
New Insights into the Mechanism of Antibacterial Action of Synthetic Peptide Mo-CBP3-PepI against Klebsiella pneumoniae

 , ,
, , 
Abstract
1. Introduction
2. Material and Methods
2.1. Biological Material
2.2. Peptide Synthesis
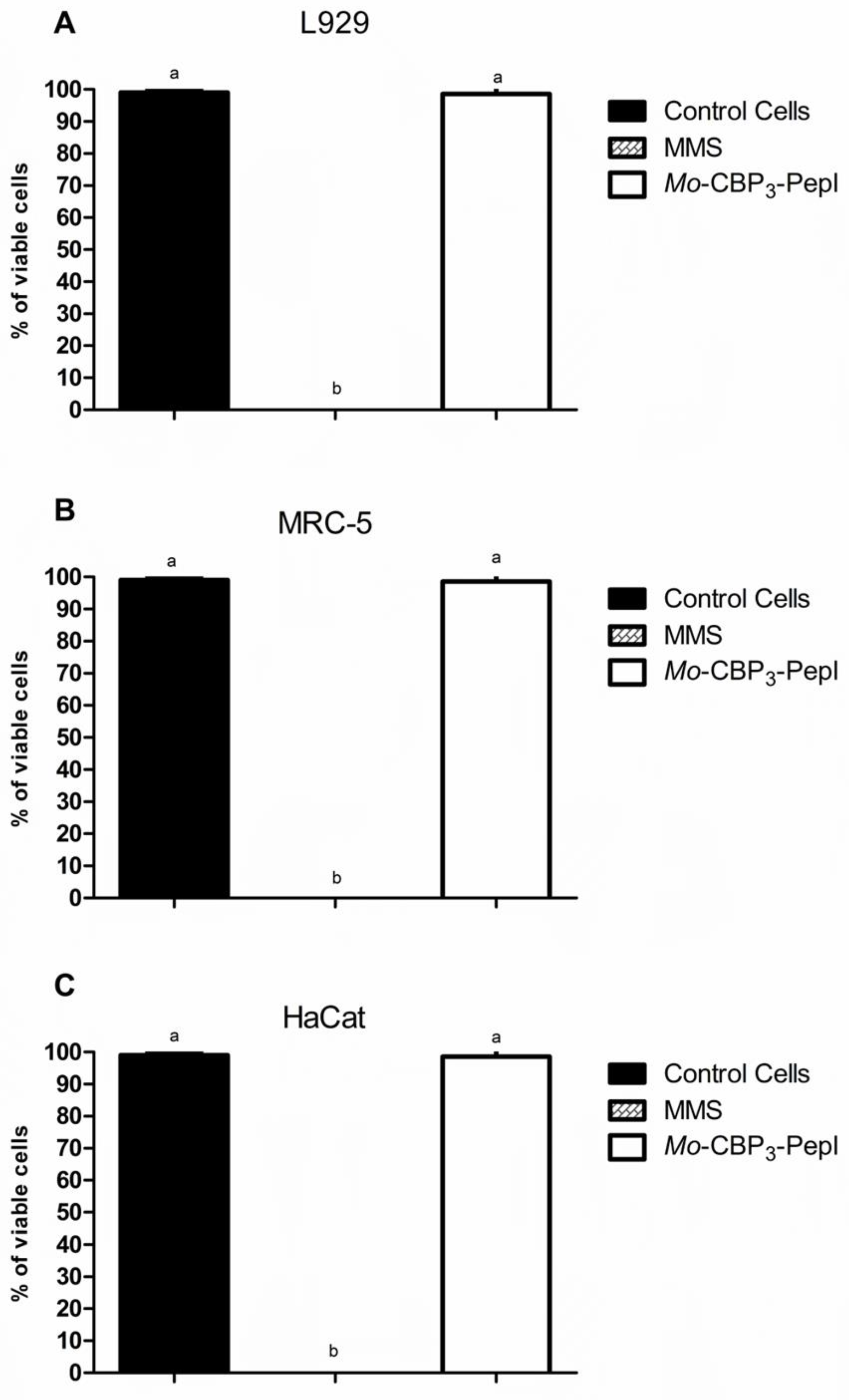
2.3. Cell Viability by MTT Assay
2.4. Antibiofilm Assay
2.5. Mechanism of Action Evaluation by Fluorescence Microscopy
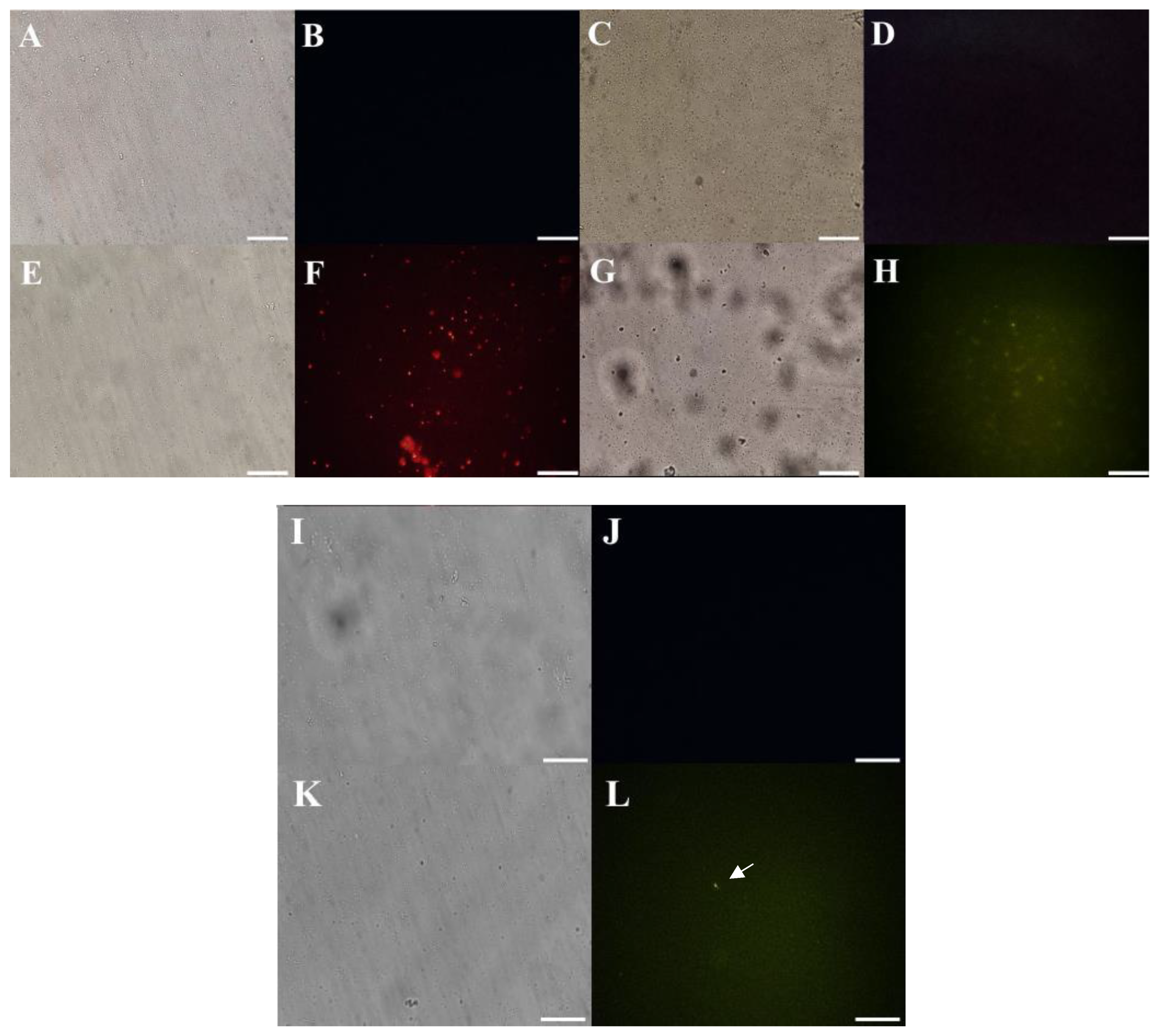
2.5.1. Cell Membrane Integrity by Propidium Iodide (PI) and FITC-Dextran Uptake
