
Host Defence Peptides: A Potent Alternative to Combat Antimicrobial Resistance in the Era of the COVID-19 Pandemic


Abstract
:1. Introduction
1.1. Virus Classification and Structure
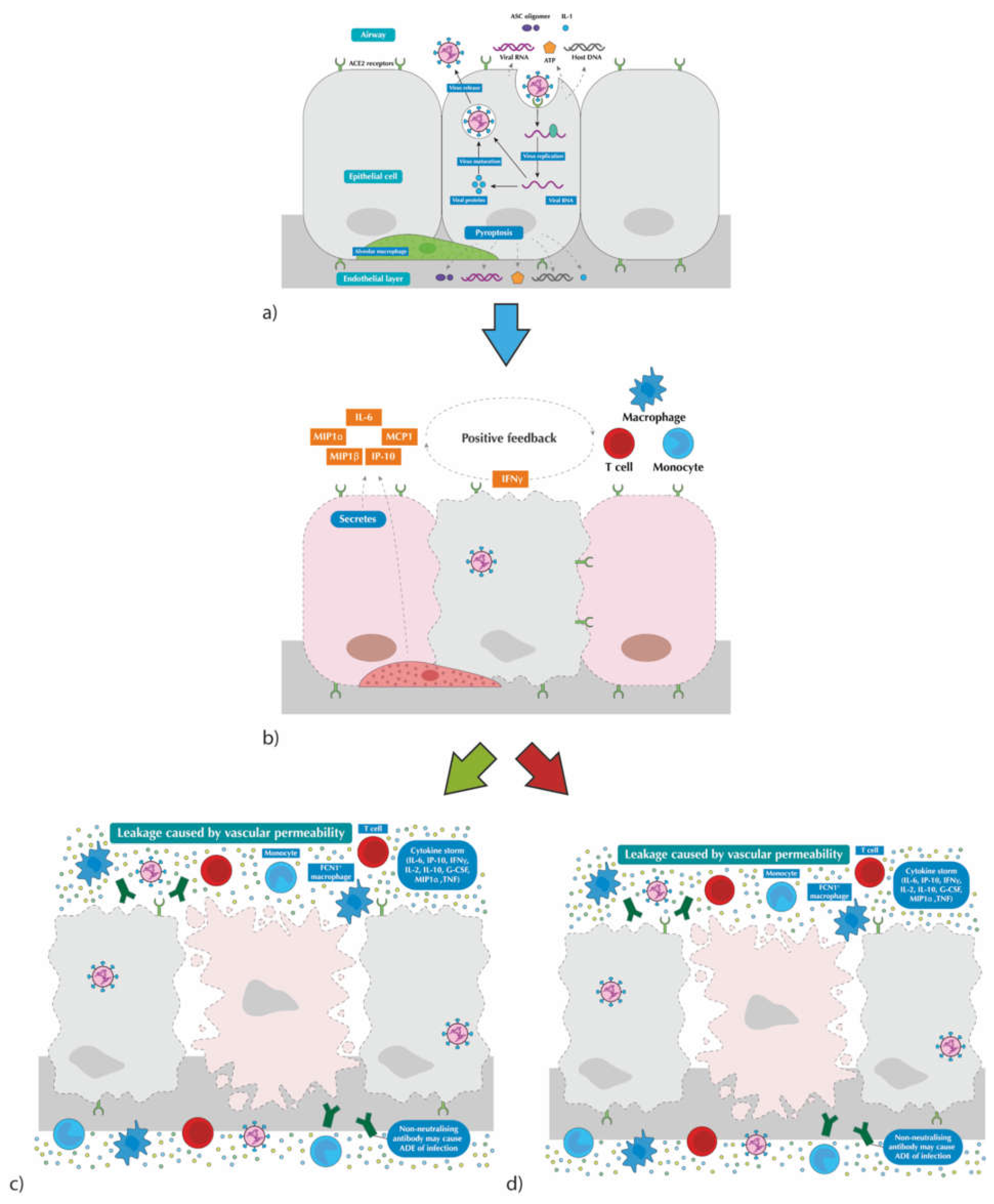
1.2. Virus Transmission and Disease Pathogenesis
1.3. Management of COVID-19
2. Antimicrobial Agents
2.1. Toxicity
2.2. Antimicrobial Resistance
3. Host defence Peptides: A Potential Solution for the Antimicrobial Resistance
3.1. History of HDPs
3.2. Structure of HDPs
3.3. HDPs Found in Humans
3.3.1. Defensins
3.3.2. Histatins
3.3.3. Cathelicidins
3.4. Mechanisms of Antimicrobial Action of HDPs
3.4.1. Membrane Targeting Mechanism
3.4.2. Intracellular Targeting Mechanism
3.5. Non-Antimicrobial Actions of HDPs
4. Use of HDPs for Combat against Antimicrobial Resistance
5. Use of HDPs as Potent Antibiofilm Agents
6. Application of HDPs for the Management of COVID-19
7. Other Applications of HDPs
8. Limitations of HDPs
8.1. Cost of Synthesis
8.2. Toxicity
8.3. Instability
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Director-General’s opening remarks at the technical briefing on 2019 novel coronavirus: 146th session of the Executive Board. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-technical-briefing-on-2019-novel-coronavirus (accessed on 21 December 2021).
- Frater, J.L.; Zini, G.; d’Onofrio, G.; Rogers, H.J. COVID-19 and the clinical hematology laboratory. Int. J. Lab. Hematol. 2020, 42 (Suppl. S1), 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Dubey, S.; Biswas, P.; Ghosh, R.; Chatterjee, S.; Dubey, M.J.; Chatterjee, S.; Lahiri, D.; Lavie, C.J. Psychosocial impact of COVID-19. Diabetes Metab. Syndr. 2020, 14, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E. Epidemiology, virology, and clinical features of severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2; Coronavirus Disease-19). Clin. Exp. Pediatr. 2020, 63, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, Y.A. Properties of Coronavirus and SARS-CoV-2. Malays. J. Pathol. 2020, 42, 3–11. [Google Scholar] [PubMed]
- Tyrrel, D.A.J. Coronaviruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.Y.; Poon, R.W.S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. 2020, 133, 1015–1024. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Cai, X.P. An overview of COVID-19. J. Zhejiang Univ. Sci. B 2020, 21, 343–360. [Google Scholar] [CrossRef]
- Ren, S.Y.; Wang, W.B.; Hao, Y.G.; Zhang, H.R.; Wang, Z.C.; Chen, Y.L.; Gao, R.D. Stability and infectivity of coronaviruses in inanimate environments. World J. Clin. Cases 2020, 8, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.L.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Mazinani, M.; Rude, B.J. The novel zoonotic Coronavirus disease 2019 (COVID-19) pandemic: Health perspective on the outbreak. J. Healthc. Qual. Res. 2021, 36, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Rochwerg, B.; Agoritsas, T.; Lamontagne, F.; Agoritsas, T.; Lamontagne, F.; Askie, L.; Lytvyn, L.; Leo, Y.S.; Macdonald, H.; Zeng, L.; et al. A living WHO guideline on drugs for COVID-19. BMJ 2020, 370, m3379. [Google Scholar]
- National Institute for Health and Care Excellence (NICE). COVID-19 rapid guideline: Antibiotics for pneumonia in adults in hospital. Available online: https://www.nice.org.uk/guidance/ng191 (accessed on 22 December 2021).
