
Stapling of Peptides Potentiates the Antibiotic Treatment of Acinetobacter baumannii In Vivo

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Solid-Phase Peptide Synthesis
2.3. Olefin Crosslink
2.4. Peptide Cleavage and Purification
2.5. Characterization
2.6. Circular Dichroism Assay
2.7. Minimum Inhibitory Concentration Assay
2.8. Checkerboard Synergy Assay
2.9. Microinjection of Zebrafish Larvae
2.10. Hemolysis Assay
2.11. Peptide Stability Assay
3. Results
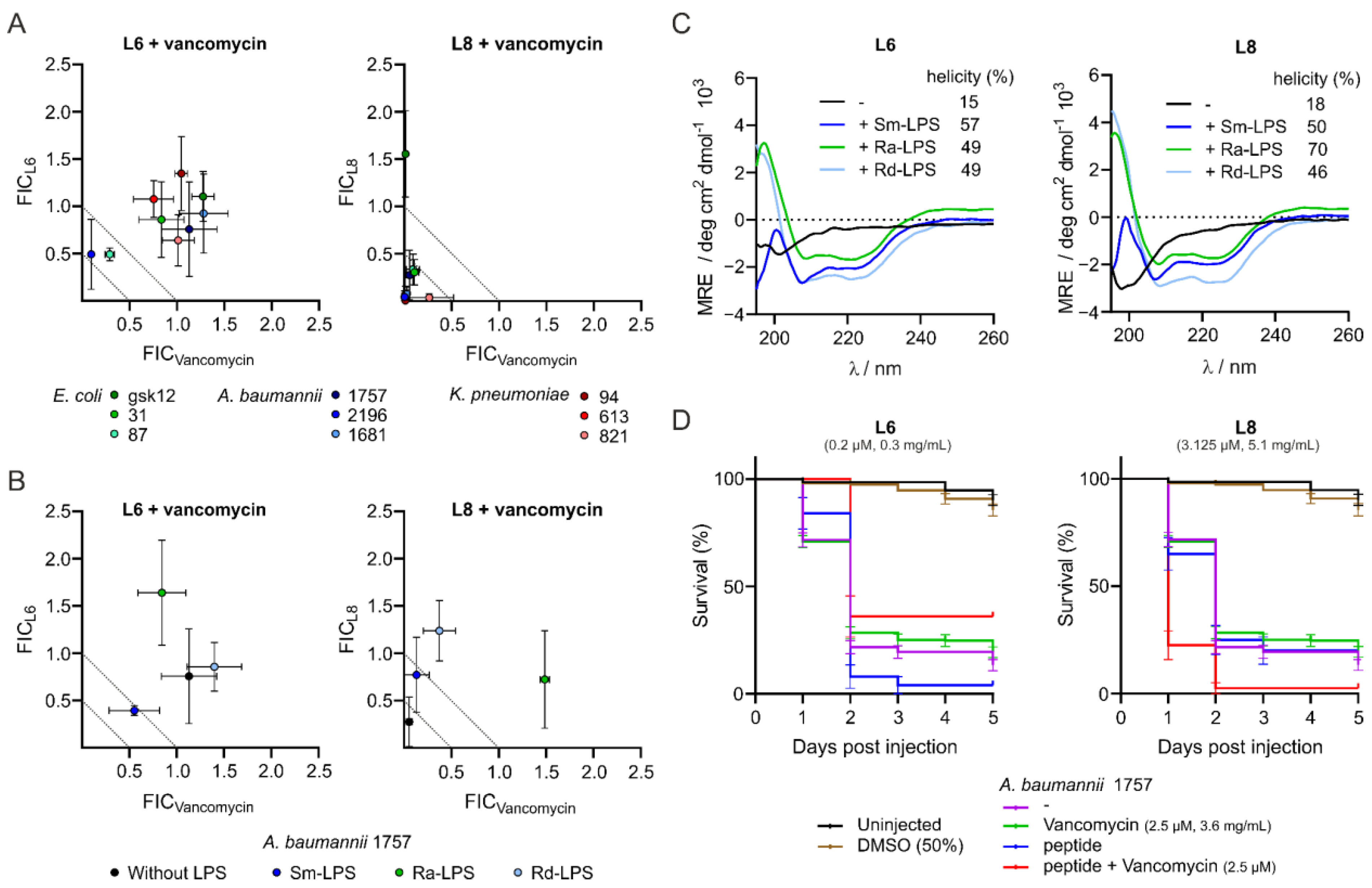
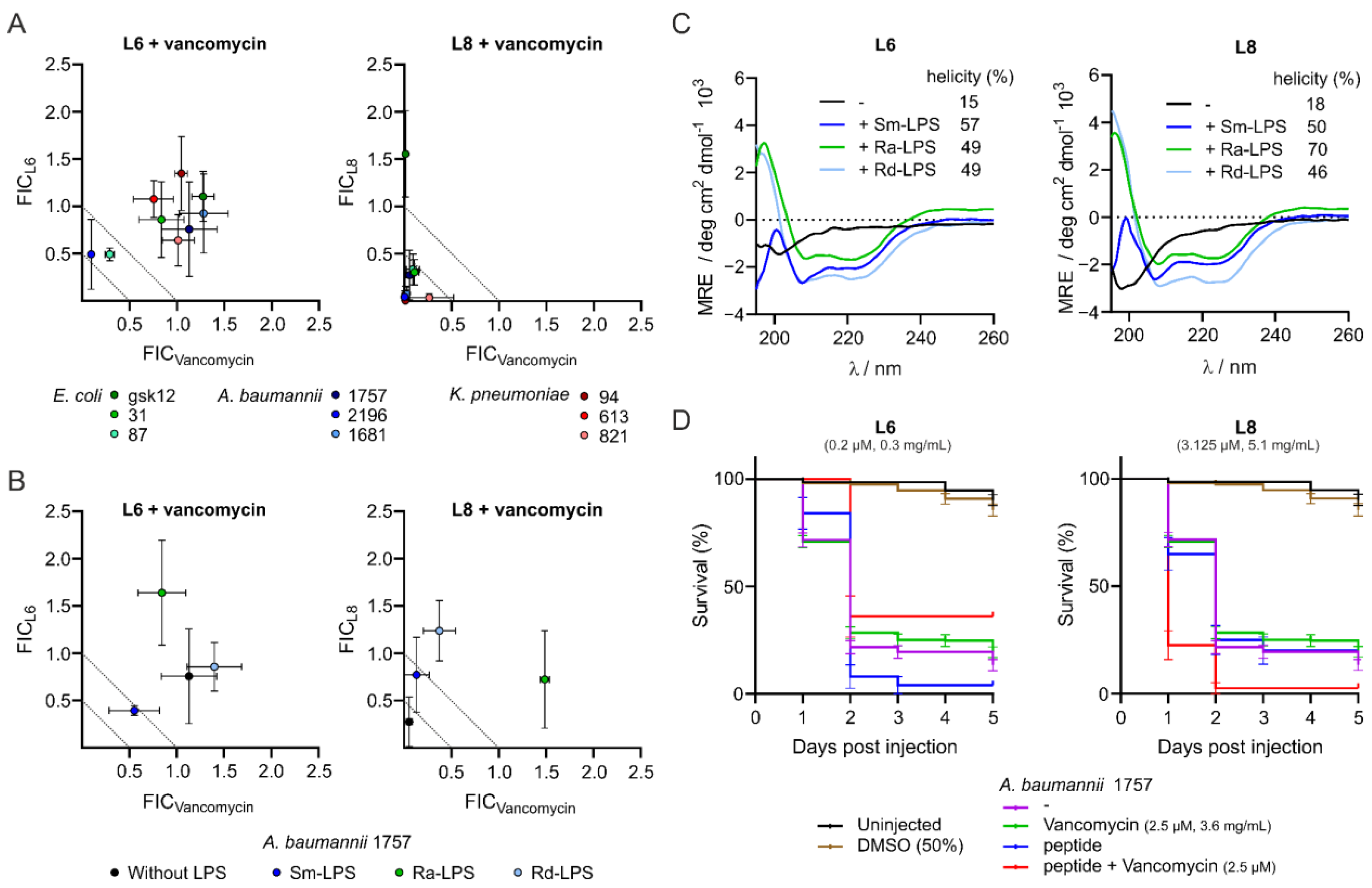
3.1. L8 Acts Synergistically with Antibiotics against Clinical Isolates In Vitro
3.2. Interaction of the Peptides with Lipopolysaccharides Abolishes Synergistic Activity and Increases α-Helicity
3.3. L8 Exhibits Toxicity in Zebrafish Larvae
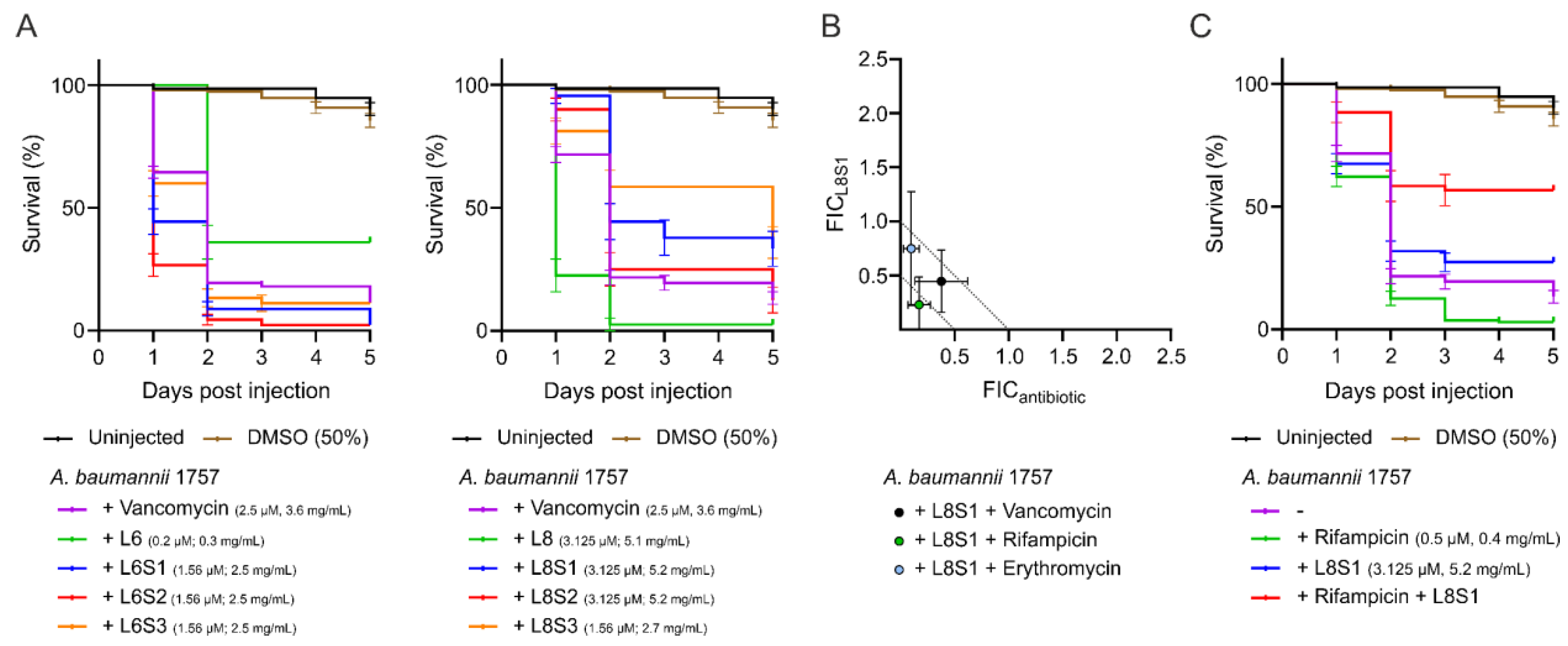
3.4. Peptide L6 Acts Additively with Vancomycin against A. baumannii In Vivo
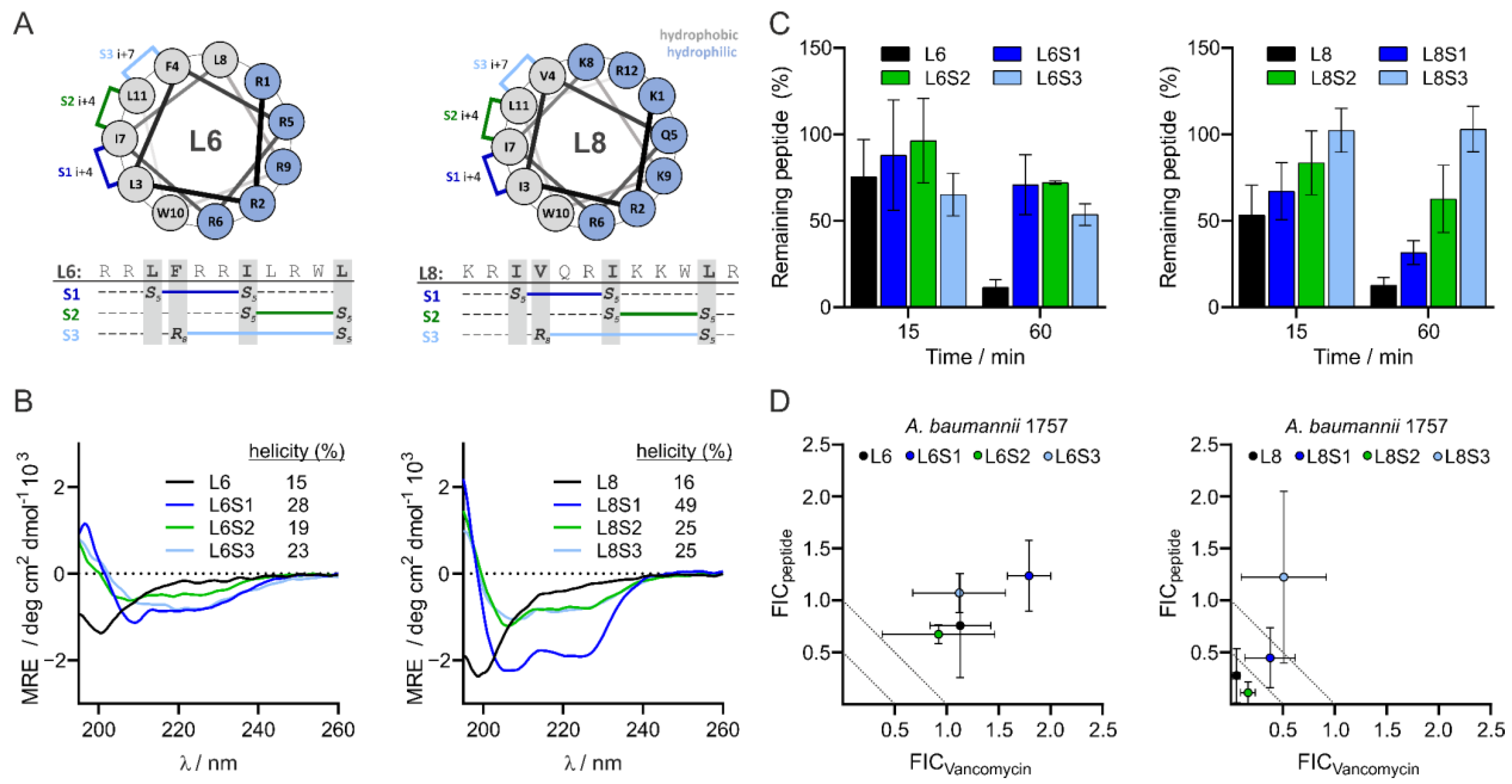
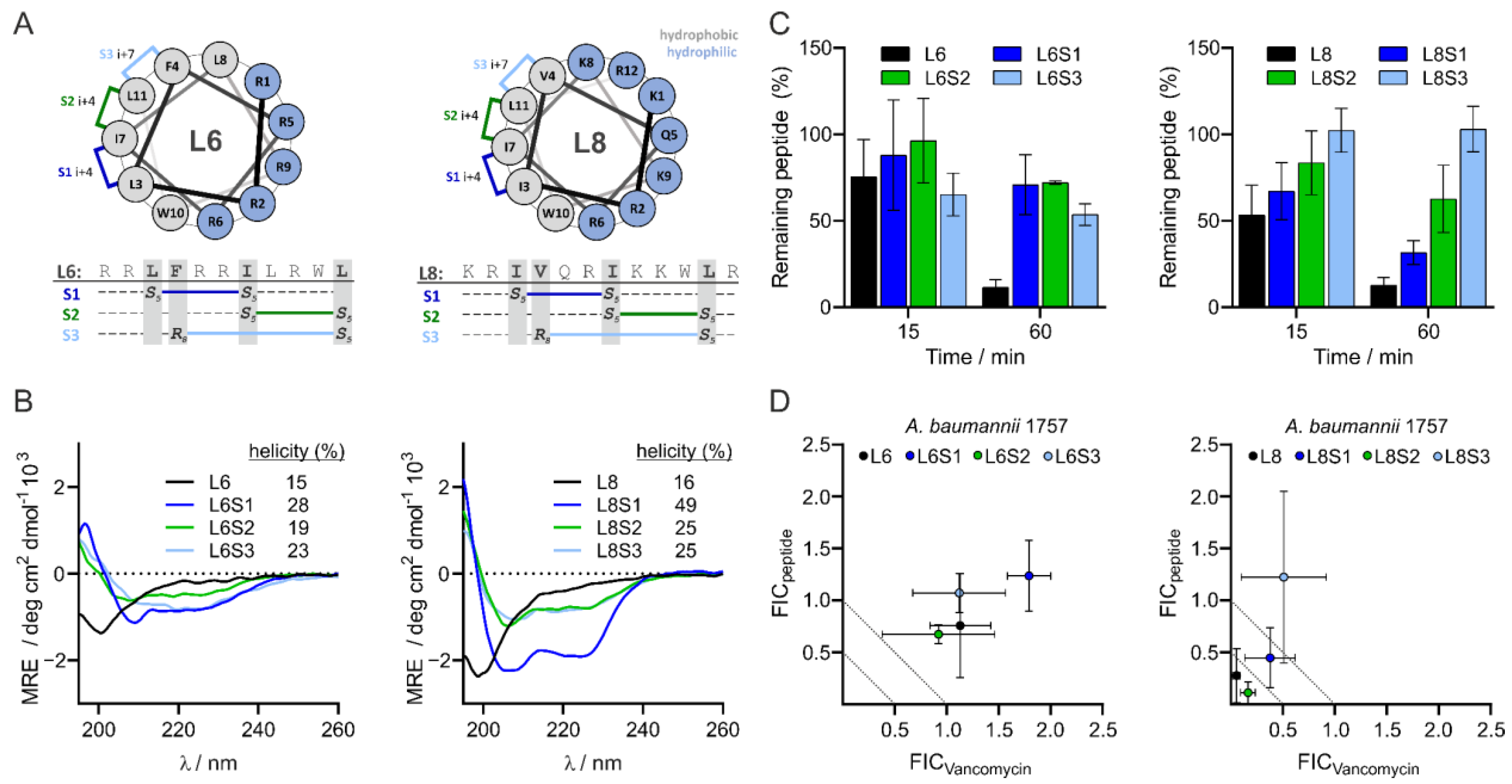
3.5. Design and Characteristics of Stapled Peptides
3.6. L8S2 Retains In Vitro Synergy with Vancomycin against A. baumannii
3.7. Most Stapled Peptides Show Little to No Adverse Effects
3.8. L8S1 Has a Synergistic Effect on In Vivo Antimicrobial Effect of Rifampicin against A. baumannii
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durao, P.; Balbontin, R.; Gordo, I. Evolutionary Mechanisms Shaping the Maintenance of Antibiotic Resistance. Trends Microbiol. 2018, 26, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, L.L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Med. Chem. 2014, 6, 25–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blot, S.; Depuydt, P.; Vandewoude, K.; De Bacquer, D. Measuring the impact of multidrug resistance in nosocomial infection. Curr. Opin Infect. Dis. 2007, 20, 391–397. [Google Scholar] [CrossRef]