2.5.2. Detection of Peptide-Induced Overproduction of Reactive Oxygen Species (ROS)
2.5.3. Scanning Electronic Microscopy (SEM) Analysis
2.6. Protein Extraction and Gel-Free Proteomic Analysis
2.7. Protein Identification
2.8. Cytotoxicity Assay
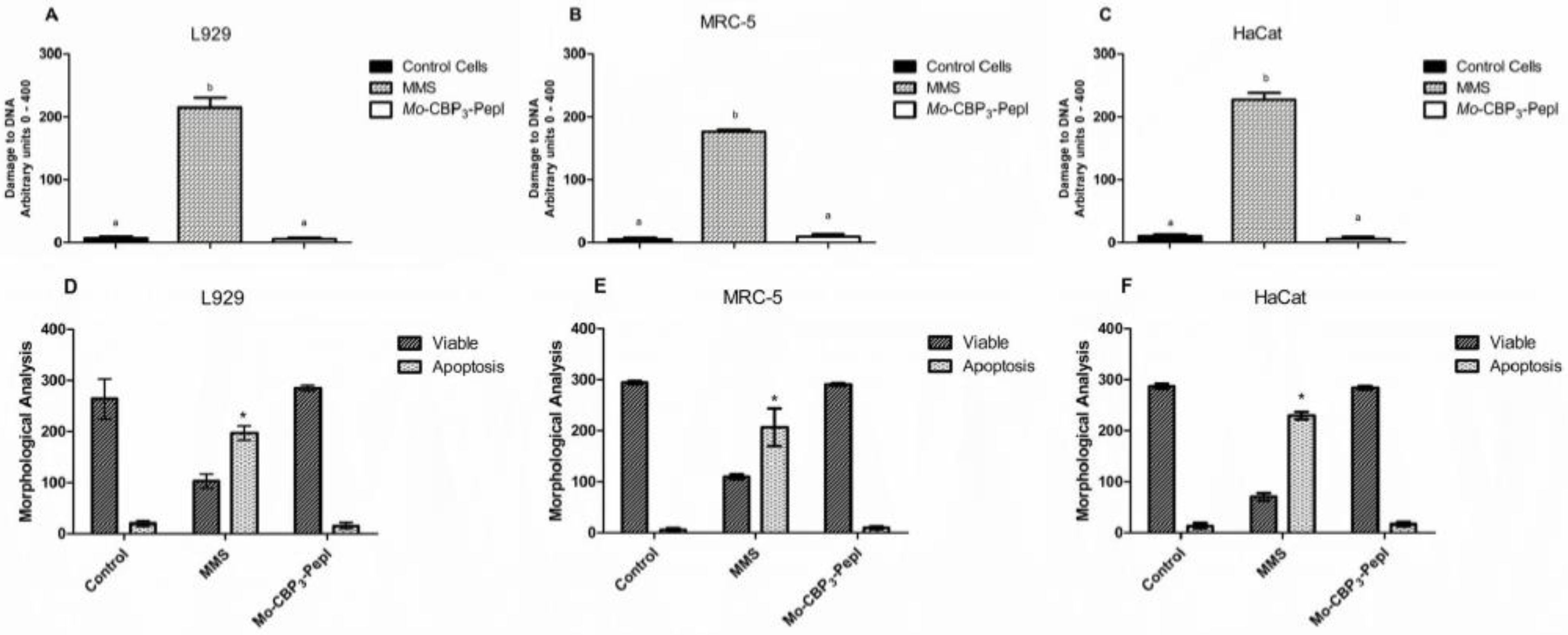
2.9. Comet Assay
- Class 0: undamaged, without a tail;
- Class 1: with a tail shorter than the diameter of the head nucleus;
- Class 2: with tail length 1–2× the diameter of the head;
- Class 3: with a tail longer than 2× the diameter of the head;
- Class 4: comets with no heads.