- Masterton, R.G.; Galloway, A.; French, G.; Street, M.; Armstrong, J.; Brown, E.; Cleverley, J.; Dilworth, P.; Fry, C.; Gascoigne, A.D.; et al. Guidelines for the management of hospital-acquired pneumonia in the UK: Report of the working party on hospital-acquired pneumonia of the British Society for Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 2008, 62, 5–34. [Google Scholar] [CrossRef]
- Vaillancourt, M.; Jorth, P. The Unrecognized Threat of Secondary Bacterial Infections with COVID-19. mBio 2020, 11, e01806-20. [Google Scholar] [CrossRef]
- Lansbury, L.; Lim, B.; Baskaran, V.; Lim, W.S. Co-infections in people with COVID-19: A systematic review and meta-analysis. J. Infect. 2020, 81, 266–275. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Du, R.H.; Liu, L.M.; Yin, W.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Hospitalization and Critical Care of 109 Decedents with COVID-19 Pneumonia in Wuhan, China. Ann. Am. Thorac. Soc. 2020, 17, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankey, G.A.; Sabath, L.D. Clinical Relevance of Bacteriostatic versus Bactericidal Mechanisms of Action in the Treatment of Gram-Positive Bacterial Infections. Clin. Infect. Dis. 2004, 38, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinner, S.H. Antibiotic use: Present and future. New Microbiol. 2007, 30, 321–325. [Google Scholar] [PubMed]
- Mückter, H. What is toxicology and how does toxicity occur? Best Pract. Res. Clin. Anaesthesiol. 2003, 17, 5–27. [Google Scholar] [CrossRef]
- Scott, A.K. Stereoisomers and Drug Toxicity. Drug Saf. 1993, 8, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Hutt, A.G.; O’Grady, J. Drug chirality: A consideration of the significance of the stereochemistry of antimicrobial agents. J. Antimicrob. Chemother. 1996, 37, 7–32. [Google Scholar] [CrossRef] [Green Version]
- Rouveix, B. Antibiotic Safety Assessment. Int. J. Antimicrob. Agents 2003, 21, 215–221. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Madden, S.; Park, B.K. Idiosyncratic Drug Reactions. Clin. Pharm. 1996, 31, 215–230. [Google Scholar] [CrossRef]
- Rouveix, B.; Coulombel, L.; Aymard, J.P.; Chau, F.; Abel, L. Amodiaquine-induced immune agranulocytosis. Br. J. Haematol. 1989, 71, 7–11. [Google Scholar] [CrossRef]
- Rolain, J.M.; Baquero, F. The refusal of the Society to accept antibiotic toxicity: Missing opportunities for therapy of severe infections. Clin. Microbiol. Infect. 2016, 22, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Yılmaz, Ç.; Özcengiz, G. Antibiotics: Pharmacokinetics, toxicity, resistance and multidrug efflux pumps. Biochem. Pharmacol. 2017, 133, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Available online: https://wellcomecollection.org/works/rdpck35v (accessed on 22 December 2021).
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsburg, A.S.; Klugman, K.P. COVID-19 pneumonia and the appropriate use of antibiotics. Lancet Glob. Health 2020, 8, e1453–e1454. [Google Scholar] [CrossRef]
- Cheng, G.; Dai, M.; Ahmed, S.; Hao, H.; Wang, X.; Yuan, Z. Antimicrobial Drugs in Fighting against Antimicrobial Resistance. Front. Microbiol. 2016, 7, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute of Medicine (US) Committee on Conflict of Interest in Medical Research, Education, and Practice. E, The Pathway from Idea to Regulatory Approval: Examples for Drug Development. In Conflict of Interest in Medical Research, Education, and Practice; Lo, B., Field, M.J., Eds.; National Academies Press (US): Washington, DC, USA, 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK22930/?report=classic (accessed on 22 December 2021).
- World Health Organization. 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis. Available online: https://www.who.int/publications/i/item/9789240021303 (accessed on 28 December 2021).
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Fleming, A.; Allison, V.D. Observations on a Bacteriolytic Substance (“Lysozyme”) Found in Secretions and Tissues. Br. J. Exp. Pathol. 1922, 3, 252–260. [Google Scholar]
- Sarges, R.; Witkop, B. Gramicidin AV The structure of valine- and isoleucine-gramicidin A. J. Am. Chem. Soc. 1965, 87, 2011–2020. [Google Scholar] [CrossRef]
- Dubos, R.J. Studies on a bactericidal agent extracted from a soil bacillus: I. preparation of the agent. its activity in vitro. J. Exp. Med. 1939, 70, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Stephens, J.M.; Marshall, J.H. Some properties of an immune factor isolated from the blood of actively immunized wax moth larvae. Can. J. Microbiol. 1962, 8, 719–725. [Google Scholar] [CrossRef]
- Broekaert, W.F.; Terras, F.R.; Cammue, B.P.; Osborn, R.W. Plant defensins: Novel antimicrobial peptides as components of the host defense system. Plant. Physiol. 1995, 108, 1353–1358. [Google Scholar] [CrossRef]
- De Coninck, B.M.; Sels, J.; Venmans, E.; Thys, W.; Goderis, I.J.; Carron, D.; Delauré, S.L.; Cammue, B.P.; De Bolle, M.F.; Mathys, J. Arabidopsis thaliana plant defensin AtPDF1.1 is involved in the plant response to biotic stress. New Phytol. 2010, 187, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.S.J.; Beuerman, R.W.; Dua, H.S.; Lakshminarayanan, R.; Mohammed, I. Strategies in Translating the Therapeutic Potentials of Host Defense Peptides. Front. Immunol. 2020, 11, 983. [Google Scholar] [CrossRef]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.C.; Sun, J.; Chen, J.X.; Li, H.Q.; Xu, H.; Lao, X.H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2015, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Biragyn, A.; Hoover, D.M.; Lubkowski, J.; Oppenheim, J.J. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 2004, 22, 181–215. [Google Scholar] [CrossRef]
- Wang, G. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol. Biol. 2015, 1268, 43–66. [Google Scholar]
- Powers, J.P.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Fruitwala, S.; El-Naccache, D.W.; Chang, T.L. Multifaceted immune functions of human defensins and underlying mechanisms. Semin. Cell Dev. Biol. 2019, 88, 163–172. [Google Scholar] [CrossRef]
- Xu, D.; Lu, W. Defensins: A Double-Edged Sword in Host Immunity. Front. Immunol. 2020, 11, 764. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Harwig, S.S.; Ganz, T.; Schilling, J.W.; Lehrer, R.I. Primary structures of three human neutrophil defensins. J. Clin. Investig. 1985, 76, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.E.; Bevins, C.L. Paneth cells of the human small intestine express an antimicrobial peptide gene. J. Biol. Chem. 1992, 267, 23216–23225. [Google Scholar] [CrossRef]
