
Antibiotic Discovery and Resistance: The Chase and the Race

 ,
,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Antimicrobial Resistance Is Ancient
2.1. The Environmental Resistome and Its Relation to Antimicrobial Resistance
2.2. Resistance, Tolerance and Persistence
- Tolerance by slow growth that is either inherited or not, occurring at a steady state.
- Tolerance by lag that is a transient state induced by starvation or stress.
2.3. Intrinsic, Phenotypic and Acquired Resistance
2.3.1. Intrinsic Resistance
2.3.2. Phenotypic Resistance
2.3.3. Acquired Resistance
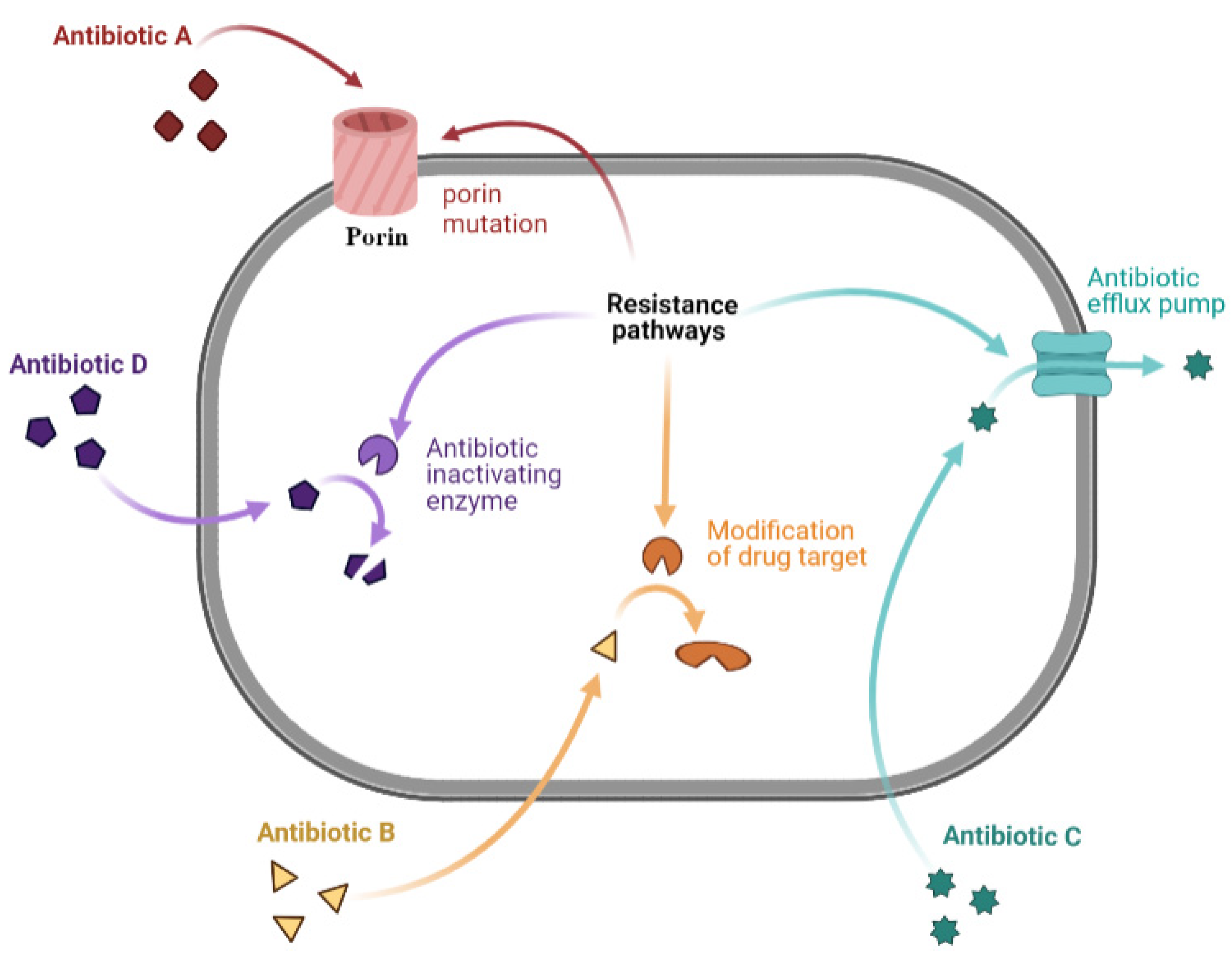
2.4. Mechanisms of Antibiotic Resistance
2.4.1. Limiting Drug Uptake
2.4.2. Modification of Drug Target
2.4.3. Drug Efflux
2.4.4. Drug Inactivation
2.5. The Spread of AMR: The Known and the Unknown
2.6. Drivers of Antimicrobial Resistance
2.7. Priority Pathogens
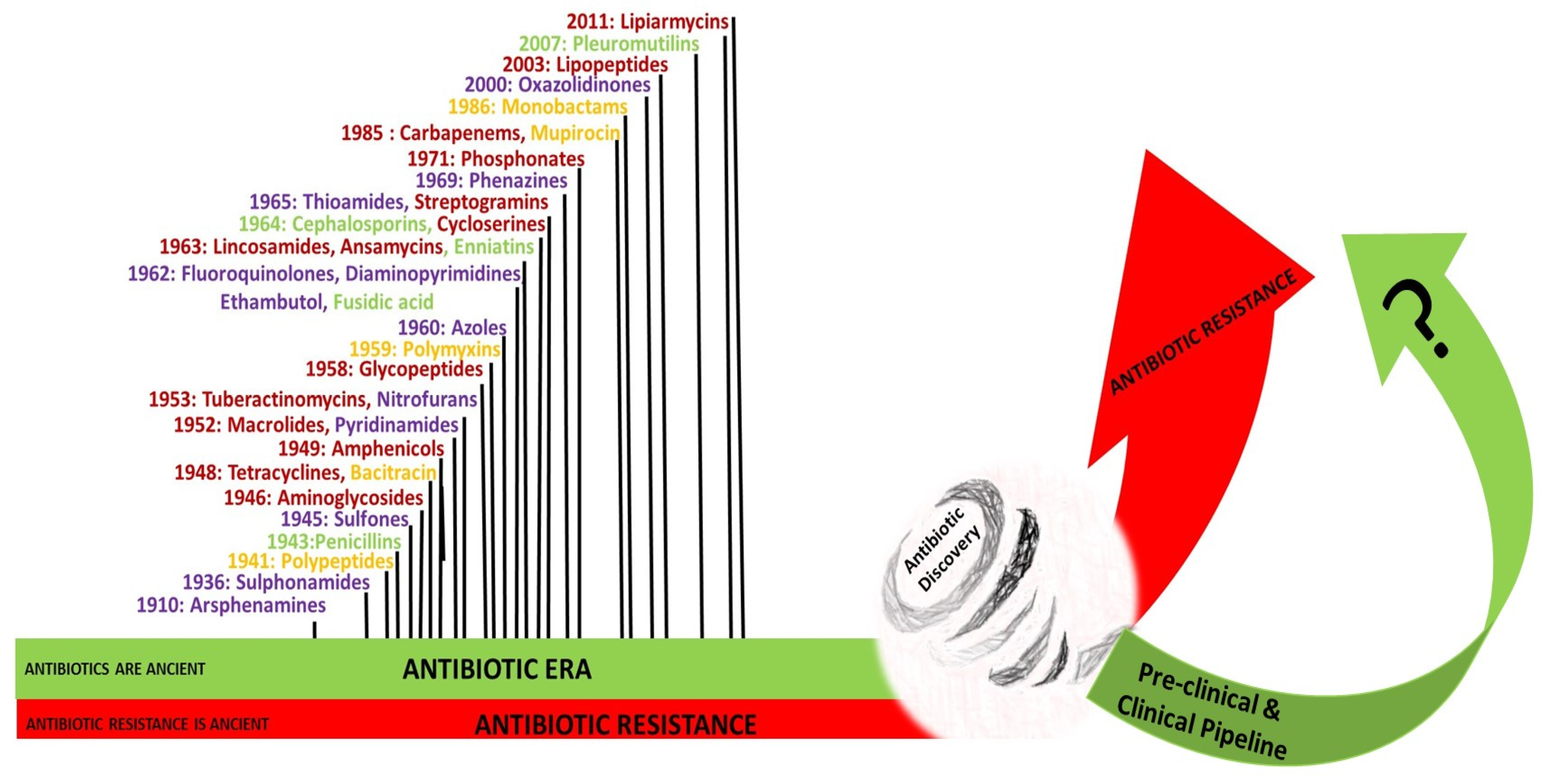
3. History of Antibiotics
3.1. The Pre-Antibiotic Era
3.2. The Antibiotic Era
4. Lists of Critically Important Antibiotics for Human Medicine
5. The Antibiotic Pipeline and the Discovery Void
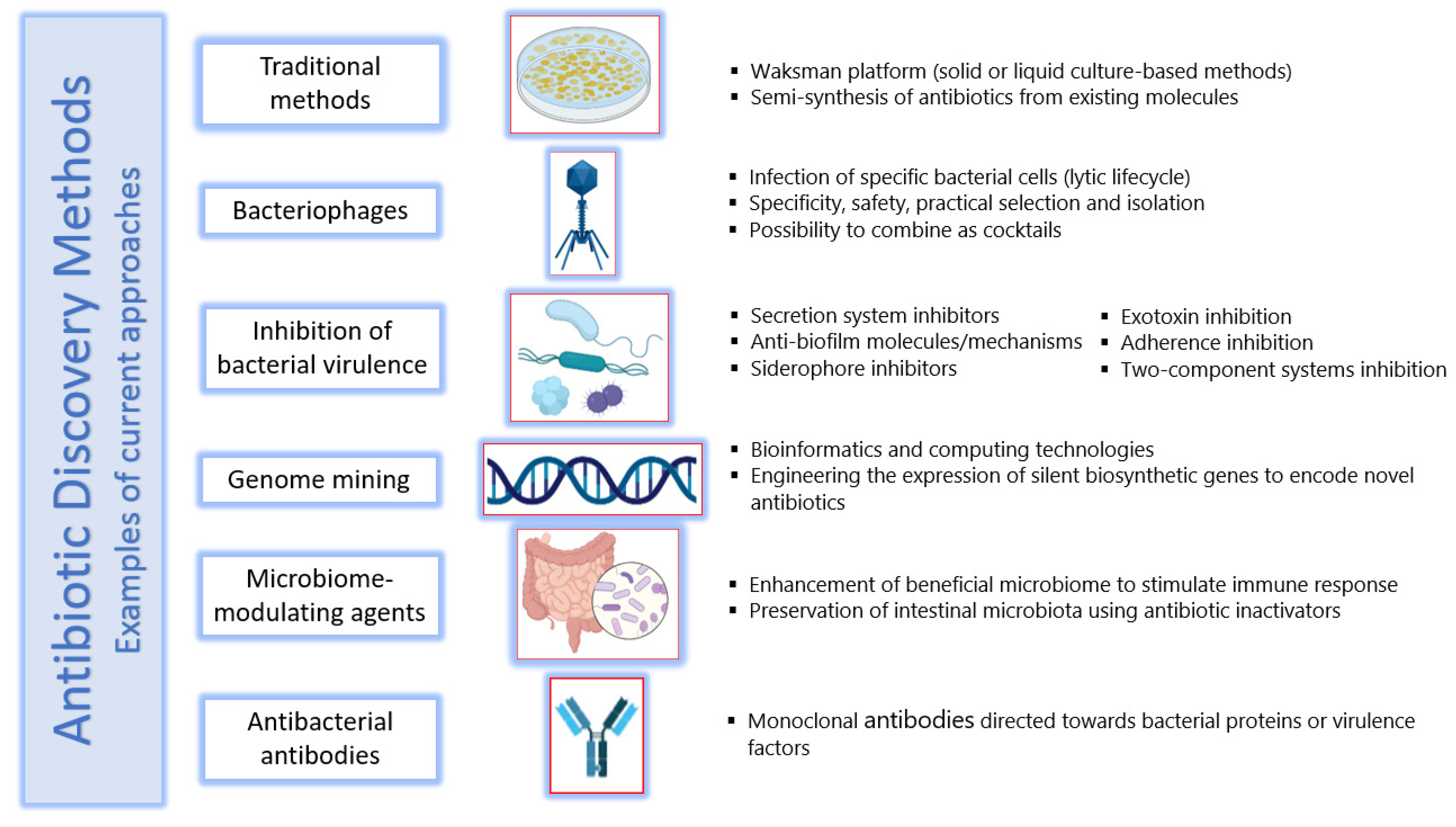
6. Methods of Antibiotic Discovery
6.1. Traditional Methods
6.2. Bacteriophages
6.3. Inhibition of Bacterial Virulence
6.4. Genome Mining
6.5. Microbiome-Modulating Agents
6.6. Antibacterial Antibodies
7. Antibiotics Research and Development: Incentives and Barriers
7.1. Incentives to Research and Development
7.2. Challenges to Antibiotic Discovery
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wilson, L.A.; Rogers Van Katwyk, S.; Fafard, P.; Viens, A.M.; Hoffman, S.J. Lessons learned from COVID-19 for the post-antibiotic future. Glob. Health 2020, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Getahun, H.; Smith, I.; Trivedi, K.; Paulin, S.; Balkhy, H.H. Tackling antimicrobial resistance in the COVID-19 pandemic. Bull. World Health Organ. 2020, 98, 442. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- Nicholson, A.; Pavlin, J.; Buckley, G.; Amponsah, E. 5, Systems Approaches to Spur Innovations in Tackling Antimicrobial Resistance. In Exploring the Frontiers of Innovation to Tackle Microbial Threats: Proceedings of a Workshop; National Academies Press (US): Washington, DC, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560426/ (accessed on 11 December 2021).
- Lv, J.; Deng, S.; Zhang, L. A review of artificial intelligence applications for antimicrobial resistance. Biosaf. Health 2021, 3, 22–31. [Google Scholar] [CrossRef]
- Joshi, M.P.; Chintu, C.; Mpundu, M.; Kibuule, D.; Hazemba, O.; Andualem, T.; Embrey, M.; Phulu, B.; Gerba, H. Multidisciplinary and multisectoral coalitions as catalysts for action against antimicrobial resistance: Implementation experiences at national and regional levels. Glob. Public Health 2018, 13, 1781–1795. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, M.; Matsoso, M.P. The World Health Organization Global Action Plan for antimicrobial resistance. S. Afr. Med. J. 2015, 105, 325. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.A.A.; Rahman, S.; Cohall, D.; Bharatha, A.; Singh, K.; Haque, M.; Gittens-St Hilaire, M. Antimicrobial Stewardship: Fighting Antimicrobial Resistance and Protecting Global Public Health. Infect. Drug Resist. 2020, 13, 4713–4738. [Google Scholar] [CrossRef]
- Dutescu, I.A.; Hillier, S.A. Encouraging the Development of New Antibiotics: Are Financial Incentives the Right Way Forward? A Systematic Review and Case Study. Infect. Drug Resist. 2021, 14, 415–434. [Google Scholar] [CrossRef]
- Jackson, N.; Czaplewski, L.; Piddock, L.J.V. Discovery and development of new antibacterial drugs: Learning from experience? J. Antimicrob. Chemother. 2018, 73, 1452–1459. [Google Scholar] [CrossRef]
- WHO. Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis. 2020. Available online: https://www.who.int/publications/i/item/9789240021303 (accessed on 11 December 2021).
- Miethke, M.; Pieroni, M.; Weber, T.; Bronstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlen, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef]
- Kumar, M.; Sarma, D.K.; Shubham, S.; Kumawat, M.; Verma, V.; Nina, P.B.; Jp, D.; Kumar, S.; Singh, B.; Tiwari, R.R. Futuristic Non-antibiotic Therapies to Combat Antibiotic Resistance: A Review. Front. Microbiol. 2021, 12, 609459. [Google Scholar] [CrossRef] [PubMed]
- WHO. Critically Important Antimicrobials for Human Medicine, 6th Revision. 2020. Available online: https://www.who.int/publications/i/item/9789241515528 (accessed on 11 December 2021).
- Clift, C. Review of Progress on Antimicrobial Resistance: Background and Analysis; The Royal Institute of International Affairs: London, UK, 2019. [Google Scholar]
- Simpkin, V.L.; Renwick, M.J.; Kelly, R.; Mossialos, E. Incentivising innovation in antibiotic drug discovery and development: Progress, challenges and next steps. J. Antibiot. 2017, 70, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Plackett, B. Why big pharma has abandoned antibiotics. Nature 2020, 586, S50–S52. [Google Scholar] [CrossRef]
- Perry, J.; Waglechner, N.; Wright, G. The Prehistory of Antibiotic Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025197. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A. History of drug-resistant microbes. In Antimicrobial Drug Resistance; Springer: Berlin/Heidelberg, Germany, 2017; pp. 3–8. [Google Scholar]
- Donadio, S.; Maffioli, S.; Monciardini, P.; Sosio, M.; Jabes, D. Antibiotic discovery in the twenty-first century: Current trends and future perspectives. J. Antibiot. 2010, 63, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Barlow, M.; Hall, B.G. Phylogenetic analysis shows that the OXA beta-lactamase genes have been on plasmids for millions of years. J. Mol. Evol. 2002, 55, 314–321. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; Turroni, F.; Ferrario, C.; Duranti, S.; van Sinderen, D.; Ventura, M. Ancient bacteria of the Otzi’s microbiome: A genomic tale from the Copper Age. Microbiome 2017, 5, 5. [Google Scholar] [CrossRef]
- Van Goethem, M.W.; Pierneef, R.; Bezuidt, O.K.I.; Van De Peer, Y.; Cowan, D.A.; Makhalanyane, T.P. A reservoir of ’historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 2018, 6, 40. [Google Scholar] [CrossRef]
- Fajardo, A.; Martinez-Martin, N.; Mercadillo, M.; Galan, J.C.; Ghysels, B.; Matthijs, S.; Cornelis, P.; Wiehlmann, L.; Tummler, B.; Baquero, F.; et al. The neglected intrinsic resistome of bacterial pathogens. PLoS ONE 2008, 3, e1619. [Google Scholar] [CrossRef] [PubMed]
- Olivares, J.; Bernardini, A.; Garcia-Leon, G.; Corona, F.B.; Sanchez, M.; Martinez, J.L. The intrinsic resistome of bacterial pathogens. Front. Microbiol. 2013, 4, 103. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. The antibiotic resistome. Expert Opin. Drug Discov. 2010, 5, 779–788. [Google Scholar] [CrossRef]