2.10. Morphological Analysis of Apoptotic Cells
3. Results and Discussion
3.1. Cell Viability and Antibiofilm Activity of Mo-CBP3-PepI against K. pneumoniae
3.2. Toxicity of Mo-CBP3-PepI to Human Cells
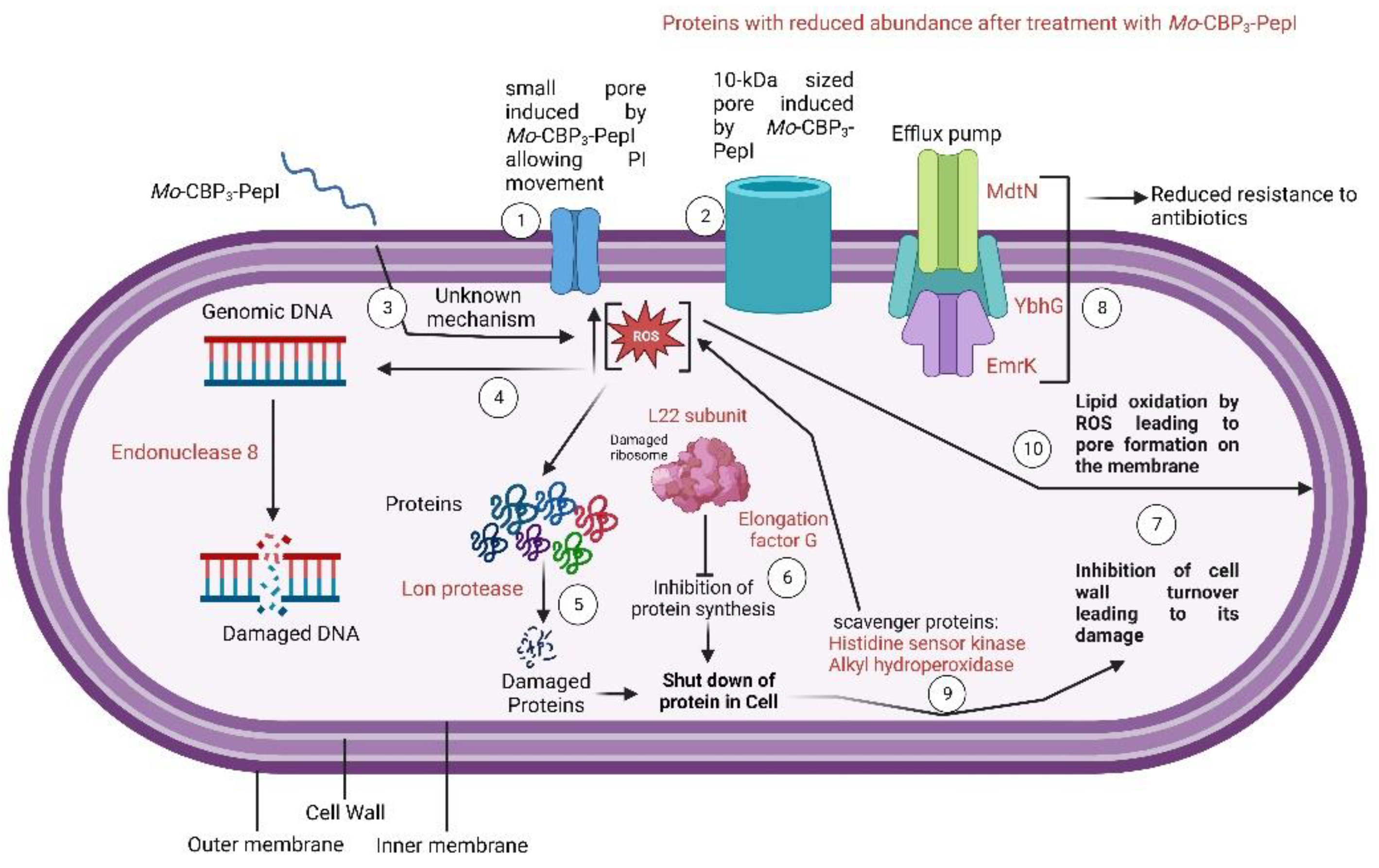
3.3. Mechanism of Action of Mo-CBP3-PepI against K. pneumoniae
3.3.1. Membrane Pore Formation and ROS Overproduction
3.3.2. Scanning Electron Microscopy (SEM)
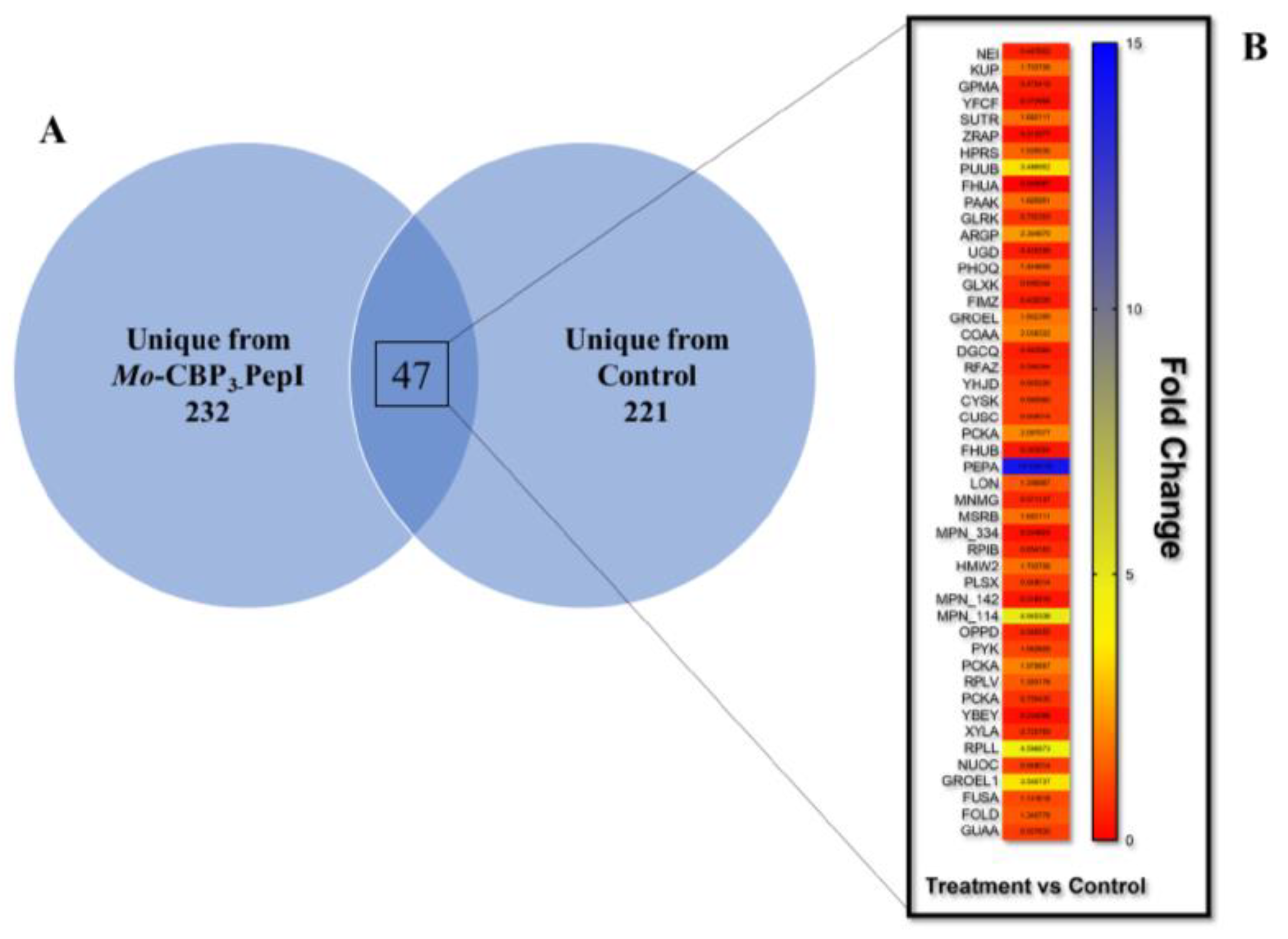
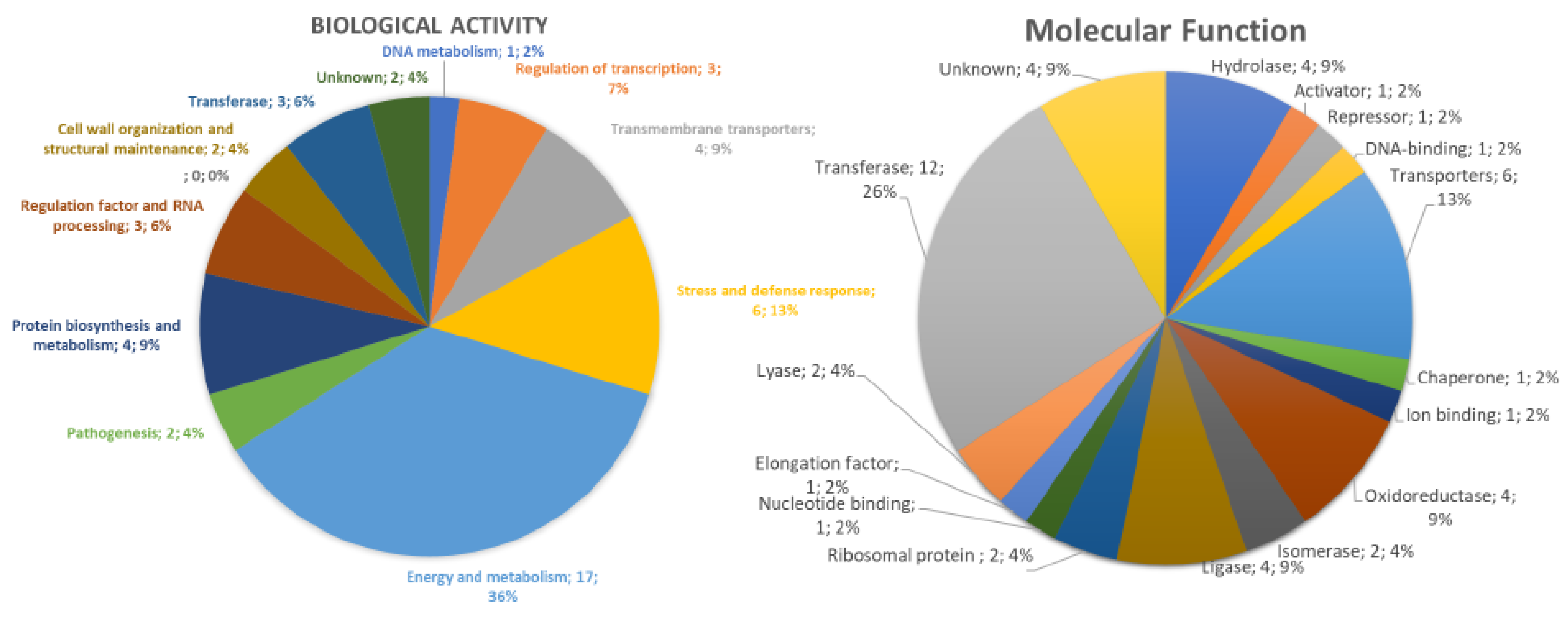
3.4. Proteomic Profile of K. pneumoniae Cells Treated with Mo-CBP3-PepI
3.4.1. Overview
3.4.2. DNA Metabolism-Related Proteins
3.4.3. Stress and Defense Response Related Proteins
3.4.4. Protein Biosynthesis and Metabolism Related Proteins
3.4.5. Regulation Factor and RNA Processing Related Proteins
3.4.6. Cell Wall Organization and Structure Maintenance Related Proteins
3.4.7. Transferase-Related Proteins
3.4.8. Cell Redox Homeostasis-Related Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- About Antibiotic Resistance|CDC. Available online: https://www.cdc.gov/drugresistance/about.html (accessed on 11 November 2022).
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Klebsiella Pneumoniae in Healthcare Settings|HAI|CDC. Available online: https://www.cdc.gov/hai/organisms/klebsiella/klebsiella.html (accessed on 11 November 2022).
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Livermore, D.M.; Macgowan, A.P.; Wale, M.C.J. Surveillance of Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 1998; Volume 317, ISBN 9789294983879. [Google Scholar]
- Tzouvelekis, L.S.; Markogiannakis, A.; Piperaki, E.; Souli, M.; Daikos, G.L. Treating Infections Caused by Carbapenemase-Producing Enterobacteriaceae. Clin. Microbiol. Infect. 2014, 20, 862–872. [Google Scholar] [CrossRef]
- Zowawi, H.M.; Forde, B.M.; Alfaresi, M.; Alzarouni, A.; Farahat, Y.; Chong, T.M.; Yin, W.F.; Chan, K.G.; Li, J.; Schembri, M.A.; et al. Stepwise Evolution of Pandrug-Resistance in Klebsiella Pneumoniae. Sci. Rep. 2015, 5, 15082. [Google Scholar] [CrossRef]
- McNeil, J.C.; Vallejo, J.G.; Hultén, K.G.; Kaplan, S.L. Osteoarticular Infections Following Open or Penetrating Trauma in Children in the Post-Community-Acquired Methicillin-resistant Staphylococcus aureus Era: The Impact of Enterobacter cloacae. Pediatr. Infect. Dis. J. 2018, 37, 1204–1210. [Google Scholar] [CrossRef]
- Corey, G.R.; Arhin, F.F.; Wikler, M.A.; Sahm, D.F.; Kreiswirth, B.N.; Mediavilla, J.R.; Good, S.; Fiset, C.; Jiang, H.; Moeck, G.; et al. Pooled Analysis of Single-Dose Oritavancin in the Treatment of Acute Bacterial Skin and Skin-Structure Infections Caused by Gram-Positive Pathogens, Including a Large Patient Subset with Methicillin-Resistant Staphylococcus Aureus. Int. J. Antimicrob. Agents 2016, 48, 528–534. [Google Scholar] [CrossRef]
- Rubinstein, E.; Kollef, M.H.; Nathwani, D. Pneumonia Caused by Methicillin-Resistant Staphylococcus Aureus. Clin. Infect. Dis. 2008, 46, S378–S385. [Google Scholar] [CrossRef]
- Lima, A.M.; Azevedo, M.I.G.; Sousa, L.M.; Oliveira, N.S.; Andrade, C.R.; Freitas, C.D.T.; Souza, P.F.N. Plant Antimicrobial Peptides: An Overview about Classification, Toxicity and Clinical Applications. Int. J. Biol. Macromol. 2022, 214, 10–21. [Google Scholar] [CrossRef]
- Souza, P.F.N.; Marques, L.S.M.; Oliveira, J.T.A.; Lima, P.G.; Dias, L.P.; Neto, N.A.S.; Lopes, F.E.S.; Sousa, J.S.; Silva, A.F.B.; Caneiro, R.F.; et al. Synthetic Antimicrobial Peptides: From Choice of the Best Sequences to Action Mechanisms. Biochimie 2020, 175, 132–145. [Google Scholar] [CrossRef]