- Jones, D.E.; Bevins, C.L. Defensin-6 mRNA in human Paneth cells: Implications for antimicrobial peptides in host defense of the human bowel. FEBS Lett. 1993, 315, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Wilde, C.G.; Griffith, J.E.; Marra, M.N.; Snable, J.L.; Scott, R.W. Purification and characterization of human neutrophil peptide 4, a novel member of the defensin family. J. Biol. Chem. 1989, 264, 11200–11203. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, I.; Lehrer, R.I. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS Lett. 1996, 396, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Scheetz, T.; Bartlett, J.A.; Walters, J.D.; Schutte, B.C.; Casavant, T.L.; McCray, P.B., Jr. Genomics-based approaches to gene discovery in innate immunity. Immunol. Rev. 2002, 190, 137–145. [Google Scholar] [CrossRef]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Stenger, S.; Hanson, D.A.; Teitelbaum, R.; Dewan, P.; Niazi, K.R.; Froelich, C.J.; Ganz, T.; Thoma-Uszynski, S.; Melián, A.; Bogdan, C.; et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 1998, 282, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Abedin, A.; Mohammed, I.; Hopkinson, A.; Dua, H.S. A novel antimicrobial peptide on the ocular surface shows decreased expression in inflammation and infection. Investig. Ophthalmol. Vis. Sci. 2008, 49, 28–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, I.; Suleman, H.; Otri, A.M.; Kulkarni, B.B.; Chen, P.; Hopkinson, A.; Dua, H.S. Localization and gene expression of human beta-defensin 9 at the human ocular surface epithelium. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4677–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dua, H.S.; Otri, A.M.; Hopkinson, A.; Mohammed, I. In vitro studies on the antimicrobial peptide human beta-defensin 9 (HBD9): Signalling pathways and pathogen-related response (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 2014, 112, 50–73. [Google Scholar]
- Mohammed, I.; Mohanty, D.; Said, D.G.; Barik, M.R.; Reddy, M.M.; Alsaadi, A.; Das, S.; Dua, H.S.; Mittal, R. Antimicrobial peptides in human corneal tissue of patients with fungal keratitis. Br. J. Ophthalmol. 2020, 105, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Abedin, A.; Tsintzas, K.; Abedin, S.A.; Otri, A.M.; Hopkinson, A.; Mathew, M.; Dua, H.S. Increased expression of hepcidin and toll-like receptors 8 and 10 in viral keratitis. Cornea 2011, 30, 899–904. [Google Scholar] [CrossRef]
- Premratanachai, P.; Joly, S.; Johnson, G.K.; McCray, P.B., Jr.; Jia, H.P.; Guthmiller, J.M. Expression and regulation of novel human beta-defensins in gingival keratinocytes. Oral Microbiol. Immunol. 2004, 19, 111–117. [Google Scholar] [CrossRef]
- Oppenheim, F.G.; Xu, T.; McMillian, F.M.; Levitz, S.M.; Diamond, R.D.; Offner, G.; Troxler, R.F. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J. Biol. Chem. 1988, 263, 7472–7477. [Google Scholar] [CrossRef]
- Van der Spek, J.C.; Wyandt, H.E.; Skare, J.C.; Milunsky, A.; Oppenheim, F.G.; Troxler, R.F. Localization of the genes for histatins to human chromosome 4q13 and tissue distribution of the mRNAs. Am. J. Hum. Genet. 1989, 45, 381–387. [Google Scholar]
- Alford, M.A.; Baquir, B.; Santana, F.L.; Haney, E.F.; Hancock, R.E.W. Cathelicidin Host Defense Peptides and Inflammatory Signaling: Striking a Balance. Front. Microbiol. 2020, 11, 1902. [Google Scholar] [CrossRef]
- Lee, P.H.A.; Ohtake, T.; Zaiou, M.; Murakami, M.; Rudisill, J.A.; Lin, K.H.; Gallo, R.L. Expression of an additional cathelicidin antimicrobial peptide protects against bacterial skin infection. Proc. Natl. Acad. Sci. USA 2005, 102, 3750–3755. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.; Borregaard, N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef] [Green Version]
- Romeo, D.; Skerlavaj, B.; Bolognesi, M.; Gennaro, R. Structure and bactericidal activity of an antibiotic dodecapeptide purified from bovine neutrophils. J. Biol. Chem. 1988, 263, 9573–9575. [Google Scholar] [CrossRef]
- Lai, Y.; Adhikarakunnathu, S.; Bhardwaj, K.; Ranjith-Kumar, C.T.; Wen, Y.; Jordan, J.L.; Wu, L.H.; Dragnea, B.; Mateo, L.S.; Kao, C.C. LL37 and Cationic Peptides Enhance TLR3 Signaling by Viral Double-stranded RNAs. PLoS ONE 2011, 6, e26632. [Google Scholar] [CrossRef] [Green Version]
- Crane-Godreau, M.A.; Clem, K.J.; Payne, P.; Fiering, S. Vitamin D Deficiency and Air Pollution Exacerbate COVID-19 through Suppression of Antiviral Peptide LL37. Front. Public Health 2020, 8, 232. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [Green Version]
- Elsbach, P. What is the real role of antimicrobial polypeptides that can mediate several other inflammatory responses? J. Clin. Investig. 2003, 111, 1643–1645. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.M.; Dewan, P.; Ganz, T. Innate Antimicrobial Activity of Nasal Secretions. Infect. Immun. 1999, 67, 3267–3275. [Google Scholar] [CrossRef] [Green Version]
- Elphick, D.; Liddell, S.; Mahida, Y.R. Impaired luminal processing of human defensin-5 in Crohn’s disease: Persistence in a complex with chymotrypsinogen and trypsin. Am. J. Pathol. 2008, 172, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K. The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Sochacki, K.A.; Barns, K.J.; Bucki, R.; Weisshaar, J.C. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc. Natl. Acad. Sci. USA 2011, 108, E77–E81. [Google Scholar] [CrossRef] [Green Version]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Chikushi, A.; Tougu, S.; Imura, Y.; Nishida, M.; Yano, A.Y.; Matsuzaki, K. Membrane Translocation Mechanism of the Antimicrobial Peptide Buforin 2. Biochemistry 2004, 43, 15610–15616. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar]
- Hendrick, J.P.; Hartl, F.U. The role of molecular chaperones in protein folding. FASEB J. 1995, 9, 1559–1569. [Google Scholar] [CrossRef]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [Green Version]
- Mardirossian, M.; Pérébaskine, N.; Benincasa, M.; Gambato, S.; Hofmann, S.; Huter, P.; Müller, C.; Hilpert, K.; Innis, C.A.; Tossi, A.; et al. The Dolphin Proline-Rich Antimicrobial Peptide Tur1A Inhibits Protein Synthesis by Targeting the Bacterial Ribosome. Cell Chem. Biol. 2018, 25, 530–539.e7. [Google Scholar] [CrossRef] [Green Version]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef]