- Martinez, J.L. General principles of antibiotic resistance in bacteria. Drug Discov. Today Technol. 2014, 11, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE 2012, 7, e34953. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.D.; Wright, G.D. Lessons from the Environmental Antibiotic Resistome. Annu. Rev. Microbiol. 2017, 71, 309–329. [Google Scholar] [CrossRef]
- Martinez, J.L. Ecology and Evolution of Chromosomal Gene Transfer between Environmental Microorganisms and Pathogens. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Brauner, A.; Ronin, I.; Balaban, N.Q. Epistasis between antibiotic tolerance, persistence, and resistance mutations. Proc. Natl. Acad. Sci. USA 2019, 116, 14734–14739. [Google Scholar] [CrossRef]
- Spencer, D.C.; Paton, T.F.; Mulroney, K.T.; Inglis, T.J.J.; Sutton, J.M.; Morgan, H. A fast impedance-based antimicrobial susceptibility test. Nat. Commun. 2020, 11, 5328. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Mattie, H. Antibiotic efficacy in vivo predicted by in vitro activity. Int. J. Antimicrob. Agents 2000, 14, 91–98. [Google Scholar] [CrossRef]
- Kester, J.C.; Fortune, S.M. Persisters and beyond: Mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Rosenberg, E.Y. Effect on solute size on diffusion rates through the transmembrane pores of the outer membrane of Escherichia coli. J. Gen. Physiol. 1981, 77, 121–135. [Google Scholar] [CrossRef]
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef]
- Arzanlou, M.; Chai, W.C.; Venter, H. Intrinsic, adaptive and acquired antimicrobial resistance in Gram-negative bacteria. Essays Biochem. 2017, 61, 49–59. [Google Scholar] [CrossRef]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef]
- Gefen, O.; Balaban, N.Q. The importance of being persistent: Heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol. Rev. 2009, 33, 704–717. [Google Scholar] [CrossRef]
- De la Fuente-Nunez, C.; Reffuveille, F.; Fernandez, L.; Hancock, R.E. Bacterial biofilm development as a multicellular adaptation: Antibiotic resistance and new therapeutic strategies. Curr. Opin. Microbiol. 2013, 16, 580–589. [Google Scholar] [CrossRef]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar] [CrossRef]
- Hoiby, N.; Ciofu, O.; Johansen, H.K.; Song, Z.J.; Moser, C.; Jensen, P.O.; Molin, S.; Givskov, M.; Tolker-Nielsen, T.; Bjarnsholt, T. The clinical impact of bacterial biofilms. Int. J. Oral Sci. 2011, 3, 55–65. [Google Scholar] [CrossRef]
- Defoirdt, T. Quorum-Sensing Systems as Targets for Antivirulence Therapy. Trends Microbiol. 2018, 26, 313–328. [Google Scholar] [CrossRef]
- Irazoki, O.; Campoy, S.; Barbe, J. The Transient Multidrug Resistance Phenotype of Salmonella enterica Swarming Cells Is Abolished by Sub-inhibitory Concentrations of Antimicrobial Compounds. Front. Microbiol. 2017, 8, 1360. [Google Scholar] [CrossRef]
- Liu, L.; Wu, R.; Zhang, J.; Shang, N.; Li, P. D-Ribose Interferes with Quorum Sensing to Inhibit Biofilm Formation of Lactobacillus paraplantarum L-ZS9. Front. Microbiol. 2017, 8, 1860. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Li, X.; Ma, S.; Hu, H.; Wu, B.; Zhang, X.X.; Ren, H. In-situ monitoring AHL-mediated quorum-sensing regulation of the initial phase of wastewater biofilm formation. Environ. Int. 2020, 135, 105326. [Google Scholar] [CrossRef]
- Dantas, G.; Sommer, M.O.; Oluwasegun, R.D.; Church, G.M. Bacteria subsisting on antibiotics. Science 2008, 320, 100–103. [Google Scholar] [CrossRef]
- Martinez, J.L. Natural antibiotic resistance and contamination by antibiotic resistance determinants: The two ages in the evolution of resistance to antimicrobials. Front. Microbiol. 2012, 3, 1. [Google Scholar] [CrossRef]
- Barlow, M. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol. Biol. 2009, 532, 397–411. [Google Scholar] [CrossRef]
- Johnston, C.; Martin, B.; Fichant, G.; Polard, P.; Claverys, J.P. Bacterial transformation: Distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 2014, 12, 181–196. [Google Scholar] [CrossRef]
- Claverys, J.P.; Prudhomme, M.; Martin, B. Induction of competence regulons as a general response to stress in gram-positive bacteria. Annu. Rev. Microbiol. 2006, 60, 451–475. [Google Scholar] [CrossRef]
- Prudhomme, M.; Attaiech, L.; Sanchez, G.; Martin, B.; Claverys, J.P. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 2006, 313, 89–92. [Google Scholar] [CrossRef]
- Haaber, J.; Penades, J.R.; Ingmer, H. Transfer of Antibiotic Resistance in Staphylococcus aureus. Trends Microbiol. 2017, 25, 893–905. [Google Scholar] [CrossRef]
- Ubukata, K.; Konno, M.; Fujii, R. Transduction of drug resistance to tetracycline, chloramphenicol, macrolides, lincomycin and clindamycin with phages induced from Streptococcus pyogenes. J. Antibiot. 1975, 28, 681–688. [Google Scholar] [CrossRef]
- Mazaheri Nezhad Fard, R.; Barton, M.D.; Heuzenroeder, M.W. Bacteriophage-mediated transduction of antibiotic resistance in enterococci. Lett. Appl. Microbiol. 2011, 52, 559–564. [Google Scholar] [CrossRef]
- Varga, M.; Kuntova, L.; Pantucek, R.; Maslanova, I.; Ruzickova, V.; Doskar, J. Efficient transfer of antibiotic resistance plasmids by transduction within methicillin-resistant Staphylococcus aureus USA300 clone. FEMS Microbiol. Lett. 2012, 332, 146–152. [Google Scholar] [CrossRef]
- Kenzaka, T.; Tani, K.; Sakotani, A.; Yamaguchi, N.; Nasu, M. High-frequency phage-mediated gene transfer among Escherichia coli cells, determined at the single-cell level. Appl. Environ. Microbiol. 2007, 73, 3291–3299. [Google Scholar] [CrossRef]
- Von Wintersdorff, C.J.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.; Wolffs, P.F. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef]
- Volkova, V.V.; Lu, Z.; Besser, T.; Grohn, Y.T. Modeling the infection dynamics of bacteriophages in enteric Escherichia coli: Estimating the contribution of transduction to antimicrobial gene spread. Appl. Environ. Microbiol. 2014, 80, 4350–4362. [Google Scholar] [CrossRef]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Update 2010, 13, 151–171. [Google Scholar] [CrossRef]
- Carattoli, A. Plasmids and the spread of resistance. Int. J. Med. Microbiol. 2013, 303, 298–304. [Google Scholar] [CrossRef]
- Weigel, L.M.; Clewell, D.B.; Gill, S.R.; Clark, N.C.; McDougal, L.K.; Flannagan, S.E.; Kolonay, J.F.; Shetty, J.; Killgore, G.E.; Tenover, F.C. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. [Google Scholar] [CrossRef]
- Savage, V.J.; Chopra, I.; O’Neill, A.J. Staphylococcus aureus biofilms promote horizontal transfer of antibiotic resistance. Antimicrob. Agents Chemother. 2013, 57, 1968–1970. [Google Scholar] [CrossRef]