- Oliveira, J.T.A.; Souza, P.F.N.; Vasconcelos, I.M.; Dias, L.P.; Martins, T.F.; Van Tilburg, M.F.; Guedes, M.I.F.; Sousa, D.O.B. Mo-CBP3-PepI, Mo-CBP3-PepII, and Mo-CBP3-PepIII Are Synthetic Antimicrobial Peptides Active against Human Pathogens by Stimulating ROS Generation and Increasing Plasma Membrane Permeability. Biochimie 2019, 157, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Lima, P.G.; Souza, P.F.N.; Freitas, C.D.T.; Oliveira, J.T.A.; Dias, L.P.; Neto, J.X.S.; Vasconcelos, I.M.; Lopes, J.L.S.; Sousa, D.O.B. Anticandidal Activity of Synthetic Peptides: Mechanism of Action Revealed by Scanning Electron and Fluorescence Microscopies and Synergism Effect with Nystatin. J. Pept. Sci. 2020, 26, e3249. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.F.N.; Lima, P.G.; Freitas, C.D.T.; Sousa, D.O.B.; Neto, N.A.S.; Dias, L.P.; Vasconcelos, I.M.; Freitas, L.B.N.; Silva, R.G.G.; Sousa, J.S.; et al. Antidermatophytic Activity of Synthetic Peptides: Action Mechanisms and Clinical Application as Adjuvants to Enhance the Activity and Decrease the Toxicity of Griseofulvin. Mycoses 2020, 63, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Neto, N.A.S.; Oliveira, J.T.A.; Aguiar, T.K.B.; Bezerra, L.P.; Branco, L.A.C.; Mesquita, F.P.; Freitas, C.D.T.; Souza, P.F.N. Synergistic Antibiofilm Activity between Synthetic Peptides and Ciprofloxacin against Staphylococcus Aureus. Pathogens 2022, 11, 995. [Google Scholar] [CrossRef] [PubMed]
- Lima, P.G.; Freitas, C.D.T.; Oliveira, J.T.A.; Neto, N.A.S.; Amaral, J.L.; Silva, A.F.B.; Sousa, J.S.; Franco, O.L.; Souza, P.F.N. Synthetic Antimicrobial Peptides Control Penicillium Digitatum Infection in Orange Fruits. Food Res. Int. 2021, 147, 110582. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, M.; Bondaryk, M.; Swoboda-Kopec, E.; Siennicka, K.; Sygitowicz, G.; Kurzatkowski, W. Candida Albicans Morphologies Revealed by Scanning Electron Microscopy Analysis. Braz. J. Microbiol. 2013, 44, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Fleeman, R.M.; Macias, L.A.; Brodbelt, J.S.; Davies, B.W. Defining Principles That Influence Antimicrobial Peptide Activity against Capsulated Klebsiella Pneumoniae. Proc. Natl. Acad. Sci. USA 2020, 117, 27620–27626. [Google Scholar] [CrossRef]
- Tincho, M.B.; Morris, T.; Meyer, M.; Pretorius, A. Antibacterial Activity of Rationally Designed Antimicrobial Peptides. Int. J. Microbiol. 2020, 2020, 2131535. [Google Scholar] [CrossRef]
- Jindal, H.M.; Le, C.F.; Yusof, M.Y.M.; Velayuthan, R.D.; Lee, V.S.; Zain, S.M.; Isa, D.M.; Sekaran, S.D. Antimicrobial Activity of Novel Synthetic Peptides Derived from Indolicidin and Ranalexin against Streptococcus Pneumoniae. PLoS ONE 2015, 10, e0128532. [Google Scholar] [CrossRef]
- Parra, A.L.C.; Freitas, C.D.T.; Souza, P.F.N.; von Aderkas, P.; Borchers, C.H.; Beattie, G.A.; Silva, F.D.A.; Thornburg, R.W. Ornamental Tobacco Floral Nectar Is a Rich Source of Antimicrobial Peptides. Plant Sci. 2022, 324, 111427. [Google Scholar] [CrossRef]
- Aguiar, T.K.B.; Neto, N.A.S.; Freitas, C.D.T.; Silva, A.F.B.; Bezerra, L.P.; Malveira, E.A.; Branco, L.A.C.; Mesquita, F.P.; Goldman, G.H.; Alencar, L.M.R.; et al. Antifungal Potential of Synthetic Peptides against Cryptococcus Neoformans: Mechanism of Action Studies Reveal Synthetic Peptides Induce Membrane–Pore Formation, DNA Degradation, and Apoptosis. Pharmaceutics 2022, 14, 1678. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Dang, W.; Xie, J.; Zhu, R.; Sun, M.; Jia, F.; Zhao, Y.; An, X.; Qiu, S.; Li, X.; et al. Antimicrobial Peptide Protonectin Disturbs the Membrane Integrity and Induces ROS Production in Yeast Cells. Biochim. Biophys. Acta-Biomembr. 2015, 1848, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Hung, W.C.; Chen, F.Y.; Huang, H.W. Mechanism and Kinetics of Pore Formation in Membranes by Water-Soluble Amphipathic Peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 5087–5092. [Google Scholar] [CrossRef]
