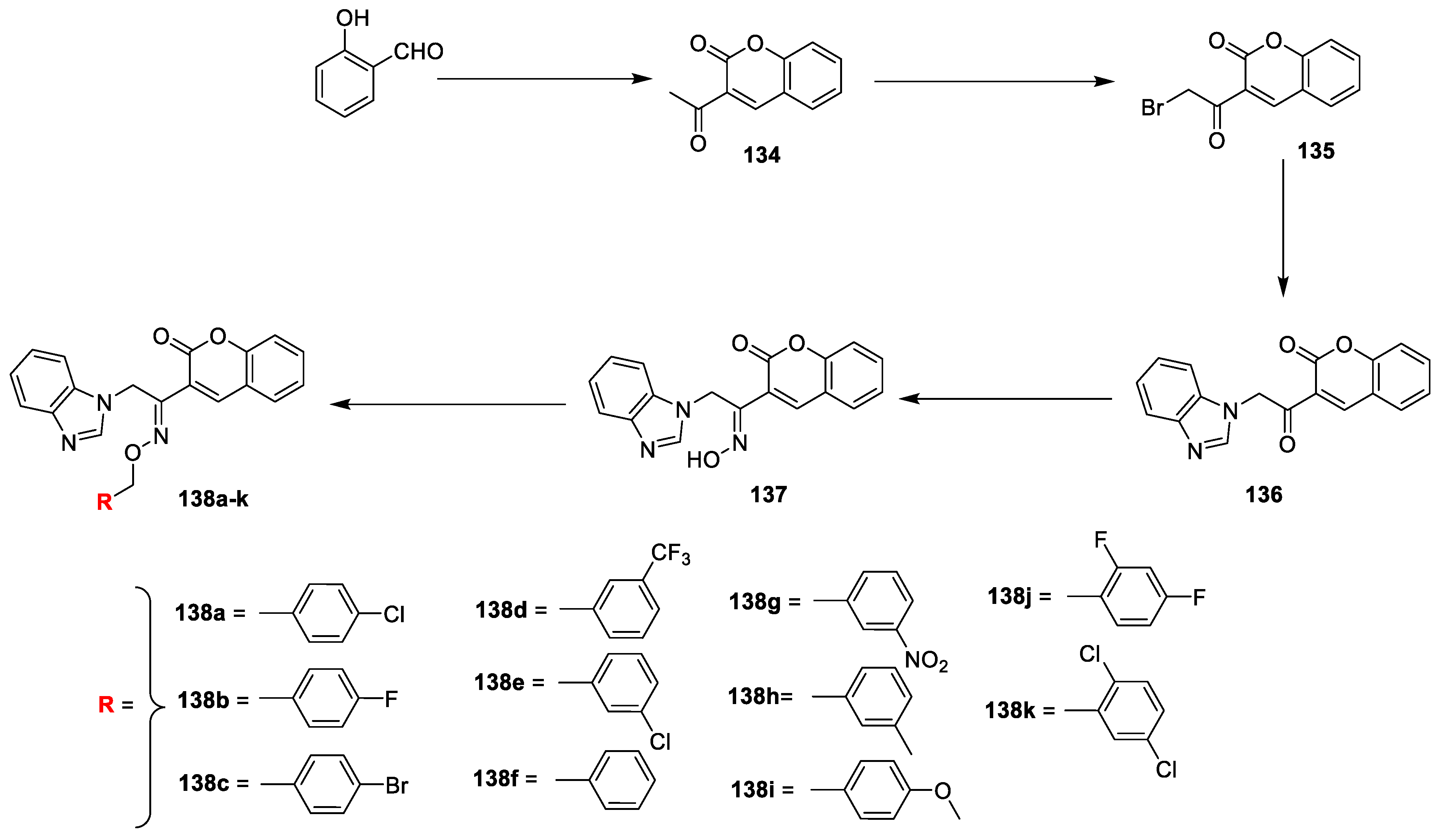{kind=link}
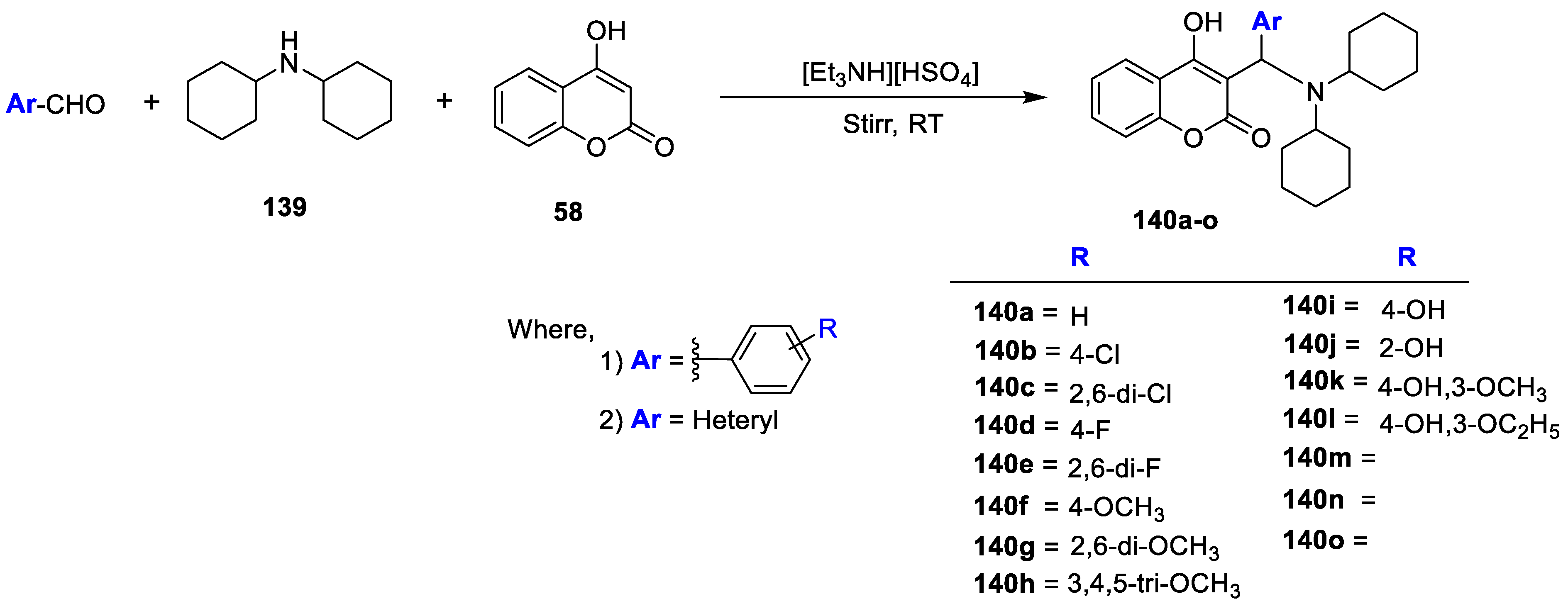