- Brusselaers, N.; Vogelaers, D.; Blot, D. The rising problem of antimicrobial resistance in the intensive care unit. Ann. Intensive Care 2011, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Rice, L.B. Progress and Challenges in Implementing the Research on ESKAPE Pathogens. Infect. Control. Hosp. Epidemiol. 2010, 31, 7–10. [Google Scholar] [CrossRef]
- Agaba, P.; Tumukunde, J.; Tindimwebwa, J.V.B.; Kwizera, A. Nosocomial bacterial infections and their antimicrobial susceptibility patterns among patients in Ugandan intensive care units: A cross sectional study. BMC Res. Notes 2017, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–657. [Google Scholar] [CrossRef] [Green Version]
- Sarshar, M.; Behzadi, P.; Scribano, D.; Palamara, A.T.; Ambrosi, C. Acinetobacter baumannii: An Ancient Commensal with Weapons of a Pathogen. Pathogens 2021, 10, 387. [Google Scholar] [CrossRef]
- Boll, J.M.; Tucker, A.T.; Klein, D.R.; Beltran, A.M.; Brodbelt, J.S.; Davies, B.W.; Trent, M.S. Reinforcing Lipid A Acylation on the Cell Surface of Acinetobacter baumannii Promotes Cationic Antimicrobial Peptide Resistance and Desiccation Survival. mBio 2015, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Monem, S.; Furmanek-Blaszk, B.; Łupkowska, A.; Kuczy ´nska-Wi´snik, D.; Stojowska-Swędrzyńska, K.; Laskowska, E. Mechanisms Protecting Acinetobacter baumannii against Multiple Stresses Triggered by the Host Immune Response, Antibiotics and Outside-Host Environment. Int. J. Mol. Sci. 2020, 21, 5498. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, C.-F.; Fang, C.-M.; Sekara, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.A.; Cole, G.B.; Ravamehr-Lake, D.; Nguyen, H.Q.; Khan, F.; Sharpe, S.; Deber, C.M. Positive Charge Patterning and Hydrophobicity of MembraneActive Antimicrobial Peptides as Determinants of Activity, Toxicity, and Pharmacokinetic Stability. Am. Chem. Soc. 2019, 62, 6276–6286. [Google Scholar] [CrossRef]
