Distinct Changes in Microbiota-Mediated Intestinal Metabolites and Immune Responses Induced by Different Antibiotics


Abstract
1. Introduction
2. Results
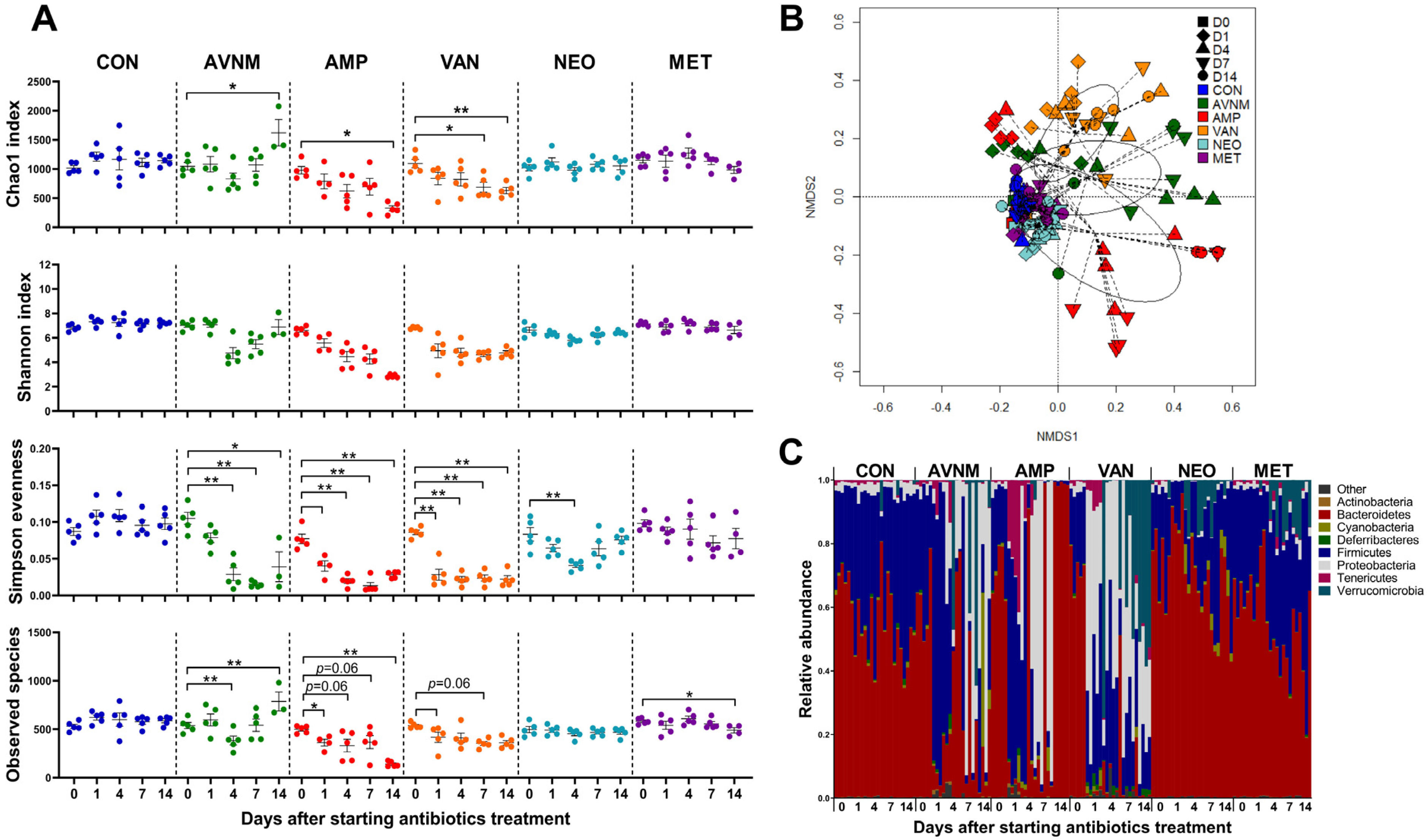
2.1. Antibiotic-Induced Changes in the Diversity and Structure of the Fecal Bacteria Community
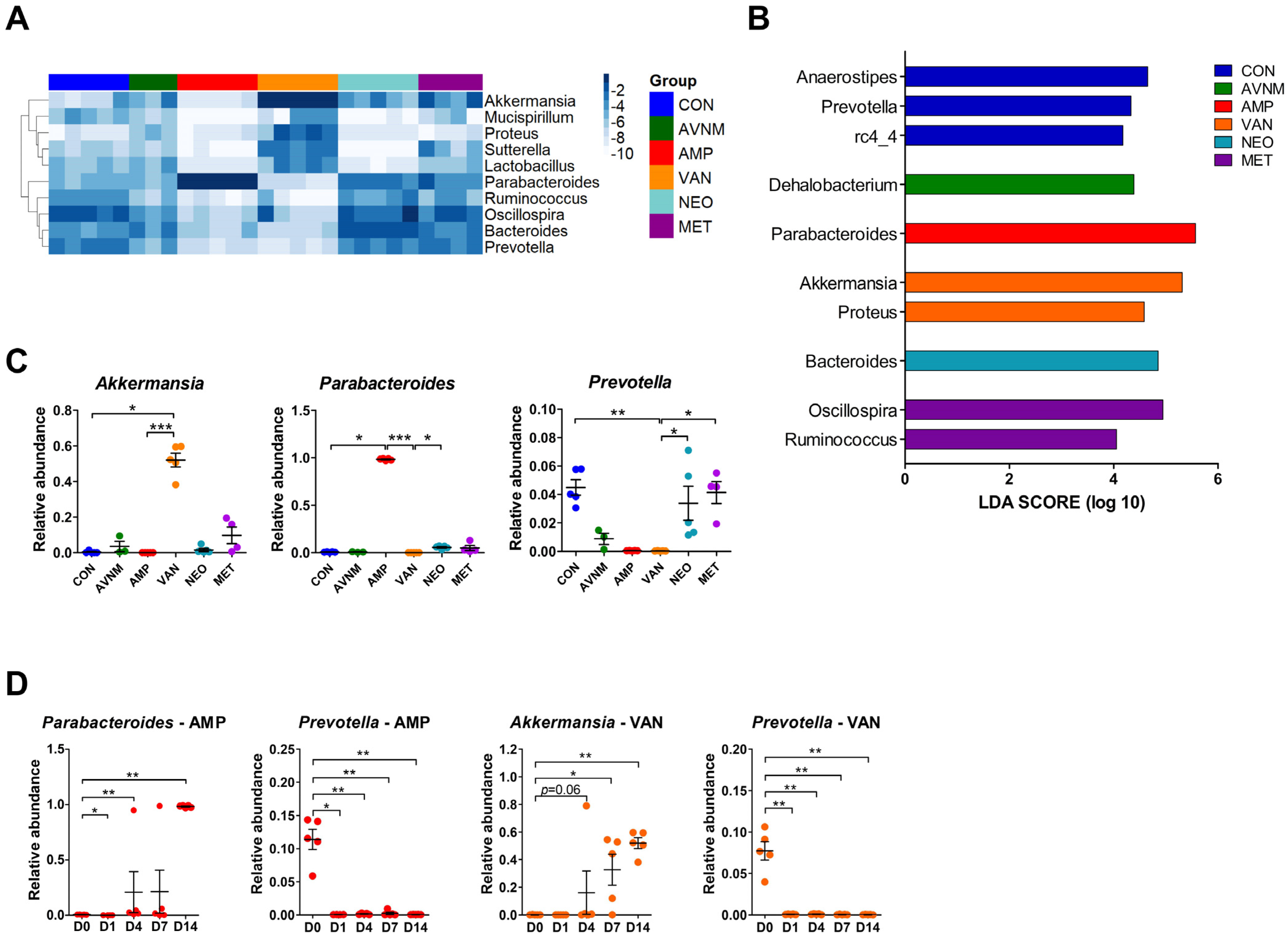
2.2. Major Bacterial Alterations under Antibiotic Treatment
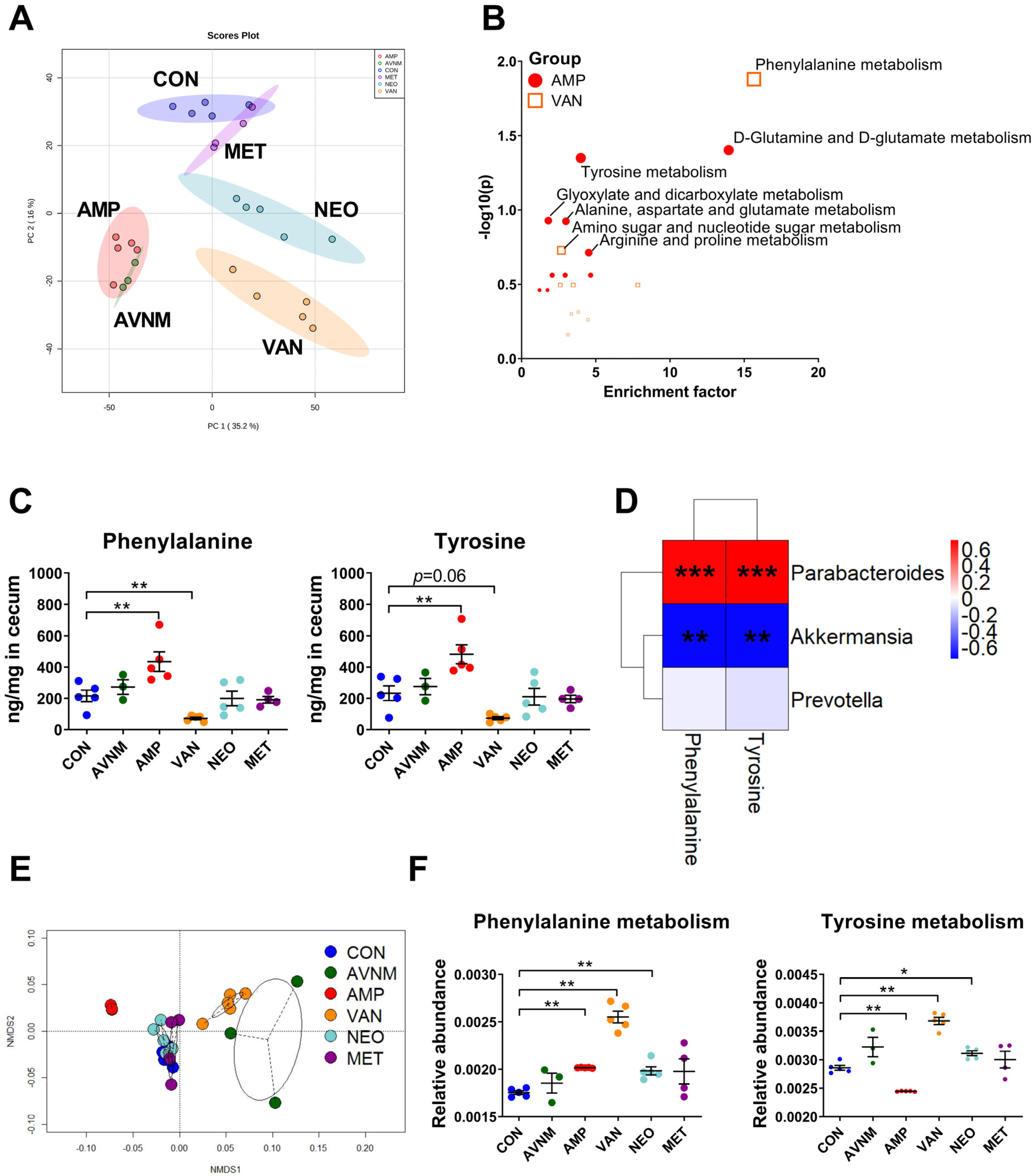
2.3. Antibiotic-Induced Changes in Gut Metabolites
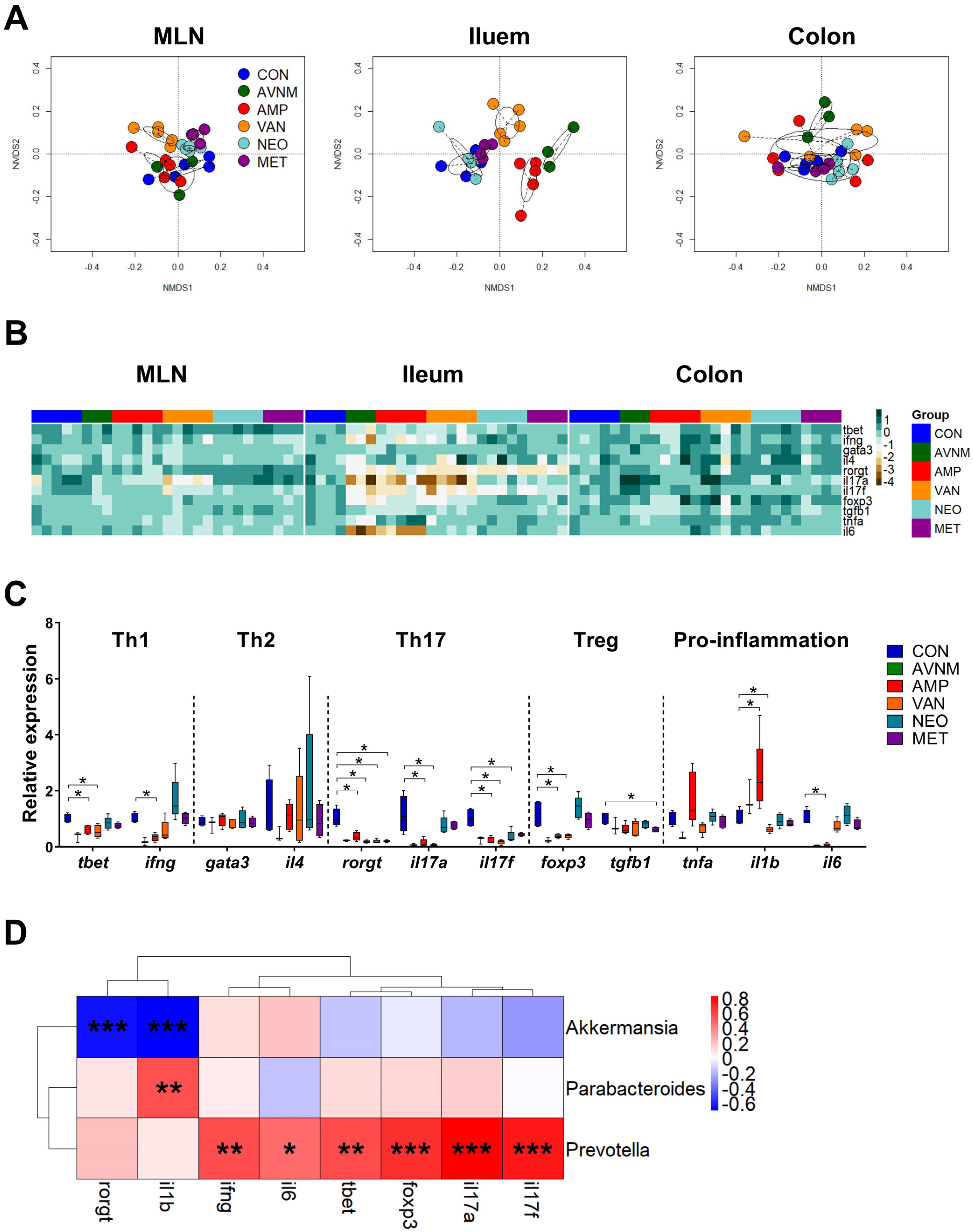
2.4. Changes in Expression of Immune-Related Genes in the Gut in Response to Antibiotic Treatment
3. Discussion
4. Materials and Methods
4.1. Mice and Antibiotic Treatment
4.2. Microbiome Analysis Using 16S rRNA Gene Amplicon Sequencing
4.3. LC-QTOF-MS/MS Analysis
4.4. Gene Expression Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Round, J.L.; Mazmanian, S.K. The Gut Microbiota Shapes Intestinal Immune Responses during Health and Disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; de Llanos Frutos, R.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Östman, S.; Rask, C.; Wold, A.E.; Hultkrantz, S.; Telemo, E. Impaired Regulatory T Cell Function in Germ-Free Mice. Eur. J. Immunol. 2006, 36, 2336–2346. [Google Scholar] [CrossRef]
- Wang, X.Q.; Zhang, A.H.; Miao, J.H.; Sun, H.; Yan, G.L.; Wu, F.F.; Wang, X.J. Gut Microbiota as Important Modulator of Metabolism in Health and Disease. RSC Adv. 2018, 8, 42380–42389. [Google Scholar] [CrossRef] [PubMed]