{kind=link}
{kind=link}
{kind=link}
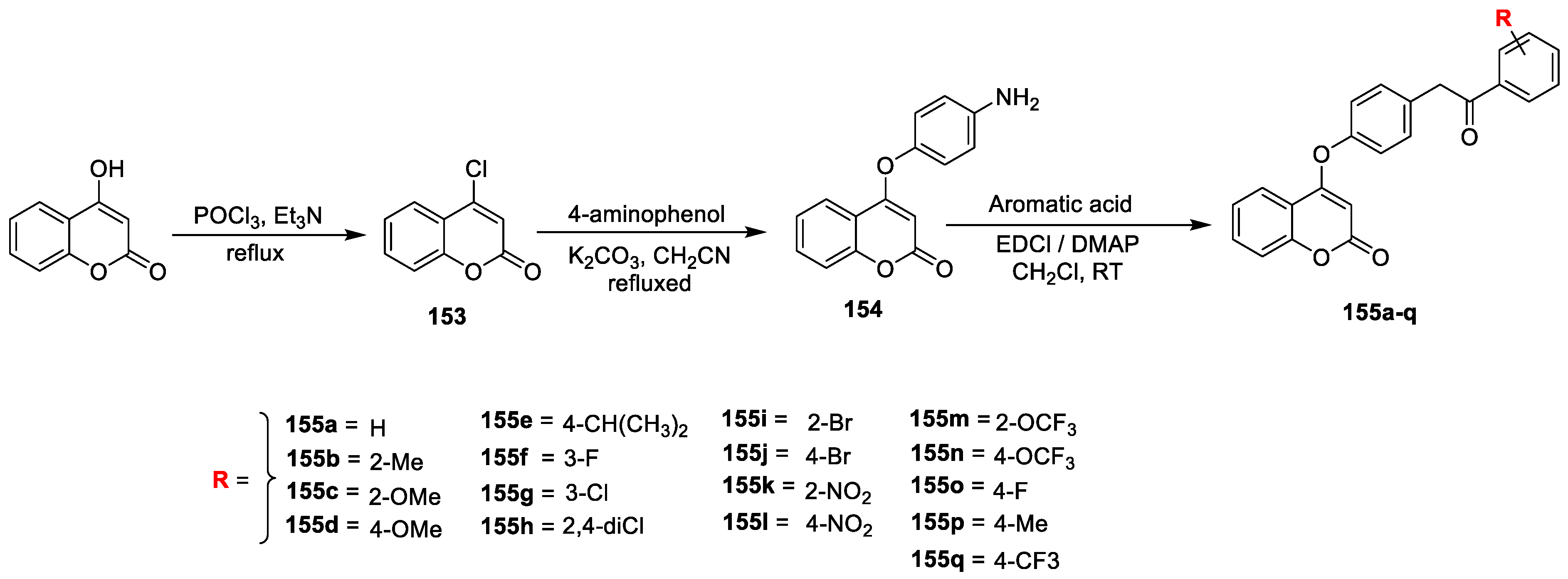