- Huddleston, J.R. Horizontal gene transfer in the human gastrointestinal tract: Potential spread of antibiotic resistance genes. Infect. Drug Resist. 2014, 7, 167–176. [Google Scholar] [CrossRef]
- Watford, S.; Warrington, S.J. Bacterial DNA Mutations. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2017. Available online: https://pubmed.ncbi.nlm.nih.gov/29083710/ (accessed on 24 December 2021).
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Pages, J.M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef]
- Ndagi, U.; Falaki, A.A.; Abdullahi, M.; Lawald, M.M.; Soliman, M.E. Antibiotic resistance: Bioinformatics-based understanding as a functional strategy for drug design. RSC Adv. 2020, 10, 18451–18468. [Google Scholar] [CrossRef]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef]
- Weisblum, B. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 1995, 39, 577–585. [Google Scholar] [CrossRef]
- Roberts, M.C. Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 2005, 245, 195–203. [Google Scholar] [CrossRef]
- Piddock, L.J. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 2006, 19, 382–402. [Google Scholar] [CrossRef]
- Duraes, F.; Pinto, M.; Sousa, E. Medicinal Chemistry Updates on Bacterial Efflux Pump Modulators. Curr. Med. Chem. 2018, 25, 6030–6069. [Google Scholar] [CrossRef]
- WHO Regional Office for South-East Asia. Guidelines on Prevention and Control of Hospital Associated Infections. Available online: https://apps.who.int/iris/handle/10665/205187 (accessed on 21 December 2021).
- Sengupta, S.; Barman, P.; Lo, J. Opportunities to Overcome Implementation Challenges of Infection Prevention and Control in Low-Middle Income Countries. Curr. Treat. Opt. Infect. Dis. 2019, 11, 267–280. [Google Scholar] [CrossRef]
- Schmidt, J.S.; Kuster, S.P.; Nigg, A.; Dazio, V.; Brilhante, M.; Rohrbach, H.; Bernasconi, O.J.; Budel, T.; Campos-Madueno, E.I.; Gobeli Brawand, S.; et al. Poor infection prevention and control standards are associated with environmental contamination with carbapenemase-producing Enterobacterales and other multidrug-resistant bacteria in Swiss companion animal clinics. Antimicrob. Resist. Infect. Control 2020, 9, 93. [Google Scholar] [CrossRef]
- Wang, R.; van Dorp, L.; Shaw, L.P.; Bradley, P.; Wang, Q.; Wang, X.; Jin, L.; Zhang, Q.; Liu, Y.; Rieux, A.; et al. The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat. Commun. 2018, 9, 1179. [Google Scholar] [CrossRef]
- Casadevall, A.; Kontoyiannis, D.P.; Robert, V. Environmental Candida auris and the Global Warming Emergence Hypothesis. mBio 2021, 12, e00360-21. [Google Scholar] [CrossRef]
- Arora, P.; Singh, P.; Wang, Y.; Yadav, A.; Pawar, K.; Singh, A.; Padmavati, G.; Xu, J.; Chowdhary, A. Environmental Isolation of Candida auris from the Coastal Wetlands of Andaman Islands, India. mBio 2021, 12, e03181-20. [Google Scholar] [CrossRef]
- Ramsamy, Y.; Mlisana, K.P.; Amoako, D.G.; Abia, A.L.K.; Ismail, A.; Allam, M.; Mbanga, J.; Singh, R.; Essack, S.Y. Mobile genetic elements-mediated Enterobacterales-associated carbapenemase antibiotic resistance genes propagation between the environment and humans: A One Health South African study. Sci. Total Environ. 2022, 806, 150641. [Google Scholar] [CrossRef]
- Knight, G.M.; Costelloe, C.; Murray, K.A.; Robotham, J.V.; Atun, R.; Holmes, A.H. Addressing the Unknowns of Antimicrobial Resistance: Quantifying and Mapping the Drivers of Burden. Clin. Infect. Dis. 2018, 66, 612–616. [Google Scholar] [CrossRef]
- Fallach, N.; Dickstein, Y.; Silberschein, E.; Turnidge, J.; Temkin, E.; Almagor, J.; Carmeli, Y.; Consortium, D.-A. Utilising sigmoid models to predict the spread of antimicrobial resistance at the country level. Eurosurveillance 2020, 25, 1900387. [Google Scholar] [CrossRef]
- Van Camp, P.J.; Haslam, D.B.; Porollo, A. Prediction of Antimicrobial Resistance in Gram-Negative Bacteria from Whole-Genome Sequencing Data. Front. Microbiol. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Chowdhury, A.S.; Call, D.R.; Broschat, S.L. PARGT: A software tool for predicting antimicrobial resistance in bacteria. Sci. Rep. 2020, 10, 11033. [Google Scholar] [CrossRef]
- White, A.; Hughes, J.M. Critical Importance of a One Health Approach to Antimicrobial Resistance. Ecohealth 2019, 16, 404–409. [Google Scholar] [CrossRef]
- Thakur, S.; Gray, G.C. The Mandate for a Global “One Health” Approach to Antimicrobial Resistance Surveillance. Am. J. Trop. Med. Hyg. 2019, 100, 227–228. [Google Scholar] [CrossRef]
- Robinson, T.P.; Bu, D.P.; Carrique-Mas, J.; Fevre, E.M.; Gilbert, M.; Grace, D.; Hay, S.I.; Jiwakanon, J.; Kakkar, M.; Kariuki, S.; et al. Antibiotic resistance is the quintessential One Health issue. Trans. R. Soc. Trop. Med. Hyg. 2016, 110, 377–380. [Google Scholar] [CrossRef]
- McEwen, S.A.; Collignon, P.J. Antimicrobial Resistance: A One Health Perspective. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Collignon, P.J.; McEwen, S.A. One Health-Its Importance in Helping to Better Control Antimicrobial Resistance. Trop. Med. Infect. Dis. 2019, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Frost, I.; Van Boeckel, T.P.; Pires, J.; Craig, J.; Laxminarayan, R. Global geographic trends in antimicrobial resistance: The role of international travel. J. Travel Med. 2019, 26, taz036. [Google Scholar] [CrossRef] [PubMed]
- Nellums, L.B.; Thompson, H.; Holmes, A.; Castro-Sanchez, E.; Otter, J.A.; Norredam, M.; Friedland, J.S.; Hargreaves, S. Antimicrobial resistance among migrants in Europe: A systematic review and meta-analysis. Lancet Infect. Dis. 2018, 18, 796–811. [Google Scholar] [CrossRef]
- Rousham, E.K.; Unicomb, L.; Islam, M.A. Human, animal and environmental contributors to antibiotic resistance in low-resource settings: Integrating behavioural, epidemiological and One Health approaches. Proc. Biol. Sci. 2018, 285, 20182684. [Google Scholar] [CrossRef]
- Nadimpalli, M.; Delarocque-Astagneau, E.; Love, D.C.; Price, L.B.; Huynh, B.T.; Collard, J.M.; Lay, K.S.; Borand, L.; Ndir, A.; Walsh, T.R.; et al. Combating Global Antibiotic Resistance: Emerging One Health Concerns in Lower- and Middle-Income Countries. Clin. Infect. Dis. 2018, 66, 963–969. [Google Scholar] [CrossRef]
- Van Boeckel, T.P.; Pires, J.; Silvester, R.; Zhao, C.; Song, J.; Criscuolo, N.G.; Gilbert, M.; Bonhoeffer, S.; Laxminarayan, R. Global trends in antimicrobial resistance in animals in low- and middle-income countries. Science 2019, 365, 1252–1255. [Google Scholar] [CrossRef]
- Global Spending on Health: A World in Transition, WHO—2019. Available online: https://www.who.int/publications/i/item/WHO-HIS-HGF-HFWorkingPaper-19.4 (accessed on 22 December 2021).