- He, S.-W.; Zhang, J.; Li, N.-Q.; Zhou, S.; Yue, B.; Zhang, M. A TFPI-1 peptide that induces degradation of bacterial nucleic acids, and inhibits bacterial and viral infection in half-smooth tongue sole, Cynoglossus semilaevis. Fish Shellfish Immunol. 2017, 60, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Gusman, H.; Travis, J.; Helmerhorst, E.J.; Potempa, J.; Troxler, R.F.; Oppenheim, F.G. Salivary Histatin 5 Is an Inhibitor of Both Host and Bacterial Enzymes Implicated in Periodontal Disease. Infect. Immun. 2001, 69, 1402–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gläser, R.; Harder, J.; Lange, H.; Harder, J.; Lange, H.; Bartels, J.; Christophers, E.; Schröder, J.-M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol. 2004, 6, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef] [Green Version]
- Van Wetering, S.; Mannesse-Lazeroms, S.P.; Van Sterkenburg, M.A.; Daha, M.R.; Dijkman, J.H.; Hiemstra, P.S. Effect of defensins on interleukin-8 synthesis in airway epithelial cells. Am. J. Physiol. 1997, 272, L888–L896. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Chertov, O.; Oppenheim, J.J. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J. Leukoc. Biol. 2000, 68, 9–14. [Google Scholar]
- Choi, K.-Y.; Mookherjee, N. Multiple Immune-Modulatory Functions of Cathelicidin Host Defense Peptides. Front. Immunol. 2012, 3, 149. [Google Scholar] [CrossRef] [Green Version]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef]
- Hemshekhar, M.; Choi, K.-Y.G.; Mookherjee, N. Host Defense Peptide LL-37-Mediated Chemoattractant Properties, but Not Anti-Inflammatory Cytokine IL-1RA Production, Is Selectively Controlled by Cdc42 Rho GTPase via G Protein-Coupled Receptors and JNK Mitogen-Activated Protein Kinase. Front. Immunol. 2018, 9, 1871. [Google Scholar] [CrossRef] [Green Version]
- Nijnik, A.; Pistolic, J.; Filewod, N.C.J.; Hancock, R.E.W. Signaling Pathways Mediating Chemokine Induction in Keratinocytes by Cathelicidin LL-37 and Flagellin. J. Innate Immun. 2012, 4, 377–386. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Ushio, H.; Hara, M.; Pistolic, J.; Filewod, N.C.J.; Hancock, R.E.W. Antimicrobial peptides human beta-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cells. J. Immunol. 2010, 184, 3526–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Kai-Larsen, Y.; Frithiof, R.; Sorensen, O.E.; Kenne, E.; Scharffetter-Kochanek, K.; Eriksson, E.E.; Herwald, H.; Agerberth, B.; Lindbom, L. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. J. Clin. Investig. 2008, 118, 3491–3502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjabringa, G.S.; Ninaber, D.K.; Drijfhout, J.W.; Rabe, K.F.; Hiemstra, P.S. Human Cathelicidin LL-37 Is a Chemoattractant for Eosinophils and Neutrophils That Acts via Formyl-Peptide Receptors. Int. Arch. Allergy Immunol. 2006, 140, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, I.; Tamura, H.; Hirata, M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J. Immunol. 2006, 176, 3044–3052. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Chow, L.N.Y.; Mookherjee, N. Cationic Host Defence Peptides: Multifaceted Role in Immune Modulation and Inflammation. J. Innate Immun. 2012, 4, 361–370. [Google Scholar] [CrossRef]
- Von Köckritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Bandholtz, L.; Ekman, G.J.; Vilhelmsson, M.; Buentke, E.; Agerberth, B.; Scheynius, A.; Gudmundsson, G.H. Antimicrobial Peptide LL-37 Internalized by Immature Human Dendritic Cells Alters their Phenotype. Scand. J. Immunol. 2006, 63, 410–419. [Google Scholar] [CrossRef]
- Davidson, D.J.; Currie, A.J.; Reid, G.S.; Bowdish, D.M.E.; MacDonald, K.L.; Ma, R.C.; Hancock, R.E.W.; Speert, D.P. The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J. Immunol. 2004, 172, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Kin, N.W.; Chen, Y.; Stefanov, E.K.; Bowdish, D.; Macdonald, K.L.; Ma, R.C.; Hancock, R.E.W.; Speert, D.P. Cathelin-related antimicrobial peptide differentially regulates T- and B-cell function. Eur. J. Immunol. 2011, 41, 3006–3016. [Google Scholar] [CrossRef] [Green Version]
- Hemshekhar, M.; Anaparti, V.; Mookherjee, N. Functions of Cationic Host Defense Peptides in Immunity. Pharmaceuticals 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciornei, C.D.; Sigurdardóttir, T.; Schmidtchen, A.; Bodelsson, M. Antimicrobial and chemoattractant activity, lipopolysaccharide neutralization, cytotoxicity, and inhibition by serum of analogs of human cathelicidin LL-37. Antimicrob. Agents Chemother. 2005, 49, 2845–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookherjee, N.; Brown, K.L.; Bowdish, D.M.; Doria, S.; Falsafi, R.; Hokamp, K.; Roche, F.M.; Mu, R.; Doho, G.H.; Pistolic, J.; et al. Modulation of the TLR-mediated inflammatory response by the endogenous human host defense peptide LL-37. J. Immunol. 2006, 176, 2455–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookherjee, N.; Hamill, P.; Gardy, J.; Blimkie, D.; Falsafi, R.; Chikatamarla, A.; Arenillas, D.J.; Doria, S.; Kollmann, T.R.; Hancock, R.E.W. Systems biology evaluation of immune responses induced by human host defence peptide LL-37 in mononuclear cells. Mol. BioSystems 2009, 5, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Turner-Brannen, E.; Choi, K.-Y.; Lippert, D.N.D.; Cortens, J.P.; Hancock, R.E.; El-Gabalawy, H.; Mookherjee, N. Modulation of interleukin-1β-induced inflammatory responses by a synthetic cationic innate defence regulator peptide, IDR-1002, in synovial fibroblasts. Arthritis Res. Ther. 2011, 13, R129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.V.; Fiedoruk, K.; Daniluk, T.; Piktel, E.; Bucki, R. Expression and Function of Host Defense Peptides at Inflammation Sites. Int. J. Mol. Sci. 2020, 21, 104. [Google Scholar] [CrossRef] [Green Version]
- Shaykhiev, R.; Sierigk, J.; Herr, C.; Krasteva, G.; Kummer, W.; Bals, R. The antimicrobial peptide cathelicidin enhances activation of lung epithelial cells by LPS. FASEB J. 2010, 24, 4756–4766. [Google Scholar] [CrossRef]
- Heilborn, J.D.; Nilsson, M.F.; Sørensen, O.; Weber, G.; Sørensen, O.; Borregaard, N.; Ståhle-Bäckdahl, M. The Cathelicidin Anti-Microbial Peptide LL-37 is Involved in Re-Epithelialization of Human Skin Wounds and is Lacking in Chronic Ulcer Epithelium. J. Investig. Dermatol. 2003, 120, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Niyonsaba, F.; Ushio, H.; Nakano, N.; Ng, W.; Sayama, K.; Hashimoto, K.; Nagaoka, I.; Okumura, K.; Ogawa, H. Antimicrobial Peptides Human β-Defensins Stimulate Epidermal Keratinocyte Migration, Proliferation and Production of Proinflammatory Cytokines and Chemokines. J. Investig. Dermatol. 2007, 127, 594–604. [Google Scholar] [CrossRef] [Green Version]
- European Surveillance of Veterinary Antimicrobial Consumption (ESVAC) (2018). Sales of Veterinary Antimicrobial Agents in 30 European Countries in 2016. Available online: https://www.ema.europa.eu/en/documents/report/sales-veterinary-antimicrobial-agents-30-european-countries-2016-trends-2010-2016-eighth-esvac_en.pdf (accessed on 15 October 2020).