{kind=link}
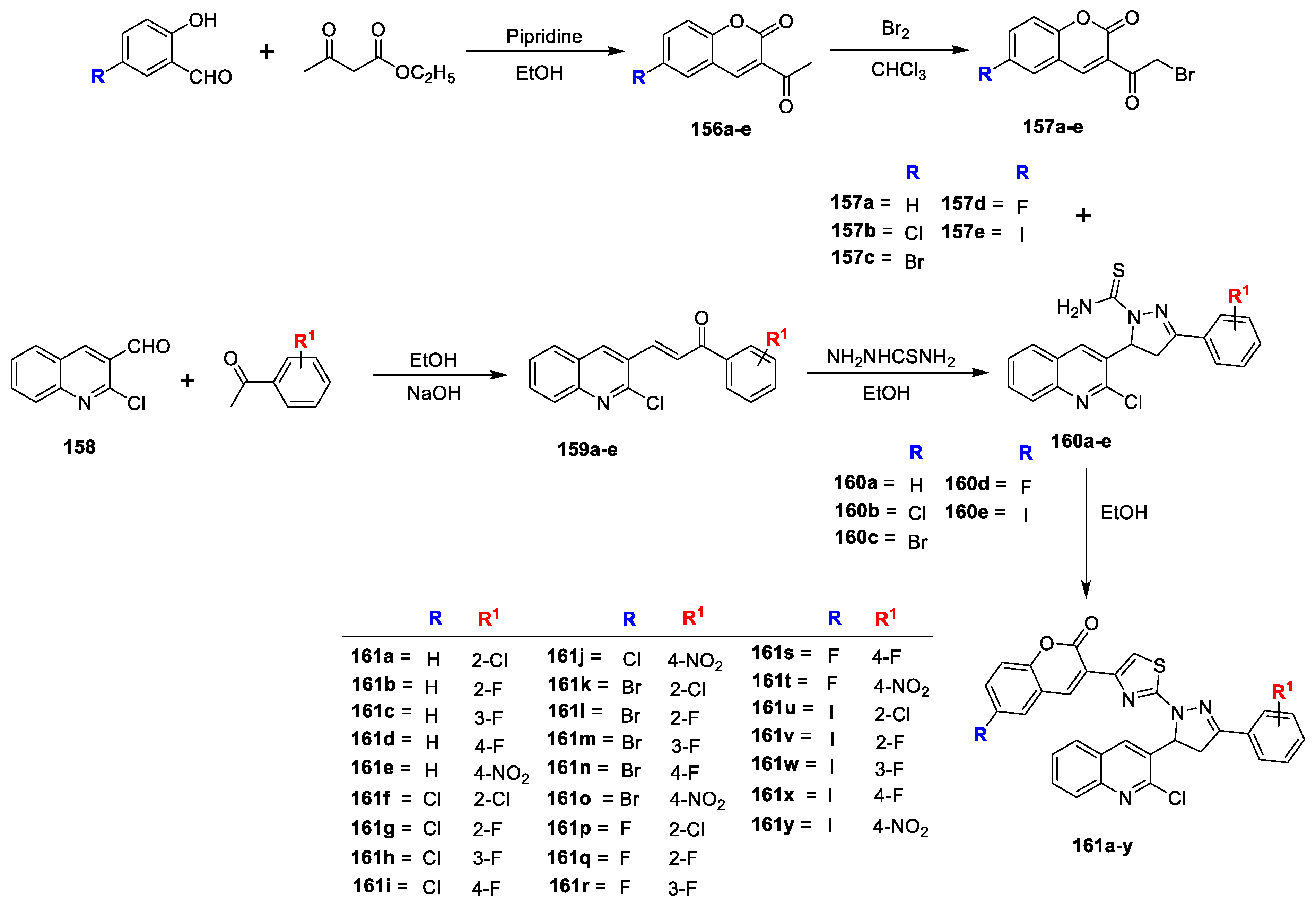{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