- World Bank Group. Operational Framework for Strengthening Human, Animal and Environmental Public Health Systems at Their Interface. Available online: https://reliefweb.int/report/world/operational-framework-strengthening-human-animal-and-environmental-public-health (accessed on 24 December 2021).
- Collignon, P.; Beggs, J.J.; Walsh, T.R.; Gandra, S.; Laxminarayan, R. Anthropological and socioeconomic factors contributing to global antimicrobial resistance: A univariate and multivariable analysis. Lancet Planet Health 2018, 2, e398–e405. [Google Scholar] [CrossRef]
- O’Neil, J. Tackling drug-resistant infections globally: Final report and recommendations. In Review on Antimicrobial Resistance; Wellcome Trust and HM Government: London, UK, 2016; Volume 1. [Google Scholar]
- Shanta, I.S.; Hasnat, M.A.; Zeidner, N.; Gurley, E.S.; Azziz-Baumgartner, E.; Sharker, M.A.Y.; Hossain, K.; Khan, S.U.; Haider, N.; Bhuyan, A.A.; et al. Raising Backyard Poultry in Rural Bangladesh: Financial and Nutritional Benefits, but Persistent Risky Practices. Transbound Emerg. Dis. 2017, 64, 1454–1464. [Google Scholar] [CrossRef]
- Karthik, L.; Kumar, G.; Keswani, T.; Bhattacharyya, A.; Chandar, S.S.; Bhaskara Rao, K.V. Protease inhibitors from marine actinobacteria as a potential source for antimalarial compound. PLoS ONE 2014, 9, e90972. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, P.; Li, J.; Sun, C.; Song, L.; Zhang, C.; Zhao, Q.; Wu, C. Prevalence and Characterization of Fluoroquinolone Resistant Salmonella Isolated from an Integrated Broiler Chicken Supply Chain. Front. Microbiol. 2019, 10, 1865. [Google Scholar] [CrossRef] [PubMed]
- Finley, R.L.; Collignon, P.; Larsson, D.G.; McEwen, S.A.; Li, X.Z.; Gaze, W.H.; Reid-Smith, R.; Timinouni, M.; Graham, D.W.; Topp, E. The scourge of antibiotic resistance: The important role of the environment. Clin. Infect. Dis. 2013, 57, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Kuster, A.; Adler, N. Pharmaceuticals in the environment: Scientific evidence of risks and its regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130587. [Google Scholar] [CrossRef] [PubMed]
- Yves Chartier, J.E.; Pieper, U.; Prüss, A.; Rushbrook, P.; Stringer, R.; Townend, W. Safe Management of Wastes from Health-Care Activities, 2nd ed.; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- You, Y.; Silbergeld, E.K. Learning from agriculture: Understanding low-dose antimicrobials as drivers of resistome expansion. Front. Microbiol. 2014, 5, 284. [Google Scholar] [CrossRef] [PubMed]
- Aminov, R.I.; Mackie, R.I. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef] [PubMed]
- SCENIHR. Scientific Comitee on Emerging and Newly Identified Health Risks, Health Effects of Exposure to EMF; SCENIHR: Brussels, Belgium, 2009; Available online: https://ec.europa.eu›docs›scenihr_o_022 (accessed on 20 December 2021).
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Davin-Regli, A.; Pages, J.M. Cross-resistance between biocides and antimicrobials: An emerging question. Rev. Sci. Tech. 2012, 31, 89–104. [Google Scholar] [CrossRef]
- Jasovsky, D.; Littmann, J.; Zorzet, A.; Cars, O. Antimicrobial resistance-a threat to the world’s sustainable development. Ups. J. Med. Sci 2016, 121, 159–164. [Google Scholar] [CrossRef]
- Delepierre, A.; Gayot, A.; Carpentier, A. Update on counterfeit antibiotics worldwide; public health risks. Med. Mal. Infect. 2012, 42, 247–255. [Google Scholar] [CrossRef]
- Kelesidis, T.; Falagas, M.E. Substandard/counterfeit antimicrobial drugs. Clin. Microbiol. Rev. 2015, 28, 443–464. [Google Scholar] [CrossRef]
- Cockburn, R.; Newton, P.N.; Agyarko, E.K.; Akunyili, D.; White, N.J. The global threat of counterfeit drugs: Why industry and governments must communicate the dangers. PLoS Med. 2005, 2, e100. [Google Scholar] [CrossRef] [PubMed]
- Sartelli, M.; Hardcastle, T.; Catena, F.; Chichom-Mefire, A.; Coccolini, F.; Dhingra, S.; Haque, M.; Hodonou, A.; Iskandar, K.; Labricciosa, F.M.; et al. Antibiotic Use in Low and Middle-Income Countries and the Challenges of Antimicrobial Resistance in Surgery. Antibiotics 2020, 9, 497. [Google Scholar] [CrossRef] [PubMed]
- Guinovart, M.C.; Figueras, A.; Llor, C. Selling antimicrobials without prescription—Far beyond an administrative problem. Enferm. Infecc. Microbiol. Clin. Engl. Ed. 2018, 36, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Roque, F.; Soares, S.; Breitenfeld, L.; Figueiras, A.; Herdeiro, M.T. Influence of community pharmacists attitudes on antibiotic dispensing behavior: A cross-sectional study in Portugal. Clin. Ther. 2015, 37, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Auta, A.; Hadi, M.A.; Oga, E.; Adewuyi, E.O.; Abdu-Aguye, S.N.; Adeloye, D.; Strickland-Hodge, B.; Morgan, D.J. Global access to antibiotics without prescription in community pharmacies: A systematic review and meta-analysis. J. Infect. 2019, 78, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Salameh, P.; Sacre, H.; Hallit, S.; Hajj, A. Antibiotic Resistance in Lebanon. 2017. Available online: http://resistancecontrol.info/2017/antibiotic-resistance-in-lebanon/ (accessed on 20 December 2021).
- Fleming-Dutra, K.E.; Hersh, A.L.; Shapiro, D.J.; Bartoces, M.; Enns, E.A.; File, T.M., Jr.; Finkelstein, J.A.; Gerber, J.S.; Hyun, D.Y.; Linder, J.A.; et al. Prevalence of Inappropriate Antibiotic Prescriptions among US Ambulatory Care Visits, 2010–2011. JAMA 2016, 315, 1864–1873. [Google Scholar] [CrossRef]
- Teixeira Rodrigues, A.; Ferreira, M.; Roque, F.; Falcao, A.; Ramalheira, E.; Figueiras, A.; Herdeiro, M.T. Physicians’ attitudes and knowledge concerning antibiotic prescription and resistance: Questionnaire development and reliability. BMC Infect. Dis. 2016, 16, 7. [Google Scholar] [CrossRef]
- Morel, C.M.; Lindahl, O.; Harbarth, S.; de Kraker, M.E.A.; Edwards, S.; Hollis, A. Industry incentives and antibiotic resistance: An introduction to the antibiotic susceptibility bonus. J. Antibiot. 2020, 73, 421–428. [Google Scholar] [CrossRef]
- Saha, C.N.; Bhattacharya, S. Intellectual property rights: An overview and implications in pharmaceutical industry. J. Adv. Pharm. Technol. Res. 2011, 2, 88–93. [Google Scholar] [CrossRef]
- Tarrant, C.; Colman, A.M.; Chattoe-Brown, E.; Jenkins, D.R.; Mehtar, S.; Perera, N.; Krockow, E.M. Optimizing antibiotic prescribing: Collective approaches to managing a common-pool resource. Clin. Microbiol Infect. 2019, 25, 1356–1363. [Google Scholar] [CrossRef]
- World Health Organization. Global Priority List of Antibiotic Resistant Bacteria. 2017. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 21 December 2021).