- Baars, A.; Oosting, A.; Lohuis, M.; Koehorst, M.; El Aidy, S.; Hugenholtz, F.; Smidt, H.; Mischke, M.; Boekschoten, M.V.; Verkade, H.J.; et al. Sex Differences in Lipid Metabolism Are Affected by Presence of the Gut Microbiota. Sci. Rep. 2018, 8, 13426. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Metabolic Interactions. Science 2012, 108, 1262–1268. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut Microbial Metabolites as Multi-Kingdom Intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, Y.; Wang, G.; Zheng, X.; Hao, H. Gut Microbial Metabolites of Aromatic Amino Acids as Signals in Host–Microbe Interplay. Trends Endocrinol. Metab. 2020, 31, 818–834. [Google Scholar] [CrossRef]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol. 2018, 9, 534. [Google Scholar] [CrossRef]
- Hägerbrand, K.; Westlund, J.; Yrlid, U.; Agace, W.; Johansson-Lindbom, B. MyD88 Signaling Regulates Steady-State Migration of Intestinal CD103 + Dendritic Cells Independently of TNF-α and the Gut Microbiota. J. Immunol. 2015, 195, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Knoop, K.A.; McDonald, K.G.; McCrate, S.; McDole, J.R.; Newberry, R.D. Microbial Sensing by Goblet Cells Controls Immune Surveillance of Luminal Antigens in the Colon. Mucosal Immunol. 2015, 8, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Jakobsson, H.E.; Holmén-Larsson, J.; Schütte, A.; Ermund, A.; Rodríguez-Piñeiro, A.M.; Arike, L.; Wising, C.; Svensson, F.; Bäckhed, F.; et al. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe 2015, 18, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Benakis, C.; Poon, C.; Lane, D.; Brea, D.; Sita, G.; Moore, J.; Murphy, M.; Racchumi, G.; Iadecola, C.; Anrather, J. Distinct Commensal Bacterial Signature in the Gut Is Associated with Acute and Long-Term Protection from Ischemic Stroke. Stroke 2020, 51, 1844–1854. [Google Scholar] [CrossRef]