- Shai, Y. Mode of Action of Membrane Active Antimicrobial Peptides. Pept. Sci. 2002, 66, 236–249. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E.W. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–473. [Google Scholar] [CrossRef]
- Sierra, J.M.; Fusté, E.; Rabanal, F.; Vinuesa, T.; Viñas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–678. [Google Scholar] [CrossRef]
- Naafs, M.A.B. The Antimicrobial Peptides: Ready for Clinical Trials? Biomed. J. Sci. Tech. Res. 2018, 7, 6038–6043. [Google Scholar] [CrossRef] [Green Version]
- Migoń, D.; Neubauer, D.; Kamysz, W. Hydrocarbon Stapled Antimicrobial Peptides. Protein J. 2018, 37, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Hamamoto, K.; Kida, Y.; Zhang, Y.; Shimizu, T.; Kuwano, K. Antimicrobial Activity and Stability to Proteolysis of Small Linear Cationic Peptides with D-Amino Acid Substitutions. Microbiol. Immunol. 2002, 46, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8928. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Wu, T.; Yang, Z.; Zheng, X.; Chen, S.; Zhou, X.; Jiang, Z.-X. Quantitatively Fine-Tuning the Physicochemical and Biological Properties of Peptidic Polymers through Monodisperse PEGylation. Biomacromolecules 2020, 21, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Guarracino, D.A.; Riordan, J.A.; Barreto, G.M.; Oldfield, A.L.; Kouba, C.M.; Agrinsoni, D. Macrocyclic Control in Helix Mimetics. Chem. Rev. 2019, 119, 9915–9950. [Google Scholar] [CrossRef]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol. 2015, 10, 1362–1376. [Google Scholar] [CrossRef]
- Pham, T.K.; Kim, D.-H.; Lee, B.-J.; Kim, Y.-W. Truncated and constrained helical analogs of antimicrobial esculentin-2EM. Bioorganic Med. Chem. Lett. 2013, 23, 6717–6721. [Google Scholar] [CrossRef]
- Luong, H.X.; Kim, D.-H.; Lee, B.-J.; Kim, Y.-W. Antimicrobial activity and stability of stapled helices of polybia-MP1. Arch. Pharm. Res. 2017, 40, 1414–1420. [Google Scholar] [CrossRef]