- CDC. Antibiotic Resistance Threats in the United States. 2019. Available online: https://www.cdc.gov/DrugResistance/Biggest-Threats.html (accessed on 21 December 2021).
- World Health Organization. The Global Shortage of Innovative Antibiotics Fuels the Emergence and Spread of Drug Resistance. 15 April 2021. Available online: https://www.who.int/news/item/15-04-2021-global-shortage-of-innovative-antibiotics-fuels-emergence-and-spread-of-drug-resistance (accessed on 7 December 2021).
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic discovery: History, methods and perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Q.; Fu, Y.; Li, L.; Zhao, N.; Lu, A.; Liu, Q.; Jiang, M. Effectiveness of Chinese Herbal Medicine Combined with Antibiotics for Extensively Drug-Resistant Enterobacteria and Nonfermentative Bacteria Infection: Real-Life Experience in a Retrospective Cohort. Biomed. Res. Int. 2017, 2017, 2897045. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.; Molto, E.; Anderson, C. Fluorochrome labelling in Roman period skeletons from Dakhleh Oasis, Egypt. Am. J. Phys. Anthropol. 1989, 80, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Armelagos, G.J. Disease in ancient Nubia. Science 1969, 163, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Bassett, E.J.; Keith, M.S.; Armelagos, G.J.; Martin, D.L.; Villanueva, A.R. Tetracycline-labeled human bone from ancient Sudanese Nubia (A.D. 350). Science 1980, 209, 1532–1534. [Google Scholar] [CrossRef]
- Nelson, M.L.; Dinardo, A.; Hochberg, J.; Armelagos, G.J. Brief communication: Mass spectroscopic characterization of tetracycline in the skeletal remains of an ancient population from Sudanese Nubia 350–550 CE. Am. J. Phys. Anthropol. 2010, 143, 151–154. [Google Scholar] [CrossRef]
- Gould, I.M.; Bal, A.M. New antibiotic agents in the pipeline and how they can help overcome microbial resistance. Virulence 2013, 4, 185–191. [Google Scholar] [CrossRef]
- Wong, R.W.; Hagg, U.; Samaranayake, L.; Yuen, M.K.; Seneviratne, C.J.; Kao, R. Antimicrobial activity of Chinese medicine herbs against common bacteria in oral biofilm. A pilot study. Int. J. Oral Maxillofac. Surg. 2010, 39, 599–605. [Google Scholar] [CrossRef]
- Emmerich, R.; Löw, O. Bakteriolytische Enzyme als Ursache der erworbenen Immunität und die Heilung von Infectionskrankheiten durch dieselben. Z. Hyg. Infekt. 1899, 31, 1–65. [Google Scholar] [CrossRef]
- Saalim, M.; Villegas-Moreno, J.; Clark, B.R. Bacterial Alkyl-4-quinolones: Discovery, Structural Diversity and Biological Properties. Molecules 2020, 25, 5689. [Google Scholar] [CrossRef]
- Dubern, J.F.; Diggle, S.P. Quorum sensing by 2-alkyl-4-quinolones in Pseudomonas aeruginosa and other bacterial species. Mol. Biosyst. 2008, 4, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, G.F.; Sartorio, R.; Lee, S.H.; Rogers, C.J.; Meijler, M.M.; Moss, J.A.; Clapham, B.; Brogan, A.P.; Dickerson, T.J.; Janda, K.D. Revisiting quorum sensing: Discovery of additional chemical and biological functions for 3-oxo-N-acylhomoserine lactones. Proc. Natl. Acad. Sci. USA 2005, 102, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hays, E.E.; Wells, I.C.; Katzman, P.A.; Cain, C.K.; Jacobs, F.A.; Thayer, S.A.; Doisy, E.A.; Gaby, W.L.; Roberts, E.C.; Muir, R.D.; et al. Antibiotic substances produced by Pseudomonas aeruginosa. Biol. Chem. 1945, 725–750. [Google Scholar] [CrossRef]
- Valent, P.; Groner, B.; Schumacher, U.; Superti-Furga, G.; Busslinger, M.; Kralovics, R.; Zielinski, C.; Penninger, J.M.; Kerjaschki, D.; Stingl, G.; et al. Paul Ehrlich (1854–1915) and His Contributions to the Foundation and Birth of Translational Medicine. J. Innate Immun. 2016, 8, 111–120. [Google Scholar] [CrossRef]
- Domagk, G. Ein beitrag zur chemotherapie der bakteriellen infektionen. DMW-Dtsch. Med. Wochenschr. 1935, 61, 50–53. [Google Scholar] [CrossRef]
- Bentley, R. Different roads to discovery; Prontosil (hence sulfa drugs) and penicillin (hence beta-lactams). J. Ind. Microbiol. Biotechnol. 2009, 36, 775–786. [Google Scholar] [CrossRef]
- Tan, S.Y.; Tatsumura, Y. Alexander Fleming (1881–1955): Discoverer of penicillin. Singap. Med. J. 2015, 56, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Chain, E.; Florey, H.W.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Orr-Ewing, J.; Sanders, A.G. THE CLASSIC: Penicillin as a chemotherapeutic agent. 1940. Clin. Orthop. Relat. Res. 2005, 439, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Van Epps, H.L. Rene Dubos: Unearthing antibiotics. J. Exp. Med. 2006, 203, 259. [Google Scholar] [CrossRef] [PubMed]
- Schaedler, R.; Dubos, R. Friend of the Good Earth: Microbiologist, Medical Scientist, Environmentalist. Emerg. Infect. Dis. 2006, 12, 876–877. [Google Scholar] [CrossRef]
- Levine, D.P. Vancomycin: A history. Clin. Infect. Dis 2006, 42 (Suppl. 1), S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Critically Important Antibiotics for Human Medicine. 2018. Available online: https://apps.who.int/iris/bitstream/handle/10665/312266/9789241515528-eng.pdf (accessed on 21 December 2021).
- Alm, R.A.; Gallant, K. Innovation in Antimicrobial Resistance: The CARB-X Perspective. ACS Infect. Dis 2020, 6, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- McClure, N.S.; Day, T. A theoretical examination of the relative importance of evolution management and drug development for managing resistance. Proc. Biol. Sci. 2014, 281, 20141861. [Google Scholar] [CrossRef]
- Al-Tawfiq, J.A.; Momattin, H.; Al-Ali, A.Y.; Eljaaly, K.; Tirupathi, R.; Haradwala, M.B.; Areti, S.; Alhumaid, S.; Rabaan, A.A.; Al Mutair, A.; et al. Antibiotics in the pipeline: A literature review (2017–2020). Infection 2021, 1–12. [Google Scholar] [CrossRef]
- WHO. No Time to Wait: Securing the Future from Drug-Resistant Infections; World Health Organization: Geneva, Switzerland, 2019; Available online: https://ahpsr.who.int/publications/i/item/no-time-to-wait-securing-the-future-from-drug-resistant-infections (accessed on 21 December 2021).
- WHO. Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline. 2019. Available online: https://www.who.int/publications/i/item/9789240000193 (accessed on 21 December 2021).
- The Pew Charitable Trusts. Tracking the Global Pipeline of Antibiotics in Development. April 2020 (The Pew Charitable Trusts, 2020). Available online: https://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2020/04/trackingthe-global-pipeline-of-antibiotics-in-development (accessed on 21 December 2021).