- Lima, P.G.; Oliveira, J.T.A.; Amaral, J.L.; Freitas, C.D.T.; Souza, P.F.N. Synthetic Antimicrobial Peptides: Characteristics, Design, and Potential as Alternative Molecules to Overcome Microbial Resistance. Life Sci. 2021, 278, 119647. [Google Scholar] [CrossRef] [PubMed]
- Hristova, K.; Wimley, W.C. A Look at Arginine in Membranes. J. Membr. Biol. 2011, 239, 49. [Google Scholar] [CrossRef]
- Campelo, F.; McMahon, H.T.; Kozlov, M.M. The Hydrophobic Insertion Mechanism of Membrane Curvature Generation by Proteins. Biophys. J. 2008, 95, 2325–2339. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-Helical Cationic Antimicrobial Peptides: Relationships of Structure and Function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the Binding, Insertion and Destabilization of Phospholipid Bilayer Membranes by Alpha-Helical Antimicrobial and Cell Non-Selective Membrane-Lytic Peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Jean-François, F.; Elezgaray, J.; Berson, P.; Vacher, P.; Dufourc, E.J. Pore Formation Induced by an Antimicrobial Peptide: Electrostatic Effects. Biophys. J. 2008, 95, 5748–5756. [Google Scholar] [CrossRef]
- Rowe-Magnus, D.A.; Kao, A.Y.; Prieto, A.C.; Pu, M.; Kao, C. Cathelicidin Peptides Restrict Bacterial Growth via Membrane Perturbation and Induction of Reactive Oxygen Species. mBio 2019, 10, e02021-19. [Google Scholar] [CrossRef]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Krämer, U.; et al. Small Cationic Antimicrobial Peptides Delocalize Peripheral Membrane Proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Tsakou, F.; Jersie-Christensen, R.; Jenssen, H.; Mojsoska, B. The Role of Proteomics in Bacterial Response to Antibiotics. Pharmaceuticals 2020, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Moyer, T.B.; Purvis, A.L.; Wommack, A.J.; Hicks, L.M. Proteomic Response of Escherichia Coli to a Membrane Lytic and Iron Chelating Truncated Amaranthus Tricolor Defensin. BMC Microbiol. 2021, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Maaß, S.; Bartel, J.; Mücke, P.A.; Schlüter, R.; Sura, T.; Zaschke-Kriesche, J.; Smits, S.H.J.; Becher, D. Proteomic Adaptation of Clostridioides Difficile to Treatment with the Antimicrobial Peptide Nisin. Cells 2021, 10, 372. [Google Scholar] [CrossRef]
- Senges, C.H.R.; Stepanek, J.J.; Wenzel, M.; Raatschen, N.; Ay, Ü.; Märtens, Y.; Prochnow, P.; Hernández, M.V.; Yayci, A.; Schubert, B.; et al. Comparison of Proteomic Responses as Global Approach to Antibiotic Mechanism of Action Elucidation. Antimicrob. Agents Chemother. 2021, 65, e01373-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, Y.; Zhang, H.; Jin, J.; Zhang, H. Quantitative Proteomic Analysis Reveals the Influence of Plantaricin BM-1 on Metabolic Pathways and Peptidoglycan Synthesis in Escherichia Coli K12. PLoS ONE 2020, 15, e0231975. [Google Scholar] [CrossRef] [PubMed]
- Noronha Souza, P.F.; Abreu Oliveira, J.T.; Vasconcelos, I.M.; Magalhães, V.G.; Albuquerque Silva, F.D.; Guedes Silva, R.G.; Oliveira, K.S.; Franco, O.L.; Gomes Silveira, J.A.; Leite Carvalho, F.E. H2O2Accumulation, Host Cell Death and Differential Levels of Proteins Related to Photosynthesis, Redox Homeostasis, and Required for Viral Replication Explain the Resistance of EMS-Mutagenized Cowpea to Cowpea Severe Mosaic Virus. J. Plant Physiol. 2020, 245, 153110. [Google Scholar] [CrossRef]
- Burgess, S.; Jaruga, P.; Dodson, M.L.; Dizdaroglu, M.; Stephen Lloyd, R. Determination of Active Site Residues in Escherichia Coli Endonuclease VIII. J. Biol. Chem. 2002, 277, 2938–2944. [Google Scholar] [CrossRef]