- Lee, J.G.; Lee, Y.-R.; Lee, A.-R.; Park, C.H.; Han, D.S.; Eun, C.S. Role of the Global Gut Microbial Community in the Development of Colitis-Associated Cancer in a Murine Model. Biomed. Pharmacother. 2021, 135, 111206. [Google Scholar] [CrossRef]
- Huang, C.; Feng, S.; Huo, F.; Liu, H. Effects of Four Antibiotics on the Diversity of the Intestinal Microbiota. Microbiol. Spectr. 2022, 10, e0190421. [Google Scholar] [CrossRef]
- Neis, E.P.J.G.; Dejong, C.H.C.; Rensen, S.S. The Role of Microbial Amino Acid Metabolism in Host Metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut Microbiota, Metabolites and Host Immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut Microbiota Diversity and Human Diseases: Should We Reintroduce Key Predators in Our Ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef]
- Xu, L.; Surathu, A.; Raplee, I.; Chockalingam, A.; Stewart, S.; Walker, L.; Sacks, L.; Patel, V.; Li, Z.; Rouse, R. The Effect of Antibiotics on the Gut Microbiome: A Metagenomics Analysis of Microbial Shift and Gut Antibiotic Resistance in Antibiotic Treated Mice. BMC Genom. 2020, 21, 263. [Google Scholar] [CrossRef]
- Hansen, C.H.F.; Krych, L.; Nielsen, D.S.; Vogensen, F.K.; Hansen, L.H.; Sørensen, S.J.; Buschard, K.; Hansen, A.K. Early Life Treatment with Vancomycin Propagates Akkermansia Muciniphila and Reduces Diabetes Incidence in the NOD Mouse. Diabetologia 2012, 55, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Caballero, S.; Kim, S.; Carter, R.A.; Leiner, I.M.; Sušac, B.; Miller, L.; Kim, G.J.; Ling, L.; Pamer, E.G. Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enterococcus Faecium. Cell Host Microbe 2017, 21, 592–602.e4. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Cho, I.H.; Jeong, B.C.; Lee, S.H. Strategies to Minimize Antibiotic Resistance. Int. J. Environ. Res. Public Health 2013, 10, 4274–4305. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.; Moser, H.E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef]
- Caputo, A.; Dubourg, G.; Croce, O.; Gupta, S.; Robert, C.; Papazian, L.; Rolain, J.M.; Raoult, D. Whole-Genome Assembly of Akkermansia Muciniphila Sequenced Directly from Human Stool. Biol. Direct 2015, 10, 5. [Google Scholar] [CrossRef]
- Geerlings, S.; Kostopoulos, I.; de Vos, W.; Belzer, C. Akkermansia Muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef]
- Dellacecca, E.R.; Cosgrove, C.; Mukhatayev, Z.; Akhtar, S.; Engelhard, V.H.; Rademaker, A.W.; Knight, K.L.; Le Poole, I.C. Antibiotics Drive Microbial Imbalance and Vitiligo Development in Mice. J. Investig. Dermatol. 2020, 140, 676–687.e6. [Google Scholar] [CrossRef]
- Bryan, L.E.; Kowand, S.K.; Van den Elzen, H.M. Mechanism of Aminoglycoside Antibiotic Resistance in Anaerobic Bacteria: Clostridium Perfringens and Bacteroides Fragilis. Antimicrob. Agents Chemother. 1979, 15, 7–13. [Google Scholar] [CrossRef]
- Willis, A.D. Rarefaction, Alpha Diversity, and Statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef]
- Rashid, M.U.; Weintraub, A.; Nord, C.E. Effect of New Antimicrobial Agents on the Ecological Balance of Human Microflora. Anaerobe 2012, 18, 249–253. [Google Scholar] [CrossRef]