- Impey, R.E.; Hawkins, D.A.; Sutton, J.M.; Soares da Costa, T.P. Overcoming Intrinsic and Acquired Resistance Mechanisms Associated with the Cell Wall of Gram-Negative Bacteria. Antibiotics 2020, 9, 623. [Google Scholar] [CrossRef]
- Shlaes, D.M. Antibacterial Drugs: The Last Frontier. ACS Infect. Dis. 2020, 6, 1313–1314. [Google Scholar] [CrossRef]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Ribeiro da Cunha, B.; Fonseca, L.P.; Calado, C.R.C. Antibiotic Discovery: Where Have We Come from, Where Do We Go? Antibiotics 2019, 8, 45. [Google Scholar] [CrossRef]
- Townsend, C.A. Convergent biosynthetic pathways to beta-lactam antibiotics. Curr. Opin. Chem. Biol. 2016, 35, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.R.; March, J.B. Bacteriophages and biotechnology: Vaccines, gene therapy and antibacterials. Trends Biotechnol. 2006, 24, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Luo, T.; Yang, Y.; Dong, D.; Wang, R.; Wang, Y.; Xu, M.; Guo, X.; Hu, F.; He, P. Phage Therapy as a Promising New Treatment for Lung Infection Caused by Carbapenem-Resistant Acinetobacter baumannii in Mice. Front. Microbiol. 2017, 8, 2659. [Google Scholar] [CrossRef] [PubMed]
- Leitner, L.; Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Chkonia, I.; Rigvava, S.; Chkhotua, A.; Changashvili, G.; McCallin, S.; Schneider, M.P.; et al. Intravesical bacteriophages for treating urinary tract infections in patients undergoing transurethral resection of the prostate: A randomised, placebo-controlled, double-blind clinical trial. Lancet Infect. Dis. 2021, 21, 427–436. [Google Scholar] [CrossRef]
- Diard, M.; Hardt, W.D. Evolution of bacterial virulence. FEMS Microbiol. Rev. 2017, 41, 679–697. [Google Scholar] [CrossRef]
- Muhlen, S.; Dersch, P. Anti-virulence Strategies to Target Bacterial Infections. Curr. Top. Microbiol. Immunol. 2016, 398, 147–183. [Google Scholar] [CrossRef]
- Jana, B.; Cain, A.K.; Doerrler, W.T.; Boinett, C.J.; Fookes, M.C.; Parkhill, J.; Guardabassi, L. The secondary resistome of multidrug-resistant Klebsiella pneumoniae. Sci. Rep. 2017, 7, 42483. [Google Scholar] [CrossRef]
- Panta, P.R.; Kumar, S.; Stafford, C.F.; Billiot, C.E.; Douglass, M.V.; Herrera, C.M.; Trent, M.S.; Doerrler, W.T. A DedA Family Membrane Protein Is Required for Burkholderia thailandensis Colistin Resistance. Front. Microbiol. 2019, 10, 2532. [Google Scholar] [CrossRef]
- Kumar, S.; Doerrler, W.T. Members of the conserved DedA family are likely membrane transporters and are required for drug resistance in Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 923–930. [Google Scholar] [CrossRef]
- Doerrler, W.T.; Sikdar, R.; Kumar, S.; Boughner, L.A. New functions for the ancient DedA membrane protein family. J. Bacteriol. 2013, 195, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.R.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion systems in Gram-negative bacteria: Structural and mechanistic insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V. Type IV secretion machinery: Molecular architecture and function. Biochem. Soc. Trans. 2013, 41, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Robertson, M.; Gauthier, A.; Finlay, B.B.; van Soest, R.; Andersen, R.J. Caminoside A, an antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Org. Lett. 2002, 4, 4089–4092. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Uchida, R.; Yoshijima, H.; Ui, H.; Shiomi, K.; Matsumoto, A.; Takahashi, Y.; Abe, A.; Tomoda, H.; Omura, S. Guadinomines, Type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. I: Taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2008, 61, 222–229. [Google Scholar] [CrossRef] [PubMed]
- McHugh, R.E.; O’Boyle, N.; Connolly, J.P.R.; Hoskisson, P.A.; Roe, A.J. Characterization of the Mode of Action of Aurodox, a Type III Secretion System Inhibitor from Streptomyces goldiniensis. Infect. Immun. 2019, 87, e00595-18. [Google Scholar] [CrossRef] [PubMed]
- Dufour, D.; Leung, V.; Lévesque, C.M. Bacterial biofilm: Structure, function, and antimicrobial resistance. Endod. Top. 2012, 22, 2–16. [Google Scholar] [CrossRef]
- Venkatesan, N.; Perumal, G.; Doble, M. Bacterial resistance in biofilm-associated bacteria. Future Microbiol. 2015, 10, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bassler, B.L. Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe 2019, 26, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Tiwari, M.; Donelli, G.; Tiwari, V. Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence 2018, 9, 522–554. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: Current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef] [PubMed]
- McDougald, D.; Rice, S.A.; Barraud, N.; Steinberg, P.D.; Kjelleberg, S. Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 2011, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Schleheck, D.; Klebensberger, J.; Webb, J.S.; Hassett, D.J.; Rice, S.A.; Kjelleberg, S. Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels, and enhanced dispersal. J. Bacteriol. 2009, 191, 7333–7342. [Google Scholar] [CrossRef] [PubMed]
- Marvasi, M.; Durie, I.A.; McLamore, E.S.; Vanegas, D.C.; Chaturvedi, P. Salmonella enterica biofilm-mediated dispersal by nitric oxide donors in association with cellulose nanocrystal hydrogels. AMB Express 2015, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Hasan, N.; Cao, J.; Lee, J.; Hlaing, S.P.; Yoo, J.W. Chitosan-based nitric oxide-releasing dressing for anti-biofilm and in vivo healing activities in MRSA biofilm-infected wounds. Int. J. Biol. Macromol. 2020, 142, 680–692. [Google Scholar] [CrossRef]
- Kramer, J.; Ozkaya, O.; Kummerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Boiteau, R.M.; Mende, D.R.; Hawco, N.J.; McIlvin, M.R.; Fitzsimmons, J.N.; Saito, M.A.; Sedwick, P.N.; DeLong, E.F.; Repeta, D.J. Siderophore-based microbial adaptations to iron scarcity across the eastern Pacific Ocean. Proc. Natl. Acad. Sci. USA 2016, 113, 14237–14242. [Google Scholar] [CrossRef]
- Miethke, M.; Marahiel, M.A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef]
- Wencewicz, T.A. New antibiotics from Nature’s chemical inventory. Bioorg. Med. Chem. 2016, 24, 6227–6252. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.L.; Johnstone, T.C.; Nolan, E.M. Chemical Synthesis of Staphyloferrin B Affords Insight into the Molecular Structure, Iron Chelation, and Biological Activity of a Polycarboxylate Siderophore Deployed by the Human Pathogen Staphylococcus aureus. J. Am. Chem. Soc. 2015, 137, 9117–9127. [Google Scholar] [CrossRef] [PubMed]
- Fleck, C.A. Differentiating MMPs, biofilm, endotoxins, exotoxins, and cytokines. Adv. Skin Wound Care 2006, 19, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Baldwin, M.R.; Barbieri, J.T. Toxins from bacteria. In Molecular, Clinical and Environmental Toxicology; Experientia Supplementum Book Series; Springer: Berlin/Heidelberg, Germany, 2010; Volume 100, pp. 1–29. [Google Scholar] [CrossRef]
- Xiong, Y.; Wiltsie, J.; Woods, A.; Guo, J.; Pivnichny, J.V.; Tang, W.; Bansal, A.; Cummings, R.T.; Cunningham, B.R.; Friedlander, A.M.; et al. The discovery of a potent and selective lethal factor inhibitor for adjunct therapy of anthrax infection. Bioorg. Med. Chem. Lett. 2006, 16, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.B.; Turk, B.E. Inhibitors of the Metalloproteinase Anthrax Lethal Factor. Curr. Top. Med. Chem. 2016, 16, 2350–2358. [Google Scholar] [CrossRef]
- Li, F.; Terzyan, S.; Tang, J. Subsite specificity of anthrax lethal factor and its implications for inhibitor development. Biochem. Biophys. Res. Commun. 2011, 407, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chvyrkova, I.; Terzyan, S.; Wakeham, N.; Turner, R.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Inhibition of anthrax lethal factor: Lability of hydroxamate as a chelating group. Appl. Microbiol. Biotechnol. 2012, 94, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Slotwinska, S.M. Host and bacterial adhesion. Pol. J. Vet. Sci. 2013, 16, 153–156. [Google Scholar] [CrossRef]
- Epler Barbercheck, C.R.; Bullitt, E.; Andersson, M. Bacterial Adhesion Pili. Subcell Biochem. 2018, 87, 1–18. [Google Scholar] [CrossRef]
- Pinkner, J.S.; Remaut, H.; Buelens, F.; Miller, E.; Aberg, V.; Pemberton, N.; Hedenstrom, M.; Larsson, A.; Seed, P.; Waksman, G.; et al. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 17897–17902. [Google Scholar] [CrossRef]
- Wu, S.; Lin, K.; Liu, Y.; Zhang, H.; Lei, L. Two-component signaling pathways modulate drug resistance of Staphylococcus aureus (Review). Biomed. Rep. 2020, 13, 5. [Google Scholar] [CrossRef]
- Worthington, R.J.; Blackledge, M.S.; Melander, C. Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Med. Chem. 2013, 5, 1265–1284. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular Mechanisms of Two-Component Signal Transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed]
- Igo, M.M.; Ninfa, A.J.; Stock, J.B.; Silhavy, T.J. Phosphorylation and dephosphorylation of a bacterial transcriptional activator by a transmembrane receptor. Genes Dev. 1989, 3, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Sully, E.K.; Malachowa, N.; Elmore, B.O.; Alexander, S.M.; Femling, J.K.; Gray, B.M.; DeLeo, F.R.; Otto, M.; Cheung, A.L.; Edwards, B.S.; et al. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 2014, 10, e1004174. [Google Scholar] [CrossRef]
- Mahdally, N.H.; George, R.F.; Kashef, M.T.; Al-Ghobashy, M.; Murad, F.E.; Attia, A.S. Staquorsin: A Novel Staphylococcus aureus Agr-Mediated Quorum Sensing Inhibitor Impairing Virulence in vivo without Notable Resistance Development. Front. Microbiol. 2021, 12, 700494. [Google Scholar] [CrossRef]
- Goswami, M.; Espinasse, A.; Carlson, E.E. Disarming the virulence arsenal of Pseudomonas aeruginosa by blocking two-component system signaling. Chem. Sci. 2018, 9, 7332–7337. [Google Scholar] [CrossRef]
- Rasko, D.A.; Moreira, C.G.; de Li, R.; Reading, N.C.; Ritchie, J.M.; Waldor, M.K.; Williams, N.; Taussig, R.; Wei, S.; Roth, M.; et al. Targeting QseC signaling and virulence for antibiotic development. Science 2008, 321, 1078–1080. [Google Scholar] [CrossRef]
- Rooks, M.G.; Veiga, P.; Reeves, A.Z.; Lavoie, S.; Yasuda, K.; Asano, Y.; Yoshihara, K.; Michaud, M.; Wardwell-Scott, L.; Gallini, C.A.; et al. QseC inhibition as an antivirulence approach for colitis-associated bacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 142–147. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Piddock, L.J.V. Non-traditional Antibacterial Therapeutic Options and Challenges. Cell Host Microbe 2019, 26, 61–72. [Google Scholar] [CrossRef]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef]
- Albarano, L.; Esposito, R.; Ruocco, N.; Costantini, M. Genome Mining as New Challenge in Natural Products Discovery. Mar. Drugs 2020, 18, 199. [Google Scholar] [CrossRef]
- Foulston, L. Genome mining and prospects for antibiotic discovery. Curr. Opin. Microbiol. 2019, 51, 1–8. [Google Scholar] [CrossRef]
- Schneider, O.; Simic, N.; Aachmann, F.L.; Ruckert, C.; Kristiansen, K.A.; Kalinowski, J.; Jiang, Y.; Wang, L.; Jiang, C.L.; Lale, R.; et al. Genome Mining of Streptomyces sp. YIM 130001 Isolated from Lichen Affords New Thiopeptide Antibiotic. Front. Microbiol. 2018, 9, 3139. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Zhong, Z.; Zhang, W.P.; Qian, P.Y. Discovery of cationic nonribosomal peptides as Gram-negative antibiotics through global genome mining. Nat. Commun. 2018, 9, 3273. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C. The gut microbiome. Inflamm Allergy Drug Targets 2014, 13, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J. Where are we with monoclonal antibodies for multidrug-resistant infections? Drug Discov. Today 2019, 24, 1132–1138. [Google Scholar] [CrossRef]
- Nagy, E.; Nagy, G.; Power, C.A.; Badarau, A.; Szijarto, V. Anti-bacterial Monoclonal Antibodies. Adv. Exp. Med. Biol. 2017, 1053, 119–153. [Google Scholar] [CrossRef]
- Sause, W.E.; Buckley, P.T.; Strohl, W.R.; Lynch, A.S.; Torres, V.J. Antibody-Based Biologics and Their Promise to Combat Staphylococcus aureus Infections. Trends Pharmacol. Sci. 2016, 37, 231–241. [Google Scholar] [CrossRef]
- Yang, Y.; Qian, M.; Yi, S.; Liu, S.; Li, B.; Yu, R.; Guo, Q.; Zhang, X.; Yu, C.; Li, J.; et al. Monoclonal Antibody Targeting Staphylococcus aureus Surface Protein A (SasA) Protect against Staphylococcus aureus Sepsis and Peritonitis in Mice. PLoS ONE 2016, 11, e0149460. [Google Scholar] [CrossRef]
- Diago-Navarro, E.; Motley, M.P.; Ruiz-Perez, G.; Yu, W.; Austin, J.; Seco, B.M.S.; Xiao, G.; Chikhalya, A.; Seeberger, P.H.; Fries, B.C. Novel, Broadly Reactive Anticapsular Antibodies against Carbapenem-Resistant Klebsiella pneumoniae Protect from Infection. mBio 2018, 9, e00091-18. [Google Scholar] [CrossRef] [PubMed]
- Ardal, C.; Findlay, D.; Savic, M.; Carmeli, Y.; Gyssens, I.; Laxminarayan, R.; Outterson, K.; Rex, J.H. Innovative Medicines Initiative DRIVE-AB Report. Revitalizing the Antibiotic Pipeline: Stimulating Innovation While Driving Sustainable Use and Global. 2018. Available online: http://drive-ab.eu/wp-content/uploads/2018/01/CHHJ5467-Drive-AB-Main-Report-180319-WEB.pdf (accessed on 22 December 2021).