- Jiang, D.; Hatahet, Z.; Blaisdell, J.O.; Melamede, R.J.; Wallace, S.S. Escherichia Coli Endonuclease VIII: Cloning, Sequencing, and Overexpression of the Nei Structural Gene and Characterization of Nei and Nei Nth Mutants. J. Bacteriol. 1997, 179, 3773–3782. [Google Scholar] [CrossRef]
- Oshima, T.; Aiba, H.; Baba, T.; Fujita, K.; Hayashi, K.; Honjo, A.; Ikemoto, K.; Inada, T.; Itoh, T.; Kajihara, M.; et al. A 718-Kb DNA Sequence of the Escherichia Coli K-12 Genome Corresponding to the 12.7–28.0 Min Region on the Linkage Map. DNA Res. 1996, 3, 137–155. [Google Scholar] [CrossRef]
- Verhoeven, E.E.A.; Wyman, C.; Moolenaar, G.F.; Goosen, N. The Presence of Two UvrB Subunits in the UvrAB Complex Ensures Damage Detection in Both DNA Strands. EMBO J. 2002, 21, 4196–4205. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Sanchez, H.; Alonso, J.C.; Graumann, P.L. Visualization of DNA Double-Strand Break Repair in Live Bacteria Reveals Dynamic Recruitment of Bacillus Subtilis RecF, RecO and RecN Proteins to Distinct Sites on the Nucleoids. Mol. Microbiol. 2004, 52, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Delaye, L.; Becerra, A.; Orgel, L.; Lazcano, A. Molecular Evolution of Peptide Methionine Sulfoxide Reductases (MsrA and MsrB): On the Early Development of a Mechanism That Protects against Oxidative Damage. J. Mol. Evol. 2007, 64, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; McNeill, M.; Myers, T.G.; Hohman, R.J.; Levine, R.L. Lon Protease Promotes Survival of Escherichia Coli During Anaerobic Glucose Starvation. Arch. Microbiol. 2008, 189, 181. [Google Scholar] [CrossRef]
- He, L.; Luo, D.; Yang, F.; Li, C.; Zhang, X.; Deng, H.; Zhang, J.R. Multiple Domains of Bacterial and Human Lon Proteases Define Substrate Selectivity. Emerg. Microbes Infect. 2018, 7, 1–18. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Free Radical-Mediated Oxidation of Free Amino Acids and Amino Acid Residues in Proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef]
- Torres-Cabassa, A.; Gottesman, S.; Frederick, R.D.; Dolph, P.J.; Coplin, D.L. Control of Extracellular Polysaccharide Synthesis in Erwinia Stewartii and Escherichia Coli K-12: A Common Regulatory Function. J. Bacteriol. 1987, 169, 4525. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bree, A.C.; Liu, J.; Patrick, J.E.; Chien, P.; Kearns, D.B. Adaptor-Mediated Lon Proteolysis Restricts Bacillus Subtilis Hyperflagellation. Proc. Natl. Acad. Sci. USA 2015, 112, 250–255. [Google Scholar] [CrossRef]
- Bissonnette, S.A.; Rivera-Rivera, I.; Sauer, R.T.; Baker, T.A. The IbpA and IbpB Small Heat-Shock Proteins Are Substrates of the AAA+ Lon Protease. Mol. Microbiol. 2010, 75, 1539. [Google Scholar] [CrossRef]
- Shan, Y.; Gandt, A.B.; Rowe, S.E.; Deisinger, J.P.; Conlon, B.P.; Lewis, K. ATP-Dependent Persister Formation in Escherichia Coli. mBio 2017, 8, e02267-16. [Google Scholar] [CrossRef]
- Ricci, V.; Blair, J.M.A.; Piddock, L.J.V. RamA, Which Controls Expression of the MDR Efflux Pump AcrAB-TolC, Is Regulated by the Lon Protease. J. Antimicrob. Chemother. 2014, 69, 643. [Google Scholar] [CrossRef] [PubMed]
- Herbst, K.; Bujara, M.; Heroven, A.K.; Opitz, W.; Weichert, M.; Zimmermann, A.; Dersch, P. Intrinsic Thermal Sensing Controls Proteolysis of Yersinia Virulence Regulator RovA. PLoS Pathog. 2009, 5, 1000435. [Google Scholar] [CrossRef] [PubMed]
- EmrK—Probable Multidrug Resistance Protein EmrK—Escherichia Coli (Strain K12)|UniProtKB|UniProt. Available online: https://www.uniprot.org/uniprotkb/P52599/entry (accessed on 7 November 2022).