- Mattila, J.; Mannisto, P.T.; Mantyla, R.; Nykänen, S.; Lamminsivu, U. Comparative Pharmacokinetics of Metronidazole and Tinidazole as Influenced by Administration Route. Antimicrob. Agents Chemother. 1983, 23, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Löfmark, S.; Edlund, C.; Nord, C.E. Metronidazole Is Still the Drug of Choice for Treatment of Anaerobic Infections. Clin. Infect. Dis. 2010, 50 (Suppl. 1), S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed]
- Alsharhan, H.; Ficicioglu, C. Disorders of Phenylalanine and Tyrosine Metabolism. Transl. Sci. Rare Dis. 2020, 5, 3–58. [Google Scholar] [CrossRef]
- Matthews, D.E. An Overview of Phenylalanine and Tyrosine Kinetics in Humans. J. Nutr. 2007, 137 (Suppl. 1), 1549S–1555S. [Google Scholar] [CrossRef]
- Kopple, J.D. Phenylalanine and Tyrosine Metabolism in Chronic Kidney Failure. J. Nutr. 2007, 137, 3–7. [Google Scholar] [CrossRef]
- Kleerebezem, M.; Vaughan, E.E. Probiotic and Gut Lactobacilli and Bifidobacteria: Molecular Approaches to Study Diversity and Activity. Annu. Rev. Microbiol. 2009, 63, 269–290. [Google Scholar] [CrossRef]
- Shanahan, F. The Colonic Microbiota in Health and Disease. Curr. Opin. Gastroenterol. 2013, 29, 49–54. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional Specialization within the Intestinal Immune System. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef]
- McAleer, J.P.; Nguyen, N.L.H.; Chen, K.; Kumar, P.; Ricks, D.M.; Binnie, M.; Armentrout, R.A.; Pociask, D.A.; Hein, A.; Yu, A.; et al. Pulmonary Th17 Antifungal Immunity Is Regulated by the Gut Microbiome. J. Immunol. 2016, 197, 97–107. [Google Scholar] [CrossRef]
- Bradley, C.P.; Teng, F.; Felix, K.M.; Sano, T.; Naskar, D.; Block, K.E.; Huang, H.; Knox, K.S.; Littman, D.R.; Wu, H.J.J. Segmented Filamentous Bacteria Provoke Lung Autoimmunity by Inducing Gut-Lung Axis Th17 Cells Expressing Dual TCRs. Cell Host Microbe 2017, 22, 697–704.e4. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The Immune Response to Prevotella Bacteria in Chronic Inflammatory Disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 Cells in Autoimmune Diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rathore, J.S.; Wang, Y. Protective Role of Th17 Cells in Pulmonary Infection. Vaccine 2016, 34, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, D.H.; Erofeev, A.; Sandvik, A.; Grcic, V.; Jahnsen, F.L.; Gaustad, P.; McCoy, K.D.; Macpherson, A.J.; Meza-Zepeda, L.A.; Johansen, F.E. Depletion of Murine Intestinal Microbiota: Effects on Gut Mucosa and Epithelial Gene Expression. PLoS ONE 2011, 6, e17996. [Google Scholar] [CrossRef]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Ditrio, L.E.; Burroughs, A.R.; Foureau, D.M.; Haque-Begum, S.; Kasper, L.H. Role of Gut Commensal Microflora in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2009, 183, 6041–6050. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Correspondence QIIME Allows Analysis of High- Throughput Community Sequencing Data Intensity Normalization Improves Color Calling in SOLiD Sequencing. Nat. Publ. Gr. 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Kim, W.K.; Jang, Y.J.; Han, D.H.; Jeon, K.; Lee, C.; Han, H.S.; Ko, G.P. Lactobacillus Paracasei KBL382 Administration Attenuates Atopic Dermatitis by Modulating Immune Response and Gut Microbiota. Gut Microbes 2020, 12, 1–14. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. Targeted Profiling of Circulating and Hepatic Bile Acids in Human, Mouse, and Rat Using a UPLC-MRM-MS-Validated Method. J. Lipid Res. 2012, 53, 2231–2241. [Google Scholar] [CrossRef]