- Hirano, M.; Saito, C.; Yokoo, H.; Goto, C.; Kawano, R.; Demizu, Y. Development of Antimicrobial Stapled Peptides Based on Magainin 2 Sequence. Molecules 2021, 26, 444. [Google Scholar] [CrossRef]
- Li, Q.; Cebrián, R.; Montalbán-López, M.; Ren, H.; Wu, W.; Kuipers, O.P. Outer-membrane-acting peptides and lipid II-targeting antibiotics cooperatively kill Gram-negative pathogens. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of Stapled Antimicrobial Peptides that Overcome Antibiotic Resistance and In Vivo Toxicity. Nat. Biotechnol. 2019, 37, 1186–1217. [Google Scholar] [CrossRef]
- Lee, E.; Shin, A.; Kim, Y. Anti-inflammatory activities of cecropin A and its mechanism of action. Arch. Insect Biochem. Physiol. 2015, 88, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Hultmark, D.; Engstrom, A.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, A.; PeBenito, L.; Pino-Angeles, A.; Lazaridis, T. What makes a good pore former: A study of synthetic melittin derivatives. Biophys. J. 2020, 188, 1901–1914. [Google Scholar] [CrossRef]
- Memariani, H.; Memariani, M. Melittin as a promising anti-protozoan peptide: Current knowledge and future prospects. AMB Express 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Torcato, I.M.; Huang, Y.-H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.R.B.; Henriques, S.T. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-PB100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta 2012, 1828, 944–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, B.J.; King, A.T.; Katsifis, A.; Matesic, L.; Jamie, J.F. Methods to Enhance the Metabolic Stability of Peptide-Based PET Radiopharmaceuticals. Molecules 2020, 25, 2314. [Google Scholar] [CrossRef]
- Young-Woo, K.; Grossmann, T.N.; Verdine, G.L. Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011, 6, 761–771. [Google Scholar]
- Bohm, G.; Muhr, R.; Jaenicke, R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992, 5, 191–196. [Google Scholar] [CrossRef]