- Luepke, K.H.; Mohr, J.F., 3rd. The antibiotic pipeline: Reviving research and development and speeding drugs to market. Expert Rev. Anti-Infect. Ther. 2017, 15, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Renwick, M.J.; Brogan, D.M.; Mossialos, E. A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics. J. Antibiot. 2016, 69, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Outterson, K.; Rex, J.H.; Jinks, T.; Jackson, P.; Hallinan, J.; Karp, S.; Hung, D.T.; Franceschi, F.; Merkeley, T.; Houchens, C.; et al. Accelerating global innovation to address antibacterial resistance: Introducing CARB-X. Nat. Rev. Drug Discov. 2016, 15, 589–590. [Google Scholar] [CrossRef]
- Duffy, E.M.; Buurman, E.T.; Chiang, S.L.; Cohen, N.R.; Uria-Nickelsen, M.; Alm, R.A. The CARB-X Portfolio of Nontraditional Antibacterial Products. ACS Infect. Dis. 2021, 7, 2043–2049. [Google Scholar] [CrossRef]
- Ardal, C.; Baraldi, E.; Ciabuschi, F.; Outterson, K.; Rex, J.H.; Piddock, L.J.V.; Findlay, D.; Committee, D.-A.S. To the G20: Incentivising antibacterial research and development. Lancet Infect. Dis. 2017, 17, 799–801. [Google Scholar] [CrossRef]
- Ardal, C.; Rottingen, J.A.; Opalska, A.; Van Hengel, A.J.; Larsen, J. Pull Incentives for Antibacterial Drug Development: An Analysis by the Transatlantic Task Force on Antimicrobial Resistance. Clin. Infect. Dis. 2017, 65, 1378–1382. [Google Scholar] [CrossRef]
- Baraldi, E.; Lindahl, O.; Savic, M.; Findlay, D.; Ardal, C. Antibiotic Pipeline Coordinators. J. Law Med. Ethics 2018, 46, 25–31. [Google Scholar] [CrossRef]
- Drive, A.B. Driving Reinvestment in R&D for Antibiotics and Advocating their Responsible Use. 2014. Available online: http://drive-ab.eu/ (accessed on 24 December 2021).
- Stiglitz, J.E. The Stiglitz Report: Reforming the International Monetary and Financial Systems in the Wake of the Global Crisis; The New Press: New York, NY, USA, 2010. [Google Scholar]
- Horowitz, J.B.; Moehring, H.B. How property rights and patents affect antibiotic resistance. Health Econ. 2004, 12, 575–583. [Google Scholar] [CrossRef]
- Andrews, E.B. The Economic Law of Monopoly. J. Soc. Sci. 1890, 26, 1. [Google Scholar]
- Singer, A.C.; Kirchhelle, C.; Roberts, A.P. Reinventing the antimicrobial pipeline in response to the global crisis of antimicrobial-resistant infections. F1000Res 2019, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.K.; Teng, C.; Frei, C.R. Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus on Drug Repurposing. Front. Cell Infect. Microbiol. 2021, 11, 684515. [Google Scholar] [CrossRef] [PubMed]
- Renwick, M.; Mossialos, E. What are the economic barriers of antibiotic R&D and how can we overcome them? Expert Opin. Drug Discov. 2018, 13, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Brogan, D.M.; Mossialos, E. A critical analysis of the review on antimicrobial resistance report and the infectious disease financing facility. Glob. Health 2016, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Khardori, N.; Stevaux, C.; Ripley, K. Antibiotics: From the Beginning to the Future: Part 2. Indian J. Pediatr. 2020, 87, 43–47. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO [134] | CDC [135] | |||
|---|---|---|---|---|
| Organisms | Pathogens | Level of Priority | Pathogens | Level of Threat |
| Acinetobacter baumannii | Acinetobacter baumannii, carbapenem-resistant | Critical | Urgent * | |
| Pseudomonas aeruginosa | Pseudomonas aeruginosa, carbapenem-resistant | Critical | Multidrug-resistant Pseudomonas aeruginosa 1 | Serious ** |
| Enterobacteriaceae | Enterobacteriaceae, carbapenem-resistant, ESBL-producing | Critical | Enterobacteriacea, Carbapenem-resistant 2 3 | Urgent |
| Enterobacteriaceae, ESBL-producing | Critical | Extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae 4 | Serious | |
| Enterococcus faecium | Enterococcus faecium, vancomycin-resistant | High | Vancomycin-resistant Enterococci (VRE) | Serious |
| Staphylococcus aureus | Staphylococcus aureus, methicillin-resistant | High | Methicillin-resistant Staphylococcus aureus (MRSA) 5 | Serious |
| Staphylococcus aureus | Staphylococcus aureus, vancomycin-intermediate and resistant | High | ||
| Helicobacter pylori | Helicobacter pylori, clarithromycin-resistant | High | ||
| Campylobacter spp. | Campylobacter spp., fluoroquinolone-resistant | High | Drug-resistant Campylobacter | Serious |
| Salmonellae | Salmonellae, fluoroquinolone-resistant | High | Drug-resistant nontyphoidal Salmonella Drug-resistant Salmonella serotype Typhi | Serious |
| Neisseria gonorrhoeae | Neisseria gonorrhoeae, cephalosporin-resistant, fluoroquinolone-resistant | High | Drug-resistant Neisseria gonorrhoeae | Urgent |
| Streptococcus pneumoniae | Streptococcus pneumoniae, penicillin-non-susceptible | Medium | Drug-resistant Streptococcus pneumoniae | Serious |
| Erythromycin-Resistant Group A Streptococcus Clindamycin-resistant Group B Streptococcus | Concerning *** | |||
| Haemophilus influenzae | Haemophilus influenzae, ampicillin-resistant | Medium | ||
| Shigella spp. | Shigella spp., fluoroquinolone-resistant | Medium | Drug-resistant Shigella | Serious |
| Clostridioides difficile | Urgent | |||
| Mycobacterium tuberculosis | Not listed in the 2017 high priority pathogens because it is previously established as high priority | Drug-resistant tuberculosis | Serious | |
| Bordetella pertusis | Drug-resistant Bordetella pertusis | Watch | ||
| Product Name | Alternative Name | Product Type | Non-Traditional Categories | R&D Phase | Antibacterial Class | Expected Activity against Priority Pathogens | Route of Administration | Innovative | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Critical Priority Pathogens | Other Priority Pathogens | IV | Oral | Inh | |||||||||||||||||||||||||||
| Non-traditionals | Antibiotics | Antibodies | Microbiome modulating agents | Bacteriophages and phage-derived enzymes | Immunomodulating agents | Miscellaneous | Phase I | Phase II | Phase III | Unknown | Acinetobacter baumannii | Pseudomos aeruginosa | Enterobacterales | All critical priority pathogens | Gram-positive priority pathogens | Neisseria gonorrhea | Helicobacter pylori | Staphylococcus aureus | Enterococcus faecium | Streptococcus pneumoniae | Campylobacter species | Other priority pathogens | Mycobacterium tuberculosis | Clostridium difficile | |||||||
| 514G3 | True human™ Mab | • | • | • | Anti-Staphylococcus aureus IgG monoclonal Ab | • | • | • | |||||||||||||||||||||||
| AB103 | Reltecimod | • | • | • | Antagonist of superantigen exotoxins and CD28 T-cell | • | • | • | • | ||||||||||||||||||||||
| ACX-362E | - | • | • | D polymerase IIIC inhibitor | • | • (na) | • | ||||||||||||||||||||||||
| Afabicin | Debio-1450 | • | • | FabI inhibitor | No | No | No | No | • | No | No | • | No | No | No | • | • | • | • | ||||||||||||
| AR-101 | Panobacumab, Aerumab | • | • | • | Anti-Pseudomonas aeruginosa serotype O11 IgG monoclonal Ab | • | • | ||||||||||||||||||||||||
| AR-105 | Aerucin | • | • | • | Anti-P. aeruginosa IgG1 monoclol Ab | • | • | ||||||||||||||||||||||||
| AR-301 | Tosatoxumab | • | • | • | Anti-S. aureus IgM monoclol Ab | • | • | • | |||||||||||||||||||||||
| ARX-1796 | Oral Avibactam prodrug | • | • | DBO-BLI + β-lactam | No | No | • | No | No | No | No | No | No | No | No | No | • | No | |||||||||||||
| Bepenem | - | • | • | Carbapenem | No | No | No | No | • | • | No | ||||||||||||||||||||
| BT588 | Trimodulin | • | • | • | Anti-S. aureus polyvalent Ab (IgM, IgA and IgG) | • | • | • | |||||||||||||||||||||||