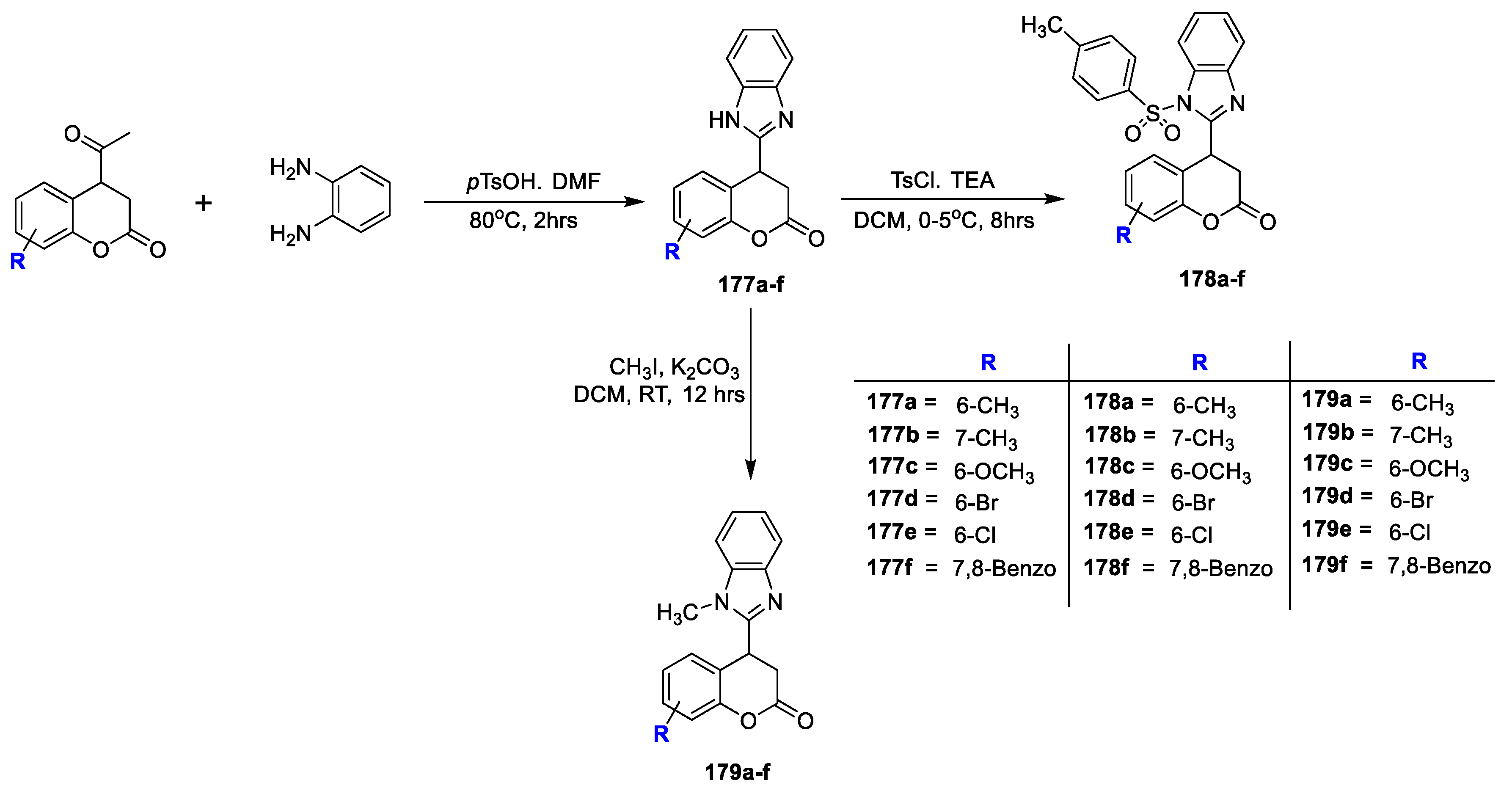{kind=link}
{kind=link}