- Wendt, M.; Bellavita, R.; Gerber, A.; Efrem, N.-L.; Van Ramshorst, T.; Pearce, N.M.; Davey, P.R.J.; Everard, I.; Vazquez-Chantada, M.; Chiarparin, E.; et al. Bicyclic β-Sheet Mimetics that Target the Transcriptional Coactivator β-Catenin and Inhibit Wnt Signaling. Angew. Chem. Int. Ed. 2021, 133, 14056–14063. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–176. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Yu, C.M.; Yu, V.L.; Chow, J.W. Synergy Assessed by Checkerboard; A Critical Analysis. Diagn. Microbiol. Infect. Dis. 1993, 16, 343–350. [Google Scholar] [CrossRef]
- Van der Sar, A.; Musters, R.J.P.; Van Eeden, F.J.M.; Appelmelk, B.J.; Vandenbroucke-Grauls, C.M.J.E.; Bitter, W. Zebrafish embryos as a model host for the real time analysis of Salmonella typhimurium infections. Cell. Microbiol. 2003, 5, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benard, E.L.; Van der Sar, A.M.; Ellett, F.; Lieschke, G.J.; Spaink, H.P.; Meijer, A.H. Infection of Zebrafish Embryos with Intracellular Bacterial Pathogens. J. Vis. Exp. 2012, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.; Van Leeuwen, L.M.; Kuijl, C.; Ummels, R.; Van Stempvoort, G.; Rubio-Canalejas, A.; Piersma, S.R.; Jimenez, C.R.; Van der Sar, A.M.; Houben, E.N.G.; et al. EspH is a hypervirulence factor for Mycobacterium marinum and essential for the secretion of the ESX-1 substrates EspE and EspF. PLoS Pathog. 2018, 14, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adihou, H.; Gopalakrishnan, R.; Forster, T.; Gueret, S.M.; Gasper, R.; Geswindner, S.; Carrillo Garcia, C.; Karatas, H.; Pobbati, A.V.; Vazquez-Chantada, M.; et al. A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Aspmo, S.I. Serum stability of peptides. Methods Mol. Biol. 2008, 494, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Doern, C.D. When Does 2 Plus 2 Equal 5? A Review of Antimicrobial Synergy Testing. J. Clin. Microbiol. 2014, 52, 4124–4129. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; McIntosh, T.J. Structure of Supported Bilayers Composed of Lipopolysaccharides and Bacterial Phospholipids: Raft Formation and Implications for Bacterial Resistance. Biophys. J. 2004, 86, 3759–3772. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Pogue, J.M.; Tam, V.H. Toxicity in Patients. In Polymyxin Antibiotics: From Laboratory Bench to Bedside, Advances in Experimental Medicine and Biology; Springer Nature Switzerland AG: Cham, Switzerland, 2019; Volume 1145, pp. 1–289. [Google Scholar]
- Danner, R.L.; Joiner, K.A.; Rubin, M.; Patterson, W.H.; Johnson, N.; Ayers, K.M.; Parillo, J.E. Purification, Toxicity, and Antiendotoxin Activity of Polymyxin B Nonapeptide. Antimicrob. Agents Chemother. 1989, 33, 1428–1434. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.D.; Azad, M.A.K.; Wang, J.; Horne, A.S.; Thompson, P.E.; Nation, R.L.; Velkov, T.; Li, J. Antimicrobial Activity and Toxicity of the Major Lipopeptide Components of Polymyxin B and Colistin: Last-line Antibiotics against Multidrug-Resistant Gram-negative Bacteria. ACS Infect. Dis. 2015, 1, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, L.; Rathmer, B.; Ewan, K.; Bange, T.; Heinrichs, S.; Dale, T.C.; Schade, D.; Grossmann, T.N. Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets b-Catenin Protein-Protein Interactions. Cell Chem. Biol. 2017, 24, 958–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–525. [Google Scholar] [CrossRef]
- Sato, H.; Feix, J.B. Peptide–membrane interactions and mechanisms of membrane destruction by amphipathic α-helical antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1245–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-W.; Kutchukian, P.S.; Verdine, G.L. Introduction of All-Hydrocarbon i,i+3 Staples into r-Helices via Ring-Closing Olefin Metathesis. Am. Chem. Soc. 2010, 12, 3046–3050. [Google Scholar] [CrossRef]