- Santé Publique France (2018) Consommation D’antibiotiques et Résistance Aux Antibiotiques en France: Une Infection Évitée, C’est un Antibiotique Préservé! Available online: https://www.santepubliquefrance.fr/maladies-et-traumatismes/infections-associees-aux-soins-et-resistance-aux-antibiotiques/resistance-aux-antibiotiques/documents/rapport-synthese/consommation-d-antibiotiques-et-resistance-aux-antibiotiques-en-france-une-infection-evitee-c-est-un-antibiotique-preserve (accessed on 15 October 2019).
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2015, 40, 133–159. [Google Scholar] [CrossRef] [Green Version]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Wang, W.; Owen, S.M.; Rudolph, D.L.; Cole, a.m.; Hong, t.; Waring, A.J.; Lal, R.B.; Lehrer, R.I. Activity of alpha- and theta-defensins against primary isolates of HIV-1. J. Immunol. 2004, 173, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Quiñones-Mateu, M.E.; Lederman, M.M.; Feng, Z.; Chakraborty, B.; Weber, J.; Rangel, H.R.; Marotta, M.L.; Mirza, M.; Jiang, B.; Kiser, P.; et al. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. Aids 2003, 17, F39–F48. [Google Scholar] [CrossRef]
- Den Hertog, A.L.; van Marle, J.; Veerman, E.C.; Valentijn-Benz, M.; Nazmi, K.; Kalay, H.; Grün, C.H.; Van’t Hof, W.; Bolscher, J.G.M. Amerongen, A.V.N. The human cathelicidin peptide LL-37 and truncated variants induce segregation of lipids and proteins in the plasma membrane of Candida albicans. Biol. Chem. 2006, 387, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Dabirian, S.; Taslimi, Y.; Zahedifard, F.; Gholami, E.; Doustdari, F.; MotamediRad, M.; Khatami, S.; Azadmanesh, K.; Nylén, S.; Rafati, S. Human neutrophil peptide-1 (HNP-1): A new anti-leishmanial drug candidate. PLoS Negl. Trop. Dis. 2013, 7, e2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.S.; Fu, C.I.; Otto, M. Bacterial strategies of resistance to antimicrobial peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Cha, D.J.; Lai, Y.; Villaruz, A.E.; Sturdevant, D.E.; Otto, M. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol. Microbiol. 2007, 66, 1136–1147. [Google Scholar] [CrossRef]
- Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics. 2014, 4, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Vester, B.; Long, K.S. Antibiotic Resistance in Bacteria Caused by Modified Nucleosides in 23S Ribosomal RNA. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000–2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6514/ (accessed on 22 December 2021).
- Baltzer, S.A.; Brown, M.H. Antimicrobial Peptides–Promising Alternatives to Conventional Antibiotics. Microb. Physiol. 2011, 20, 228–235. [Google Scholar] [CrossRef]
- Peschel, A.; Sahl, H.G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Bierbaum, G.; Sahl, H.G. Lantibiotics: Mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 2009, 10, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Perron, G.G.; Zasloff, M.; Bell, G. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 2006, 273, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Lewies, A.; du Plessis, L.; Wentzel, J.F. Antimicrobial Peptides: The Achilles’ Heel of Antibiotic Resistance? Probiotics Antimicrob. Proteins 2019, 11, 370–381. [Google Scholar] [CrossRef]
- Mohammed, I.; Said, D.G.; Nubile, M.; Said, D.G.; Nubile, M.; Mastropasqua, L.; Dua, H.S. Cathelicidin-Derived Synthetic Peptide Improves Therapeutic Potential of Vancomycin Against Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, D.S.J.; Goh, E.T.L.; Mayandi, V.; Busoy, J.M.F.; Aung, T.T.; Periayah, M.H.; Nubile, M.; Mastropasqua, L.; Said, D.G.; Htoon, H.M.; et al. Hybrid derivative of cathelicidin and human beta defensin-2 against Gram-positive bacteria: A novel approach for the treatment of bacterial keratitis. Sci. Rep. 2021, 11, 18304. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.S.J.; Li, J.; Verma, C.S.; Goh, E.T.L.; Nubile, M.; Mastropasqua, L.; Said, D.G.; Beuerman, R.W.; Lakshminarayanan, R.; Mohammed, I.; et al. Evaluation of Host Defense Peptide (CaD23)-Antibiotic Interaction and Mechanism of Action: Insights From Experimental and Molecular Dynamics Simulations Studies. Front. Pharmacol. 2021, 12, 731499. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Alford, M.A.; Haney, E.F. Antibiofilm activity of host defence peptides: Complexity provides opportunities. Nat. Rev. Microbiol. 2021, 19, 786–797. [Google Scholar] [CrossRef]
- De la Fuente-Nunez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, K.G.N.; Rodrigues, G.; Monges, B.E.D.; Cardoso, M.H.; Franco, O.L. Bioactive Peptides Against Fungal Biofilms. Front. Microbiol. 2019, 10, 2169. [Google Scholar] [CrossRef]
- Hell, É.; Giske, C.G.; Nelson, A.; RöMling, U.; Marchini, G. Human cathelicidin peptide LL37 inhibits both attachment capability and biofilm formation of Staphylococcus epidermidis. Lett. Appl. Microbiol. 2010, 50, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wu, Z.; Mao, C.; Guo, G.; Zhu, Z.; Ying, F.; Shan, W.; Jian, P.; Wu, J.W. Antimicrobial Peptide Cec4 Eradicates the Bacteria of Clinical Carbapenem-Resistant Acinetobacter baumannii Biofilm. Front. Microbiol. 2020, 11, 1532. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Nunez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libardo, M.D.; Bahar, A.A.; Ma, B.; Fu, R.; McCormick, L.; Zhao, J.; McCallum, S.A.; Nussinov, R.; Ren, D.; Angeles-Boza, A.M.; et al. Nuclease activity gives an edge to host-defense peptide piscidin 3 over piscidin 1, rendering it more effective against persisters and biofilms. FEBS J. 2017, 284, 3662–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, K.-I.; Zendo, T.; Sugimoto, S.; Iwase, T.; Tajima, A.; Yamada, S.; Sonomoto, K.; Mizunoe, Y. Effects of bacteriocins on methicillin-resistant Staphylococcus aureus biofilm. Antimicrob. Agents Chemother. 2013, 57, 5572–5579. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.C.; Yu, W.L. COVID-19 associated with pulmonary aspergillosis: A literature review. J. Microbiol. Immunol. Infect. 2021, 54, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Lackner, N.; Thomé, C.; Öfner, D.; Joannidis, M.; Mayerhöfer, T.; Arora, R.; Samardzic, E.; Posch, W.; Breitkopf, R.; Lass-Flörl, C. COVID-19 Associated Pulmonary Aspergillosis: Diagnostic Performance, Fungal Epidemiology and Antifungal Susceptibility. J. Fungi 2022, 8, 93. [Google Scholar] [CrossRef]