{kind=link}
{kind=link}
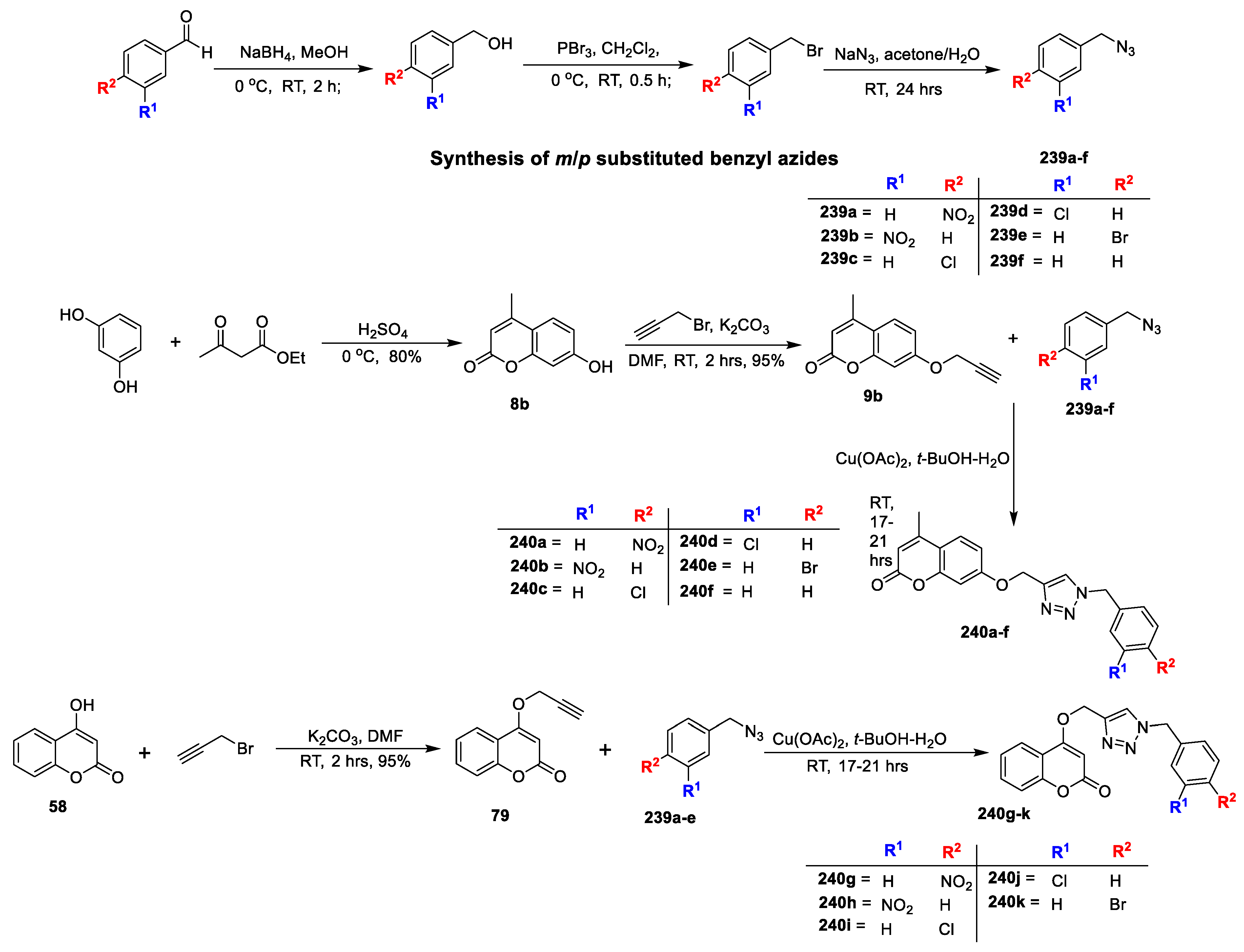{kind=link}