- Jeganathan, S.; Wendt, M.; Kiehstaller, S.; Brancaccio, D.; Kuepper, A.; Pospiech, N.; Carotenuto, A.; Novellino, E.; Hennig, S.; Grossmann, T.N. Constrained Peptides with Fine-Tuned Flexibility Inhibit NF-Y Transcription Factor Assembly. Angew. Chem. Int. Ed. 2019, 58, 17351–17359. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Grubbs, R.H. Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem. Int. Ed. 1998, 37, 3281–3285. [Google Scholar] [CrossRef]
- Patrzykat, A.; Iwama, G.K.; Zhang, L.; Hancock, R.E.W.; Mendoza, V. Synergy of Histone-Derived Peptides of Coho Salmon with Lysozyme and Flounder Pleurocidin. Antimicrob. Agents Chemother. 2001, 45, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.-Y.; Wu, J.-L.; Hui, C.-F.; Lin, C.-H.; Chen, J.-Y. Insights into the antibacterial and immunomodulatory functions of the antimicrobial peptide, epinecidin-1, against Vibrio vulnificus infection in zebrafish. Fish Shellfish. Immunol. 2011, 31, 1019–1026. [Google Scholar] [CrossRef]
- Sarkar, P.; Samaddar, S.; Ammanathan, V.; Yarlagadda, V.; Ghosh, C.; Shukla, M.; Kaul, G.; Manjithaya, R.; Chopra, S.; Haldar, J. Vancomycin Derivative Inactivates Carbapenem-Resistant Acinetobacter baumannii and Induces Autophagy. ACS Chem. Biol. 2020, 15, 884–890. [Google Scholar] [CrossRef]
- Mishra, A.K.; Choi, J.; Moon, E.; Baek, K.-H. Tryptophan-Rich and Proline-Rich Antimicrobial Peptides. Molecules 2018, 23, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.R.; Teixeira, C.; Sousa, C.F.; Bessa, L.J.; Gomes, P.; Gameiro, P. How Insertion of a Single Tryptophan in the N-Terminus of a Cecropin A-Melittin Hybrid Peptide Changes Its Antimicrobial and Biophysical Profile. Membranes 2021, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fang, Y.; Wu, J. Flexibility is a mechanical determinant of antimicrobial activity for amphipathic cationic α-helical antimicrobial peptides. Elsevier Biochim. Biophys. Acta 2013, 1828, 2479–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, L.S.; Lan, Y.; Abbate, V.; Ruh, E.; Bui, T.T.; Wilkinson, L.J.; Kanno, T.; Jumagulova, E.; Kozlowska, J.; Patel, J.; et al. Conformational Flexibility Determines Selectivity and Antibacterial, Antiplasmodial, and Anticancer Potency of Cationic alpha-Helical Peptides. J. Biol. Chem. 2012, 287, 34120–34134. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Source |
|---|---|---|
| Escherichia coli | Gsk12 | Glaxo SmithKline |
| Escherichia coli | 31 | Medical Microbiology and Infection Control (MMI), Amsterdam UMC |
| Escherichia coli | 87 | MMI, Amsterdam UMC |
| Acinetobacter baumannii | 1757 | MMI, Amsterdam UMC |
| Acinetobacter baumannii | 2196 | MMI, Amsterdam UMC |
| Acinetobacter baumannii | 1681 | MMI, Amsterdam UMC |
| Klebsiella pneumoniae | 94 | MMI, Amsterdam UMC |
| Klebsiella pneumoniae | 613 | MMI, Amsterdam UMC |
| Klebsiella pneumoniae | 821 | MMI, Amsterdam UMC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schouten, G.K.; Paulussen, F.M.; Kuipers, O.P.; Bitter, W.; Grossmann, T.N.; van Ulsen, P. Stapling of Peptides Potentiates the Antibiotic Treatment of Acinetobacter baumannii In Vivo. Antibiotics 2022, 11, 273. https://doi.org/10.3390/antibiotics11020273
Schouten GK, Paulussen FM, Kuipers OP, Bitter W, Grossmann TN, van Ulsen P. Stapling of Peptides Potentiates the Antibiotic Treatment of Acinetobacter baumannii In Vivo. Antibiotics. 2022; 11(2):273. https://doi.org/10.3390/antibiotics11020273
Chicago/Turabian StyleSchouten, Gina K., Felix M. Paulussen, Oscar P. Kuipers, Wilbert Bitter, Tom N. Grossmann, and Peter van Ulsen. 2022. "Stapling of Peptides Potentiates the Antibiotic Treatment of Acinetobacter baumannii In Vivo" Antibiotics 11, no. 2: 273. https://doi.org/10.3390/antibiotics11020273
APA StyleSchouten, G. K., Paulussen, F. M., Kuipers, O. P., Bitter, W., Grossmann, T. N., & van Ulsen, P. (2022). Stapling of Peptides Potentiates the Antibiotic Treatment of Acinetobacter baumannii In Vivo. Antibiotics, 11(2), 273. https://doi.org/10.3390/antibiotics11020273