- El-Baba, F.; Gao, Y.; Soubani, A.O. Pulmonary Aspergillosis: What the Generalist Needs to Know. Am. J. Med. 2020, 133, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Čeřovský, V.; Buděšínský, M.; Hovorka, O.; Cvacka, J.; Voburka, Z.; Slaninová, J.; Borovicková, L.; Fucík, V.; Bednárová, L.; Votruba, I.; et al. Lasioglossins: Three novel antimicrobial peptides from the venom of the eusocial bee Lasioglossum laticeps (Hymenoptera: Halictidae). Chembiochem 2009, 10, 2089–2099. [Google Scholar] [CrossRef]
- Mendes, M.A.; de Souza, B.M.; Marques, M.R.; Palma, M.S. Structural and biological characterization of two novel peptides from the venom of the neotropical social wasp Agelaia pallipes pallipes. Toxicon 2004, 44, 67–74. [Google Scholar] [CrossRef]
- Tan, L.; Bai, L.; Wang, L.; He, L.; Li, G.; Du, W.; Shen, T.; Xiang, Z.; Wu, J.; Liu, Z.; et al. Antifungal activity of spider venom-derived peptide lycosin-I against Candida tropicalis. Microbiol. Res. 2018, 216, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Freitas, C.G.; Lima, S.M.D.F.; Freire, M.S.; Cantuária, A.P.C.; Júnior, N.; Santos, T.S.; Folha, J.S.; Ribeiro, S.M.; Dias, S.C.; Rezende, T.; et al. An Immunomodulatory Peptide Confers Protection in an Experimental Candidemia Murine Model. Antimicrob. Agents Chemother. 2017, 61, e02518-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletzer, D.; Mansour, S.C.; Hancock, R.E.W. Synergy between conventional antibiotics and anti-biofilm peptides in a murine, sub-cutaneous abscess model caused by recalcitrant ESKAPE pathogens. PLoS Pathog. 2018, 14, e1007084. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Chai, J. Door to the cell for COVID-19 opened, leading way to therapies. Signal Transduct. Target Ther. 2020, 5, 104. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Weinberg, A. Ramping Up Antimicrobial Peptides Against Severe Acute Respiratory Syndrome Coronavirus-2. Front. Mol. Biosci. 2021, 8, 620806. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, D.; Wei, D.Q.; Zhao, J.; Wang, J. Human Intestinal Defensin 5 Inhibits SARS-CoV-2 Invasion by Cloaking ACE2. Gastroenterology 2020, 159, 1145–1147.e1144. [Google Scholar] [CrossRef]
- Negahdaripour, M.; Rahbar, M.R.; Mosalanejad, Z.; Gholami, A. Theta-Defensins to Counter COVID-19 as Furin Inhibitors: In Silico Efficiency Prediction and Novel Compound Design. Comput. Math. Methods Med. 2022, 2022, 9735626. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, D.; Chen, P.; Han, S.; Zhao, G.; Chen, Y.; Zhao, J.; Xiong, J.; Qiu, J.; et al. Human Cathelicidin Inhibits SARS-CoV-2 Infection: Killing Two Birds with One Stone. ACS Infect. Dis. 2021, 7, 1545–1554. [Google Scholar] [CrossRef]
- Zhang, L.; Ghosh, S.K.; Basavarajappa, S.C.; Chen, Y.; Shrestha, P.; Penfield, J.; Brewer, A.; Ramakrishnan, P.; Buck, M.; Weinberg, A. HBD-2 binds SARS-CoV-2 RBD and blocks viral entry: Strategy to combat COVID-19. iScience 2022, 25, 103856. [Google Scholar] [CrossRef]
- Brice, D.C.; Diamond, G. Antiviral Activities of Human Host Defense Peptides. Curr. Med. Chem. 2020, 27, 1420–1443. [Google Scholar] [CrossRef] [PubMed]
- Bakovic, A.; Risner, K.; Bhalla, N.; Lem, F.; Chang, T.L.; Weston, W.; Harness, J.A.; Narayanan, A. Brilacidin, a COVID-19 Drug Candidate, Exhibits Potent In Vitro Antiviral Activity Against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Young, B.; Tan, T.T.; Leo, Y.S. The place for remdesivir in COVID-19 treatment. Lancet Infect. Dis. 2021, 21, 20–21. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Idris, M.M.; Banu, S.; Siva, A.B.; Nagaraj, R. Downregulation of Defensin genes in SARS-CoV-2 infection. medRxiv 2020. [Google Scholar] [CrossRef]
- Pahar, B.; Madonna, S.; Das, A.; Albanesi, C.; Girolomoni, G. Immunomodulatory Role of the Antimicrobial LL-37 Peptide in Autoimmune Diseases and Viral Infections. Vaccines 2020, 8, 517. [Google Scholar] [CrossRef]
- Park, M.S.; Kim, J.I.; Lee, I.; Park, S.; Bae, J.Y.; Park, M.S. Towards the Application of Human Defensins as Antivirals. Biomol. Ther. 2018, 26, 242–254. [Google Scholar] [CrossRef]
- Arya, B. Defensins-Non-antibiotic Use for Vaccine Development. Curr. Protein Pept. Sci. 2005, 6, 53–60. [Google Scholar]
- Kim, J.; Yang, Y.L.; Jang, S.H.; Jang, Y.S. Human β-defensin 2 plays a regulatory role in innate antiviral immunity and is capable of potentiating the induction of antigen-specific immunity. Virol. J. 2018, 15, 124. [Google Scholar] [CrossRef] [Green Version]
- Björn, C.; Noppa, L.; Salomonsson, E.N.; Johansson, A.-L.; Nilsson, E.; Mahlapuu, M.; Håkansson, J. Efficacy and safety profile of the novel antimicrobial peptide PXL150 in a mouse model of infected burn wounds. Int. J. Antimicrob. Agents 2015, 45, 519–524. [Google Scholar] [CrossRef]
- Monteiro, C.; Costa, F.; Pirttilä, A.M.; Tejesvi, M.V.; Martins, M.C.L. Prevention of urinary catheter-associated infections by coating antimicrobial peptides from crowberry endophytes. Sci. Rep. 2019, 9, 10753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas-Santiago, B.; Trujillo, V.; Montoya, A.; Gonzalez-Curiel, I.; Castañeda-Delgado, J.; Cardenas, A.; Rincon, K.; Hernandez, M.L.; Hernán-dez-Pando, R. Expression of antimicrobial peptides in diabetic foot ulcer. J. Dermatol. Sci. 2012, 65, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.C.; Sarmento, B.; Pintado, M. The importance of antimicrobial peptides and their potential for therapeutic use in ophthalmology. Int. J. Antimicrob. Agents. 2013, 41, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Said, D.G.; Dua, H.S. Human antimicrobial peptides in ocular surface defense. Prog. Retin. Eye Res. 2017, 61, 1–22. [Google Scholar] [CrossRef]