| BTZ-043 | - | • | • | DprE1 inhibitor (benzothiazinone) | • | • | • | ||||||||||||||||||||||||
| CAL02 | - | • | • | • | Broad spectrum anti-toxin liposomal agent and noparticle | • | • | • | |||||||||||||||||||||||
| CF-301 | Exebacase | • | • | • | Phage endolysin | • | • | • | |||||||||||||||||||||||
| CP101 | – | • | • | • | Live biotherapeutic product | • | • | ||||||||||||||||||||||||
| CRS3123 | - | • | • | Methionyl-tR synthetase inhibitor (MetRS) | • | • | • | ||||||||||||||||||||||||
| DAV132 | – | • | • | • | Antibiotic ictivator and protective colon-targeted adsorbent | • | • | ||||||||||||||||||||||||
| Delpazolid | LCB01-0371 | • | • | Oxazolidinone | • | • | No | ||||||||||||||||||||||||
| DNV-3827 | MCB-3837 | • | • | Oxazolidinone-quinolone hybrid | • | • | Inconclusive | ||||||||||||||||||||||||
| DSTA4637S | RG7861 | • | • | • | Anti-S. aureus IgG mAb/rifamycin | • | • | • | |||||||||||||||||||||||
| Durlobactam + sulbactam | ETX-2514 | • | • | DBO-BLI /PBP2 binder + β-lactam-BLI/PBP1,3 binder | • | No | No | No | • | No | |||||||||||||||||||||
| EBL-1003 | Apramycin | • | • | Aminoglycoside | Pos | No | Pos | No | • | No | |||||||||||||||||||||
| Enmetazobactam + cefepime | AAl-101 + cefepime | • | • | β-lactam BLI + cephalosporin | No | No | No | No | • | No | |||||||||||||||||||||
| ETX0282 + cefpodoxime | – | • | • | DBO-BLI/PBP2 binder + cephalosporin | No | No | • | No | • | No | |||||||||||||||||||||
| Ftortiazinon + cefipime | Fluorothyazinone | • | • | • | Type III secretion system inhibition + cefepime | • | • | ||||||||||||||||||||||||
| Gepotidacin | – | • | • | Topoisomerase inhibitors (Triazaacephthylene) | No | • | • | No | • | No | No | No | • | • | • | • | |||||||||||||||
| GSK-3036656 | GSK-070 | • | • | Leu RS inhibitor (oxaborole) | • | • | • | ||||||||||||||||||||||||
| GSK3882347 | - | • | • | • | FimH antagonist | • | • | ||||||||||||||||||||||||
| IM-01 | – | • | • | • | FimH antagonist | • | • | ||||||||||||||||||||||||
| KB109 | - | • | • | • | Anti-Clostridium difficile polcyclonal Ab | • | • | • | • | ||||||||||||||||||||||
| KBP-7072 | – | • | • | Tetracycline | • | No | No | No | • | No | No | • | No | No | No | • | • | No | |||||||||||||
| LBP-EC01 | – | • | • | • | CRISPR-Cas3 enhanced phage | • | • | ||||||||||||||||||||||||
| LMN-101 | – | • | • | • | Monoclol Ab-like recombinant protein | • | • | • | • | ||||||||||||||||||||||
| Macozinone | PBTZ-169 | • | • | DprE1 inhibitor (Benzothiazinone) | • | • | • | ||||||||||||||||||||||||
| MEDI-4893 | Suvratoxumab | • | • | • | Anti-S. aureus IgG monoclonal Ab | • | • | • | |||||||||||||||||||||||
| MET-2 | - | • | • | • | Live biotherapeutic product | • | • | ||||||||||||||||||||||||
| MGB-BP-3 | – | • | • | D minor groove binder (distamycin) | • | • (na) | • | ||||||||||||||||||||||||
| cubactam + meropenem | – | • | • | DBO-BLI/PBP2 binder + cephalosporin | No | No | • | No | • | No | |||||||||||||||||||||
| fithromycin | WCK-4873 | • | • | Macrolide | • | No | No | • | No | • | No | • | • | No | |||||||||||||||||
| OligoG | CF-5/20 | • | • | • | Algite oligosaccharide (G-block) fragment | • | • | ||||||||||||||||||||||||
| OPC-167832 | – | • | • | DprE1 inhibitor (3,4-dihydrocarbostyril) | • | • | • | ||||||||||||||||||||||||
| Phage Bank | – | • | • | • | Phage bank (process) | • | • | ||||||||||||||||||||||||
| PLG0206 | WLBU2 | • | • | Cationic peptide | Pos | Pos | Pos | Pos | • | • | • | • | • | ||||||||||||||||||
| QPX7728 + QPX2014 | - | • | • | Borote-BLI + unknown | • | Pos | • | Pos | • | Inconclusive | |||||||||||||||||||||
| RBX7455 | - | • | • | • | Live biotherapeutic product | • | • | ||||||||||||||||||||||||
| Rhu-pGSN | Rhu-plasma gelsolin | • | • | • | Recombint human plasma gelsolin protein | • | • | • | • | • | • | • | • | • | • | • | • | • | |||||||||||||
| Ridinilazole | – | • | • | Bis-benzimidazole | • | • (na) | • | ||||||||||||||||||||||||
| SAL-200 | Tobacase | • | • | • | Phage endolysin | • | • | • | |||||||||||||||||||||||
| SER-109 | - | • | • | • | Live biotherapeutic product | • | • | ||||||||||||||||||||||||
| SPR-206 | - | • | • | Polymyxin | • | • | • | • | • | No | |||||||||||||||||||||
| SPR-720 | - | • | • | GyrB inhibitor (benzimidazole ethyl urea) | • | • | • | ||||||||||||||||||||||||
| Sulopenem, sulopenem etzadroxil/probenecid | – | • | • | Penem | No | No | No | No | • | • | No | ||||||||||||||||||||
| Sutezolid | – | • | • | Oxazolidinone | • | • | No | ||||||||||||||||||||||||
| SYN-004 | Ribaxamase | • | • | • | Antibiotic inactivator | • | • | ||||||||||||||||||||||||
| Taniborbactam + cefepime | VNRX-5133 + cefepime | • | • | Borote-BLI + cephalosporin | No | Pos | • | No | • | • | |||||||||||||||||||||
| TBA-7371 | – | • | • | DprE1 inhibitor (azaindole) | • | • | • | ||||||||||||||||||||||||
| TBAJ-876 | - | • | • | Diarylquinoline | • | • | No | ||||||||||||||||||||||||
| TBI-166 | – | • | • | Riminophezine (clofazimine-alogue) | • | • | No | ||||||||||||||||||||||||
| TBI-223 | - | • | • | Oxazolidinone | • | • | No | ||||||||||||||||||||||||
| Telacebec | Q-203 | • | • | Imidazopyridine amide | • | • | • | ||||||||||||||||||||||||
| TNP-2092 | – | • | • | Rifamycin-quinolizinone hybrid | No | No | No | No | Pos | No | Pos | Pos | Pos | Pos | • | • | No | ||||||||||||||
| TNP-2198 | - | • | • | rifamycin-nitroimidazole conjugate | No | Pos | • | No | • | • | No | ||||||||||||||||||||
| TP-271 | – | • | • | Tetracycline | Pos | No | No | No | • | No | No | • | • | No | No | • | • | • | No | ||||||||||||
| TP-6076 | – | • | • | Tetracycline | • | No | Pos | No | • | No | |||||||||||||||||||||
| TXA709 | - | • | • | FtsZ inhibitor | No | No | No | No | • | No | No | • | No | No | No | • | • | • | • | ||||||||||||
| VE303 | – | • | • | • | Live biotherapeutic product | • | • | ||||||||||||||||||||||||
| VNRX-7145 + ceftibuten | – | • | • | Borote-BLI + cephalosporin | No | No | • | No | • | • | |||||||||||||||||||||
| Zidebactam + cefepime | – | • | • | DBO-BLI/ PBP2 binder + cephalosporin | • | • | • | • | • | No | |||||||||||||||||||||
| Zoliflodacin | – | • | • | Topoisomerase Inhibitors (Spiropyrimidenetrione) | No | No | No | No | No | • | No | No | No | No | No | • | • | • | |||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iskandar, K.; Murugaiyan, J.; Hammoudi Halat, D.; Hage, S.E.; Chibabhai, V.; Adukkadukkam, S.; Roques, C.; Molinier, L.; Salameh, P.; Van Dongen, M. Antibiotic Discovery and Resistance: The Chase and the Race. Antibiotics 2022, 11, 182. https://doi.org/10.3390/antibiotics11020182
Iskandar K, Murugaiyan J, Hammoudi Halat D, Hage SE, Chibabhai V, Adukkadukkam S, Roques C, Molinier L, Salameh P, Van Dongen M. Antibiotic Discovery and Resistance: The Chase and the Race. Antibiotics. 2022; 11(2):182. https://doi.org/10.3390/antibiotics11020182
Chicago/Turabian StyleIskandar, Katia, Jayaseelan Murugaiyan, Dalal Hammoudi Halat, Said El Hage, Vindana Chibabhai, Saranya Adukkadukkam, Christine Roques, Laurent Molinier, Pascale Salameh, and Maarten Van Dongen. 2022. "Antibiotic Discovery and Resistance: The Chase and the Race" Antibiotics 11, no. 2: 182. https://doi.org/10.3390/antibiotics11020182
APA StyleIskandar, K., Murugaiyan, J., Hammoudi Halat, D., Hage, S. E., Chibabhai, V., Adukkadukkam, S., Roques, C., Molinier, L., Salameh, P., & Van Dongen, M. (2022). Antibiotic Discovery and Resistance: The Chase and the Race. Antibiotics, 11(2), 182. https://doi.org/10.3390/antibiotics11020182