- Yoon, S.; Yu, J.; McDowell, A.; Kim, S.H.; You, H.J.; Ko, G.P. Bile Salt Hydrolase-Mediated Inhibitory Effect of Bacteroides Ovatus on Growth of Clostridium Difficile. J. Microbiol. 2017, 55, 892–899. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotics | Abbreviation | Class | Spectrum of Activity |
|---|---|---|---|
| Ampicillin | AMP | β-lactams | Gram-positives and -negatives |
| Vancomycin | VAN | Glycopeptide | Gram-positives |
| Neomycin | NEO | Aminoglycoside | Gram-negatives |
| Metronidazole | MET | Anaerobic DNA inhibitor | Anaerobes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.; Lee, G.; Yu, J.; Lee, K.; Lee, K.; Si, J.; You, H.J.; Ko, G. Distinct Changes in Microbiota-Mediated Intestinal Metabolites and Immune Responses Induced by Different Antibiotics. Antibiotics 2022, 11, 1762. https://doi.org/10.3390/antibiotics11121762
Yoon S, Lee G, Yu J, Lee K, Lee K, Si J, You HJ, Ko G. Distinct Changes in Microbiota-Mediated Intestinal Metabolites and Immune Responses Induced by Different Antibiotics. Antibiotics. 2022; 11(12):1762. https://doi.org/10.3390/antibiotics11121762
Chicago/Turabian StyleYoon, Sunghyun, Giljae Lee, Junsun Yu, Kiuk Lee, Kyeongju Lee, Jiyeon Si, Hyun Ju You, and GwangPyo Ko. 2022. "Distinct Changes in Microbiota-Mediated Intestinal Metabolites and Immune Responses Induced by Different Antibiotics" Antibiotics 11, no. 12: 1762. https://doi.org/10.3390/antibiotics11121762
APA StyleYoon, S., Lee, G., Yu, J., Lee, K., Lee, K., Si, J., You, H. J., & Ko, G. (2022). Distinct Changes in Microbiota-Mediated Intestinal Metabolites and Immune Responses Induced by Different Antibiotics. Antibiotics, 11(12), 1762. https://doi.org/10.3390/antibiotics11121762