{kind=link}
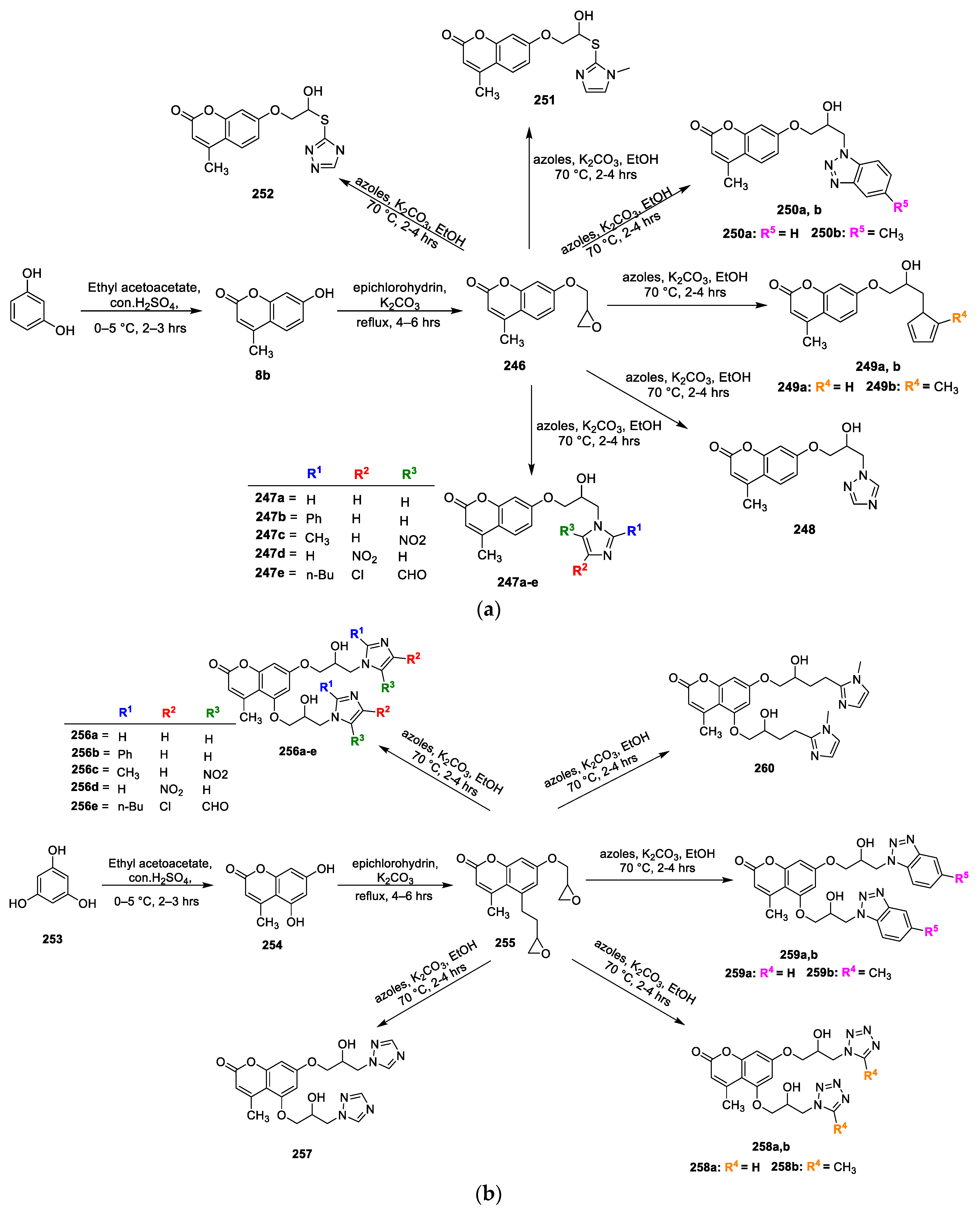{kind=link}
{kind=link}
{kind=link}