- YbhG—UPF0194 Membrane Protein YbhG—Escherichia Coli (Strain K12)|UniProtKB|UniProt. Available online: https://www.uniprot.org/uniprotkb/P75777/entry (accessed on 7 November 2022).
- Moore, S.D.; Sauer, R.T. Revisiting the Mechanism of Macrolide-Antibiotic Resistance Mediated by Ribosomal Protein L22. Proc. Natl. Acad. Sci. USA 2008, 105, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Davydova, N.; Streltsov, V.; Wilce, M.; Liljas, A.; Garber, M. L22 Ribosomal Protein and Effect of Its Mutation on Ribosome Resistance to Erythromycin. J. Mol. Biol. 2002, 322, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.A.; Haddad, B.G.; Weis, A.J.; Blackwood, C.S.; Shelton, C.D.; Wuerth, M.E.; Walter, J.D.; Spiegel, P.C. Ribosomal Protein L7/L12 Is Required for GTPase Translation Factors EF-G, RF3 and IF2 to Bind in Their GTP State to 70S Ribosomes. FEBS J. 2017, 284, 1631. [Google Scholar] [CrossRef]
- Starosta, A.L.; Lassak, J.; Jung, K.; Wilson, D.N. The Bacterial Translation Stress Response. FEMS Microbiol. Rev. 2014, 38, 1172. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cui, X.; Beausang, J.F.; Zhang, H.; Farrell, I.; Cooperman, B.S.; Goldman, Y.E. Elongation Factor G Initiates Translocation through a Power Stroke. Proc. Natl. Acad. Sci. USA 2016, 113, 7515–7520. [Google Scholar] [CrossRef]
- Divakaruni, A.V.; Baida, C.; White, C.L.; Gober, J.W. The Cell Shape Proteins MreB and MreC Control Cell Morphogenesis by Positioning Cell Wall Synthetic Complexes. Mol. Microbiol. 2007, 66, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Dik, D.A.; Marous, D.R.; Fisher, J.F.; Mobashery, S. Lytic Transglycosylases: Concinnity in Concision of the Bacterial Cell Wall. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 503–542. [Google Scholar] [CrossRef]
- Hung, W.C.; Jane, W.N.; Wong, H.C. Association of a D-Alanyl-D-Alanine Carboxypeptidase Gene with the Formation of Aberrantly Shaped Cells during the Induction of Viable but Nonculturable Vibrio Parahaemolyticus. Appl. Environ. Microbiol. 2013, 79, 7305–7312. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.A.; Levin, P.A. Bacterial Cell Wall Quality Control during Environmental Stress. mBio 2020, 11, e02456-20. [Google Scholar] [CrossRef] [PubMed]
- Szurmant, H.; Bu, L.; Brooks, C.L.; Hoch, J.A. An Essential Sensor Histidine Kinase Controlled by Transmembrane Helix Interactions with Its Auxiliary Proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 5891–5896. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Le-Khac, M.; Hendrickson, W.A. Crystal structure of a histidine kinase sensor domain with similarity to periplasmic binding proteins. Proteins 2009, 77, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Chung, C.H.; Ma, T.Y.; Wong, H.C. Roles of Alkyl Hydroperoxide Reductase Subunit C (AhpC) in Viable but Nonculturable Vibrio Parahaemolyticus. Appl. Environ. Microbiol. 2013, 79, 3734–3743. [Google Scholar] [CrossRef]
- Chen, L.; Xie, Q.W.; Nathan, C. Alkyl Hydroperoxide Reductase Subunit C (AhpC) Protects Bacterial and Human Cells against Reactive Nitrogen Intermediates. Mol. Cell 1998, 1, 795–805. [Google Scholar] [CrossRef]
- Yuan, F.; Yin, S.; Xu, Y.; Xiang, L.; Wang, H.; Li, Z.; Fan, K.; Pan, G. The Richness and Diversity of Catalases in Bacteria. Front. Microbiol. 2021, 12, 485. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a MIC50 of Mo-CBP3-PepI toward K. pneumoniae | ||||
|---|---|---|---|---|
| Treatments | Antibiofilm Potential | |||
| Cell Viability (%) | Inhibition of Biofilm Formation (%) | Degradation of Preformed Biofilm (%) | ||
| DMSO | 100 ± 0.005 | 0 | 0 | |
| Mo-CBP3-PepI | 47.54 ± 0.008 | 11.87 ± 0.001 | 0 | |
| Protein Name | ID (Uniprot) | Organism Reference | Cellular Compartment | Fold Change Mo-CBP3-PepI vs. DMSO |
|---|---|---|---|---|
| DNA metabolism | ||||
| Endonuclease 8 | P50465 | Escherichia coli (strain K12) | Cytoplasm | 0.287 |
| Regulation of transcription | ||||
| HTH-type transcriptional regulator SutR | P77626 | Escherichia coli (strain K12) | Cytoplasm | 2.566 |
| HTH-type transcriptional regulator ArgP | P0A8S1 | Escherichia coli (strain K12) | Cytoplasm | 2.364 |
| Fimbriae Z protein | P0AEL8 | Escherichia coli (strain K12) | Cytoplasm | 0.690 |
| Transmembrane transporters | ||||
| Low affinity potassium transport system protein kup | P63183 | Escherichia coli (strain K12) | Plasma membrane | 1.704 |
| Ferrichrome outer membrane transporter/phage receptor | P06971 | Escherichia coli (strain K12) | Plasma membrane | 1.063 |
| Cation efflux system protein CusC | P77211 | Escherichia coli (strain K12) | cell outer membrane | 0.920 |
| Iron (3+)-hydroxamate import system permease protein FhuB | P06972 | Escherichia coli (strain K12) | Plasma membrane | 0.948 |
| Stress and Defense Response | ||||
| Zinc resistance-associated protein | P0AAA9 | Escherichia coli (strain K12) | Periplasm space | 1.682 |
| Chaperonin GroEL | P0A6F5 | Escherichia coli (strain K12) | Cytoplasm | 1.444 |
| Undecaprenyl-diphosphatase | Q2KX31 | Bordetella avium (strain 197N) | Plasma membrane | 1.340 |
| Peptide methionine sulfoxide reductase MsrB | P75129 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Cytoplasm | 14.104 |
| Periplasmic trehalase | Q4UZ12 | Xanthomonas campestris pv. campestris (strain 8004) | Periplasmatic space | 0.937 |
| Lon protease | P78025 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Cytoplasm | 0.342 |
| Energy and Metabolism | ||||