- Mohammed, I.; Mohanty, D.; Said, D.G.; Barik, M.R.; Reddy, M.M.; Alsaadi, A.; Das, S.; Dua, H.S.; Mittal, R. Signalling pathways involved in ribonuclease-7 expression. Cell Mol. Life Sci. 2011, 68, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Otri, A.M.; Mohammed, I.; Abedin, A.; Cao, Z.; Hopkinson, A.; Panjwani, N.; Dua, H.S. Antimicrobial peptides expression by ocular surface cells in response to Acanthamoeba castellanii: An in vitro study. Br. J. Ophthalmol. 2010, 94, 1523–1527. [Google Scholar] [CrossRef] [Green Version]
- Otri, A.M.; Mohammed, I.; Al-Aqaba, M.A.; Fares, U.; Peng, C.; Hopkinson, A.; Dua, H.S. Variable expression of human Beta defensins 3 and 9 at the human ocular surface in infectious keratitis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 757–761. [Google Scholar] [CrossRef]
- Scott, M.G.; Dullaghan, E.; Mookherjee, N.; Glavas, N.; Waldbrook, M.; Thompson, A.; Wang, A.; Lee, K.; Doria, S.; Hamill, P.; et al. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 2007, 25, 465–472. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Castañeda-Delgado, J.E.; Rivas-Santiago, C.; Waldbrook, M.; Gonzalez, I.; León–Contreras, J.C.; Enciso-Moreno, J.A.; Del Villar, V.; Mendez-Ramos, J.; Hancock, R.; et al. Ability of innate defence regulator peptides IDR-1002, IDR-HH2 and IDR-1018 to protect against Mycobacterium tuberculosis infections in animal models. PLoS ONE 2013, 8, e59119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afacan, N.J.; Yeung, A.T.Y.; Pena, O.M.; Hancock, R.E.W. Therapeutic Potential of Host Defense Peptides in Antibiotic-resistant Infections. Curr. Pharm. Des. 2012, 18, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E.W. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Hirsch, T.; Schulte, M.; Kueckelhaus, M.; Jacobsen, F.; Mersch, E.A.; Stricker, I.; Afacan, N.; Jenssen, H.; Hancock, R.; et al. Innate Defense Regulator Peptide 1018 in Wound Healing and Wound Infection. PLoS ONE 2012, 7, e39373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolouri, H.; Sävman, K.; Wang, W.; Thomas, A.; Maurer, N.; Dullaghan, E.; Fjell, C.D.; Ek, C.J.; Hagberg, H.; Hancock, R.E.W.; et al. Innate defense regulator peptide 1018 protects against perinatal brain injury. Ann. Neurol. 2014, 75, 395–410. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Izumiya, N.; Kato, T.; Waki, M. Synthesis of biologically active cyclic peptides. Biopolymers 1981, 20, 1785–1791. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef]
- Yamasaki, K.; Di Nardo, A.; Bardan, A.; Murakami, M.; Ohtake, T.; Coda, A.; Dorschner, R.A.; Bonnart, C.; Descargues, P.; Hovnanian, A.; et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat. Med. 2007, 13, 975–980. [Google Scholar] [CrossRef]
- Morizane, S.; Yamasaki, K.; Mühleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin Antimicrobial Peptide LL-37 in Psoriasis Enables Keratinocyte Reactivity against TLR9 Ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Jang, J.H.; Kim, S.C.; Cho, J.H. De novo generation of short antimicrobial peptides with enhanced stability and cell specificity. J. Antimicrob. Chemother. 2013, 69, 121–132. [Google Scholar] [CrossRef]
- Van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can Animal Models of Disease Reliably Inform Human Studies? PLoS Med. 2010, 7, e1000245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A. The big and small of drug discovery. Biotech versus pharma: Advantages and drawbacks in drug development. EMBO Rep. 2003, 4, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithoor, D.; Madhu, K.M.; Dhanasekhar, R.; Ranjith, K.; Preetam, G.; Vasco, A.; Debmalya, B. Clinical Applications of Antimicrobial Peptides (AMPs): Where do we Stand Now? Protein Pept. Lett. 2020, 27, 120–134. [Google Scholar]
- Katarzyna, E.G.; Małgorzata, D. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A Review of Antimicrobial Peptides and Their Therapeutic Potential as Anti-Infective Drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Chereddy, K.K.; Vandermeulen, G.; Préat, V. PLGA based drug delivery systems: Promising carriers for wound healing activity. Wound Repair Regen. 2016, 24, 223–236. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Creran, B.; Rotello, V.M. Gold nanoparticles: Preparation, properties, and applications in bionanotechnology. Nanoscale 2012, 4, 1871–1880. [Google Scholar] [CrossRef]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef]


{kind=link}
| Severity | Clinical Symptoms |
|---|---|
| Mild | Fever Cough No radiological abnormalities |
| Moderate | Pneumonia |
| Severe | Respiratory distress—respiratory rate ≥30/min Oxygen saturation ≤93% |
| Critical | Acute Respiratory Distress Syndrome (ARDS)—use of mechanical ventilation Multi-organ failure |
| Empirical Treatment | Antibiotics | Dosage 1 |
|---|---|---|
| Oral antibiotics for moderate or severe pneumonia | Options include: | |
| Doxycycline | 200 mg on first day, then 100 mg once a day | |
| Co-amoxiclav and Clarithromycin | 500 mg/125 mg three times a day 500 mg twice a day | |
| In severe pneumonia and if the above options are unsuitable: | ||
| Levofloxacin | 500 mg once or twice a day (consideration given to the safety issues with fluoroquinolones) | |
| Intravenous antibiotics for moderate or severe pneumonia | Options include: | |
| Co-amoxiclav and Clarithromycin | 1.2 g three times a day 500 mg twice a day | |
| Cefuroxime and Clarithromycin | 750 mg three times a day 2 500 mg twice a day | |
| In severe pneumonia and if the above options are unsuitable: | ||
| Levofloxacin | 500 mg once or twice a day (consideration given to the safety issues with fluoroquinolones) | |
| Empirical Treatment | Antibiotics | Dosage 1 |
|---|---|---|
| Oral antibiotics for non-severe pneumonia when there is not a higher risk of resistance | Options include: | |
| Doxycycline | 200 mg on first day, then 100 mg once a day | |
| Co-amoxiclav | 500 mg/125 mg three times a day | |
| Co-trimoxazole | 960 mg twice a day 2 | |
| If other options are unsuitable: | ||
| Levofloxacin | 500 mg once or twice a day (consideration given to the safety issues with fluoroquinolones) | |
| Intravenous antibiotics for severe pneumonia (for example, symptoms or signs of sepsis or ventilator-associated pneumonia) or when there is a higher risk of resistance | Options include: | |
| Piperacillin with tazobactam | 4.5 g three times a day, increased to 4.5 g four times a day of infection is severe | |
| Ceftazidime | 2 g three times a day | |
| If other options are unsuitable: | ||
| Levofloxacin | 500 mg once or twice a day (consideration given to the safety issues with fluoroquinolones) | |
| Antibiotic to be added if methicillin-resistant Staphylococcus aureus infection is suspected or confirmed (dual therapy with an intravenous antibiotic listed above) | Vancomycin | 15 mg/kg to 20 mg/kg two or three times a day intravenously, adjusted according to serum vancomycin concentration. Maximum 2 g per dose 3 |
| Teicoplanin | Initially 6 mg/kg every 12 h for 3 doses intravenously, then 6 mg/kg once a day 3 | |
| Linezolid | 600 mg twice a day orally or intravenously (with specialist advice only) 2 | |
| Antibiotic Class | Dose-Related Adverse Effects |
|---|---|
| β-lactams | Hepatotoxicity Neutropenia Encephalopathy Transient increase in liver enzymes Neuronal excitation Myoclonus Seizures |
| Cephalosporins | Neutropenia Nephrotoxicity Neurotoxicity Tonic–clonic seizures |
| Carbapenems | Neurotoxicity Seizures |
| Fluoroquinolones | Cardiovascular disorders Tendinopathy Phototoxicity Neuropathy Hepatotoxicity |
| Macrolides | Cardiac toxicity Gastrointestinal disturbances Hepatotoxicity Ototoxicity |
| Glycopeptides | Nephrotoxicity Phototoxicity Auditory nerve damage Stevens-Johnson Syndrome (SJS) |
| Aminoglycosides | Ototoxicity Nephrotoxicity Interference with mitochondrial respiration, protein synthesis and sodium–potassium pump |
| Polymyxins | Neurotoxicity Nephrotoxicity |
| Name | Amino Acid Sequence | Source |
|---|---|---|
| HNP-1 | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | Neutrophils Bone marrow |
| HNP-2 | CYCRIPACIAGERRYGTCIYQGRLWAFCC | Neutrophils Bone marrow |
| HNP-3 | DCYCRIPACIAGERRYGTCIYQGRLWAFCC | Neutrophils Bone marrow |
| HNP-4 | VCSCRLVFCRRTELRVGNCLIGGVSFTYCCTRV | Neutrophils |
| HD-5 | ATCYCRTGRCATRESLSGVCEISGRLYRLCCR | Paneth Cells (intestinal epithelium) Female reproductive system |
| HD-6 | AFTCHCRRSCYSTEYSYGTCTVMGINHRFCCL | Paneth Cells (intestinal epithelium) |
| hBD-1 | DHYNCVSSGGQCLYSACPIFTKIQGTCYRGKAKCCK | Kidney Skin Salivary glands |
| Histatin 1 | DSHEKRHHGYRRKFHEKHHSHREFPFYGDYGSNYLYDN | Saliva |
| Histatin 3 | DSHAKRHHGYKRKFHEKHHSHRGYRSNYLYDN | Saliva |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | Neutrophils Skin |
| HDPs | Synergistic Interaction with Antibiotics | Microorganism |
|---|---|---|
| Cryptidin-2 | Ampicillin | Salmonella typhimurium |
| Arenicin-1 | Ampicillin | Staphylococcus aureus |
| Erythromycin | Staphylococcus epidermidis | |
| Chloramphenicol | Pseudomonas aeruginosa Escherichia coli | |
| Nisin Z | Penicillin | Pseudomonas fluorescens LRC-R73 |
| Streptomycin | Penicillin-resistant variant | |
| Leucomycin | ||
| Rifampicin | Streptomycin-resistant variant Lincomycin-resistant variant Rifampicin-resistant variant | |
| Nisin | Daptomycin | Methicillin resistant Staphylococcus aureus (MRSA) biofilms |
| Indolicidin | Teicoplanin | |
| CAMA | Ciprofloxacin | |
| Nisin | Ampicillin | Staphylococcus aureus |
| Daptomycin | Enterococcus faecalis | |
| Ampicillin | Salmonella typhimurium | |
| Cefotaxime | ||
| Ceftriaxone | ||
| Brevinin-2 CE | Levofloxacin | ESBL producing Escherichia coli |
| Amoxicillin | ||
| Chloramphenicol | Methicillin-resistant Staphylococcus aureus (MRSA) | |
| Human beta defensin 3 | Tigecycline | Clostridium difficile |
| Cathelicidin (LL-37) | Moxifloxacin | |
| Piperacillin-Tazobactam | ||
| Meropenem | ||
| Azithromycin | MDR Pseudomonas aeruginosa MDR Klebsiella pneumonia MDR Acinetobacter baumannii | |
| LL-37 derivatives | Vancomycin, Chloramphenicol | Pseudomonas aeruginosa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, W.; Elsahn, A.; Ting, D.S.J.; Dua, H.S.; Mohammed, I. Host Defence Peptides: A Potent Alternative to Combat Antimicrobial Resistance in the Era of the COVID-19 Pandemic. Antibiotics 2022, 11, 475. https://doi.org/10.3390/antibiotics11040475
Ali W, Elsahn A, Ting DSJ, Dua HS, Mohammed I. Host Defence Peptides: A Potent Alternative to Combat Antimicrobial Resistance in the Era of the COVID-19 Pandemic. Antibiotics. 2022; 11(4):475. https://doi.org/10.3390/antibiotics11040475
Chicago/Turabian StyleAli, Waqas, Ahmad Elsahn, Darren S. J. Ting, Harminder S. Dua, and Imran Mohammed. 2022. "Host Defence Peptides: A Potent Alternative to Combat Antimicrobial Resistance in the Era of the COVID-19 Pandemic" Antibiotics 11, no. 4: 475. https://doi.org/10.3390/antibiotics11040475
APA StyleAli, W., Elsahn, A., Ting, D. S. J., Dua, H. S., & Mohammed, I. (2022). Host Defence Peptides: A Potent Alternative to Combat Antimicrobial Resistance in the Era of the COVID-19 Pandemic. Antibiotics, 11(4), 475. https://doi.org/10.3390/antibiotics11040475