| 2.3-bisphosphoglycerate-dependent phosphoglycerate mutase | P62707 | Escherichia coli (strain K12) | Cytoplasm | 0.519 |
| Gamma-glutamylputrescine oxidoreductase | P37906 | Escherichia coli (strain K12) | Cytoplasm | 0.985 |
| 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase | P17109 | Escherichia coli (strain K12) | Plasma membrane | 3.486 |
| UDP-glucose 6-dehydrogenase | P76373 | Escherichia coli (strain K12) | Cytoplasm | 0.902 |
| Glycerate 3-kinase | P77364 | Escherichia coli (strain K12) | Cytoplasm | 0.745 |
| Phenylacetate-coenzyme A ligase | P76085 | Escherichia coli (strain K12) | Cytoplasm | 1.666 |
| Pantothenate kinase | P0A6I3 | Escherichia coli (strain K12) | Cytoplasm | 0.548 |
| Pyruvate kinase | P78031 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Membrane | 3.139 |
| Phosphoenolpyruvate carboxykinase (ATP) | A8AQV7 | Escherichia coli (strain K12) | Cytoplasm | 0.990 |
| Xylose isomerase | B5ZQV6 | Rhizobium leguminosarum bv. trifolii (strain WSM2304) | Cytoplasm | 1.062 |
| Bifunctional protein FolD | Q88WM8 | Lactiplantibacillus plantarum (strain ATCC BAA-793/NCIMB 8826/WCFS1) | Periplasmic space | 0.726 |
| GMP synthase (glutamine-hydrolyzing) | Q6APU2 | Desulfotalea psychrophila (strain LSv54/DSM 12343) | Cytoplasm | 4.596 |
| Sensor histidine kinase GlrK | P52101 | Escherichia coli (strain K12) | Cell inner membrane | 0.752 |
| Sensor protein PhoQ | P23837 | Escherichia coli (strain K12) | Plasma membrane | 0.823 |
| Putative ABC transporter ATP-binding protein MPN_334 | P75444 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Plasma membrane | 1.336 |
| Oligopeptide transport ATP-binding protein OppD | P75552 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Plasma membrane | 0.948 |
| Pantothenate synthetase | B8I2Z3 | Ruminiclostridium cellulolyticum (strain ATCC 35319/DSM 5812/JCM 6584/H10) | Cytoplasm | 1.142 |
| Pathogenesis | ||||
| Lipopolysaccharide core biosynthesis protein RfaZ | P27241 | Escherichia coli (strain K12) | Cytoplasm | 2.038 |
| Inner membrane protein YhjD | P37642 | Escherichia coli (strain K12) | Plasma membrane | 0.362 |
| Protein Biosynthesis and Metabolism | ||||
| 50S ribosomal protein L22 | A5IYY1 | Mycoplasmopsis agalactiae | Large ribosomal subunit | 0.187 |
| 50S ribosomal protein L7/L12 | P0A466 | Aquifex aeolicus (strain VF5) | Large ribosomal subunit | 1.979 |
| Diaminopimelate decarboxylase | Q8K9C4 | Buchnera aphidicola subsp. Schizaphis graminum (strain Sg) | Cytoplasm | 3.546 |
| Cysteine synthase A | P0ABK5 | Escherichia coli (strain K12) | Cytoplasm | 0.596 |
| Regulation Factor and RNA Processing | ||||
| tRNA uridine 5-carboxymethylaminomethyl modification enzyme MnmG | P75221 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Cytoplasm | 0.487 |
| Elongation factor G | Q7NAV3 | Mycoplasma gallisepticum (strain R(low/passage 15/clone 2)) | Cytoplasm | 0.234 |
| Methylenetetrahydrofolate--tRNA-(uracil-5-)-methyltransferase TrmFO | A4WRQ2 | Cereibacter sphaeroides (strain ATCC 17025/ATH 2.4.3) | Cytoplasm | 0.948 |
| Cell wall organization and structural maintenance | ||||
| Cytadherence high molecular weight protein 2 | P75471 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Cytoplasm | 1.682 |
| Mgp-operon protein 3 | Q50341 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Plasma membrane | 0.654 |
| Transferase | ||||
| Glutathione S-transferase YfcF | P77544 | Escherichia coli (strain K12) | Cytoplasm | 0.273 |
| Phosphate acyltransferase | P75232 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Cytoplasm | 3.096 |
| Sensor histidine kinase HprS | P76339 | Escherichia coli (strain K12) | Plasma membrane | 0.213 |
| Unknown | ||||
| Probable cytosol aminopeptidase | P75206 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Cytoplasm | 2.097 |
| Putative acetyltransferase MPN_114 | P75448 | Mycoplasma pneumoniae (strain ATCC 29342/M129) | Unknown | 1.703 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, L.A.C.; Souza, P.F.N.; Neto, N.A.S.; Aguiar, T.K.B.; Silva, A.F.B.; Carneiro, R.F.; Nagano, C.S.; Mesquita, F.P.; Lima, L.B.; Freitas, C.D.T. New Insights into the Mechanism of Antibacterial Action of Synthetic Peptide Mo-CBP3-PepI against Klebsiella pneumoniae. Antibiotics 2022, 11, 1753. https://doi.org/10.3390/antibiotics11121753
Branco LAC, Souza PFN, Neto NAS, Aguiar TKB, Silva AFB, Carneiro RF, Nagano CS, Mesquita FP, Lima LB, Freitas CDT. New Insights into the Mechanism of Antibacterial Action of Synthetic Peptide Mo-CBP3-PepI against Klebsiella pneumoniae. Antibiotics. 2022; 11(12):1753. https://doi.org/10.3390/antibiotics11121753
Chicago/Turabian StyleBranco, Levi A. C., Pedro F. N. Souza, Nilton A. S. Neto, Tawanny K. B. Aguiar, Ayrles F. B. Silva, Rômulo F. Carneiro, Celso S. Nagano, Felipe P. Mesquita, Luina B. Lima, and Cleverson D. T. Freitas. 2022. "New Insights into the Mechanism of Antibacterial Action of Synthetic Peptide Mo-CBP3-PepI against Klebsiella pneumoniae" Antibiotics 11, no. 12: 1753. https://doi.org/10.3390/antibiotics11121753
APA StyleBranco, L. A. C., Souza, P. F. N., Neto, N. A. S., Aguiar, T. K. B., Silva, A. F. B., Carneiro, R. F., Nagano, C. S., Mesquita, F. P., Lima, L. B., & Freitas, C. D. T. (2022). New Insights into the Mechanism of Antibacterial Action of Synthetic Peptide Mo-CBP3-PepI against Klebsiella pneumoniae. Antibiotics, 11(12), 1753. https://doi.org/10.3390/antibiotics11121753