
Antimicrobial Resistance Genes Analysis of Publicly Available Staphylococcus aureus Genomes





Abstract
1. Introduction
2. Results
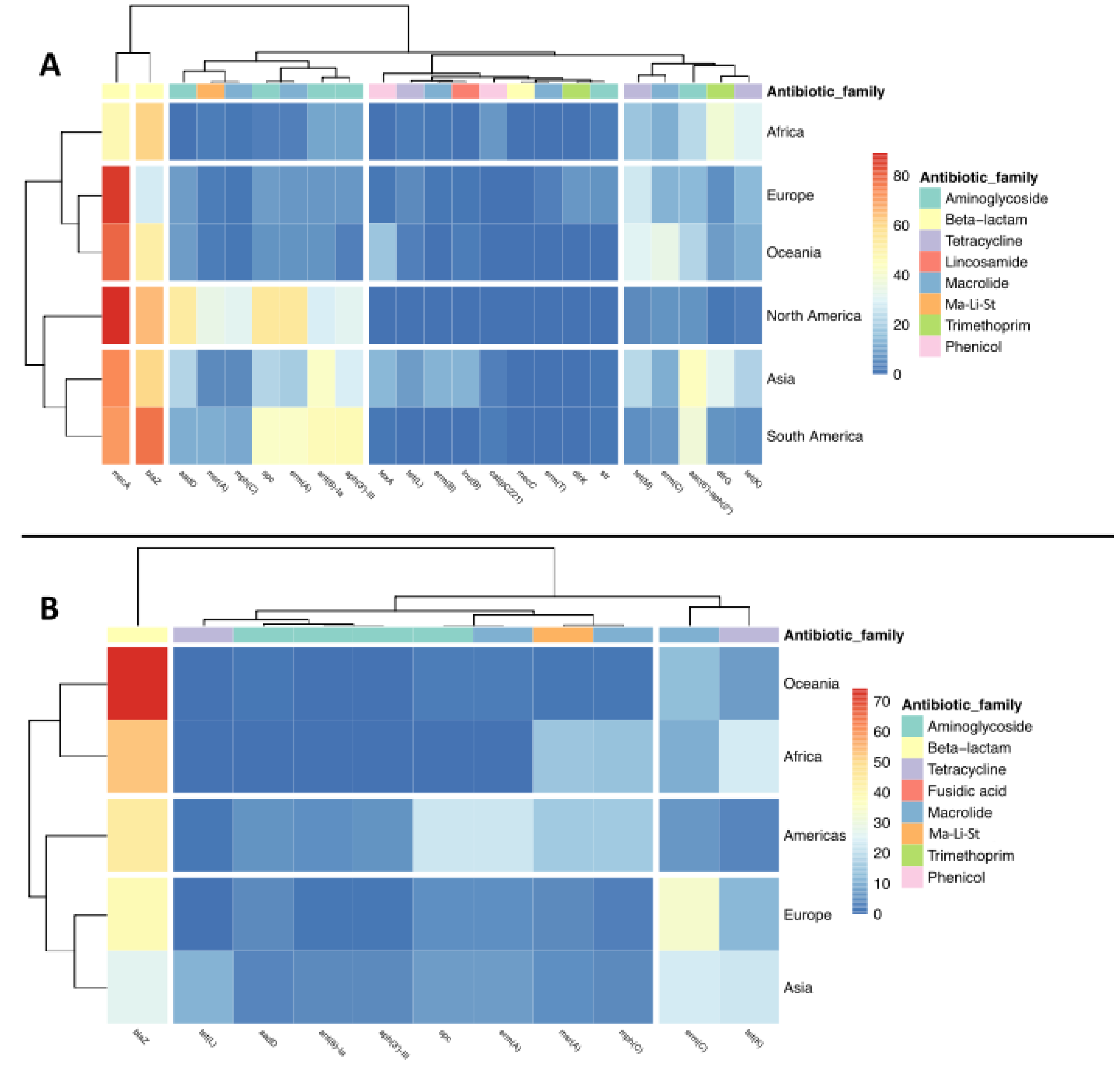
2.1. Geographical Distribution of CCs and ARGs
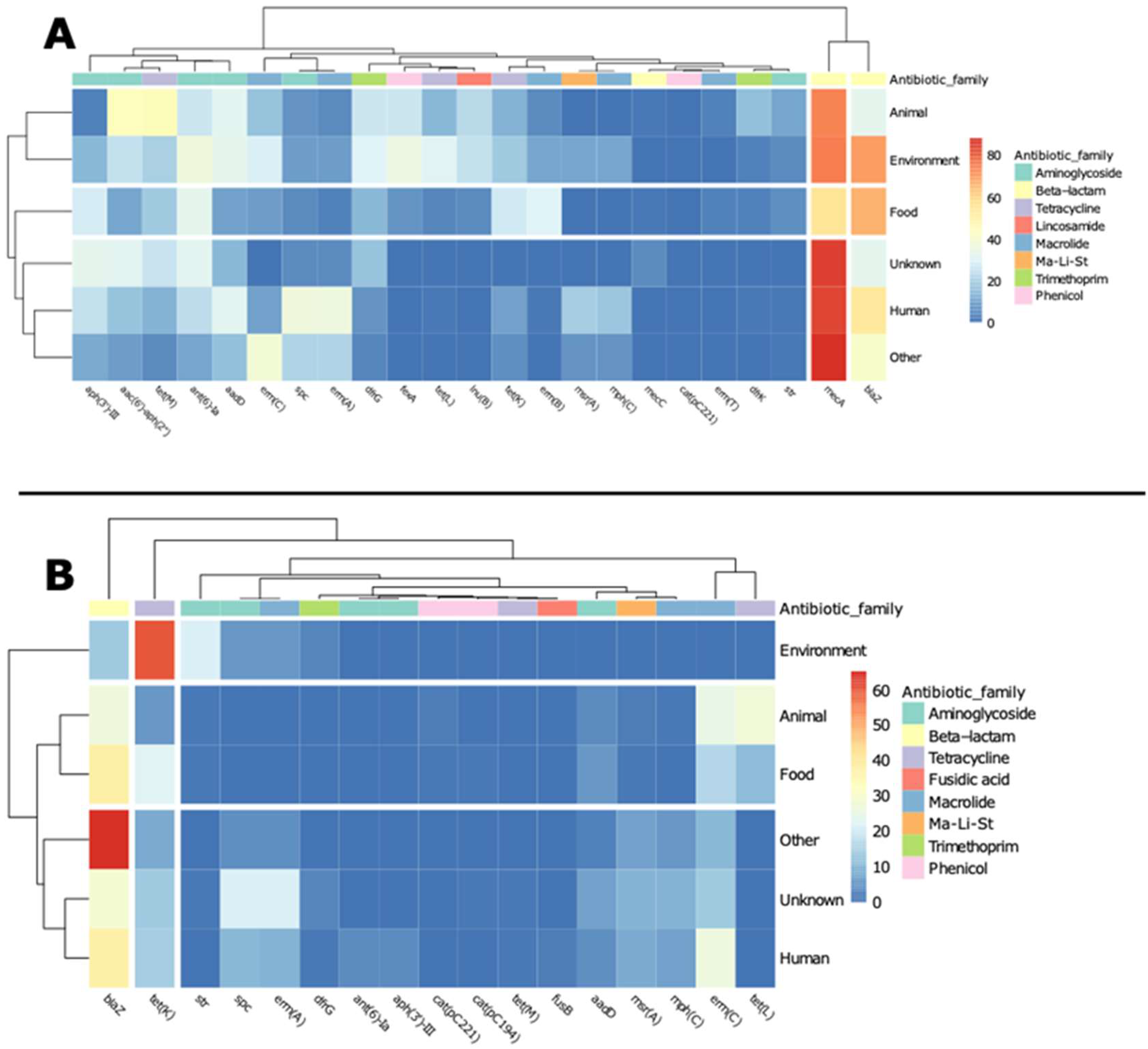
2.2. CCs and ARGs Distribution by Source of Isolation
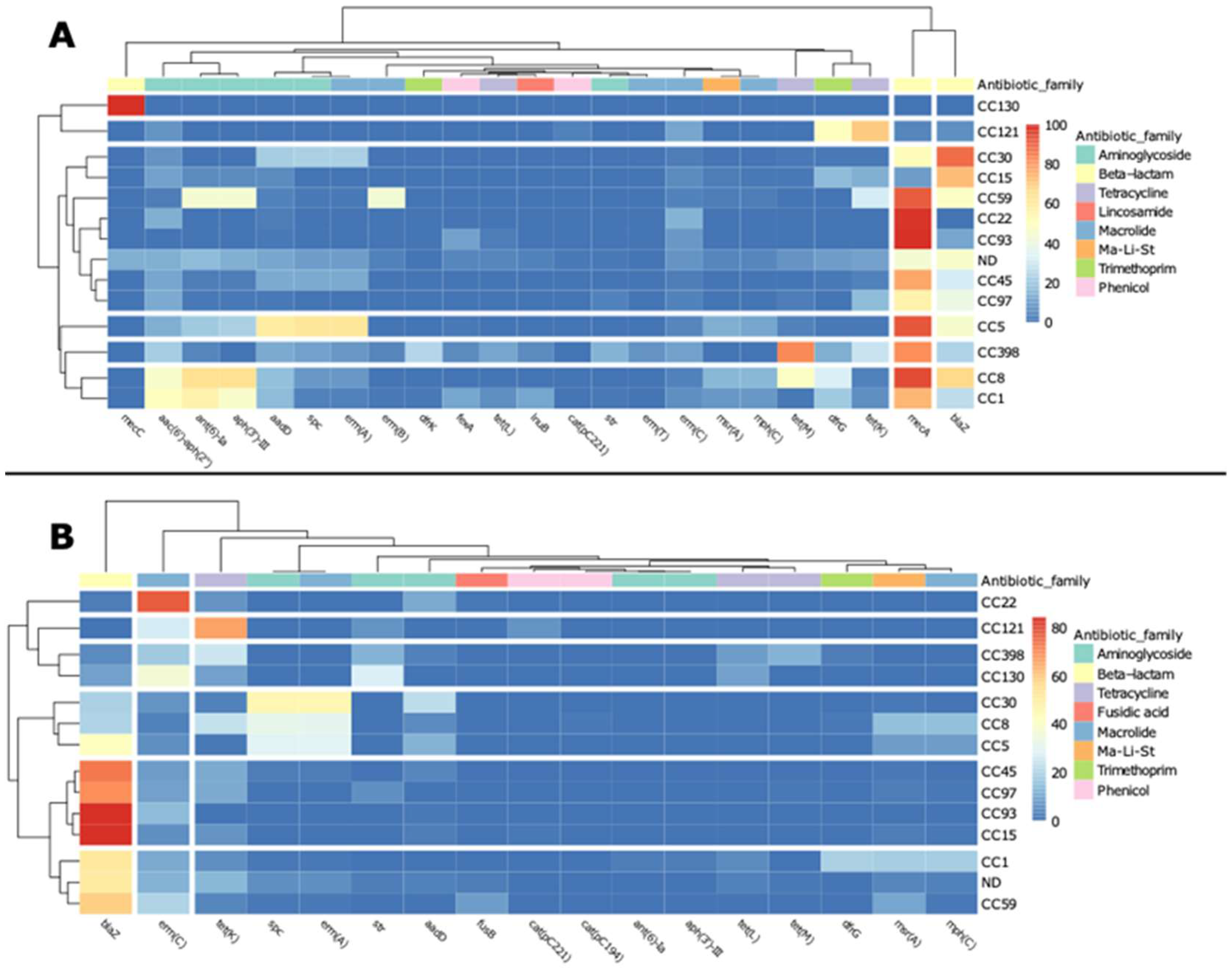
2.3. ARGs Distribution among S. aureus CCs
2.4. Temporal Changes in ARGs
3. Discussion
4. Materials and Methods
4.1. Download of Genomes and Associated Metadata
4.2. Analysis of Genomes
4.3. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Zahoor, S. Staphylococcus aureus enterotoxins: A review. J. Clin. Diagn. Res. 2007, 1, 188–197. [Google Scholar]
- Haag, A.F.; Fitzgerald, J.R.; Penadés, J.R. Staphylococcus aureus in Animals. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- CDC. 2019 AR Threats Report. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fdrugresistance%2Fbiggest_threats.html (accessed on 12 November 2021).
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. 2017. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 8 March 2022).
- WHO. Antimicrobial Resistance 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 12 November 2021).
- Harkins, C.P.; Pichon, B.; Doumith, M.; Parkhill, J.; Westh, H.; Tomasz, A.; de Lencastre, H.; Bentley, S.D.; Kearns, A.M.; Holden, M.T.G. Methicillin-resistant Staphylococcus aureus emerged long before the introduction of methicillin into clinical practice. Genome Biol. 2017, 18, 130. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Durand, G.; Javerliat, F.; Bes, M.; Veyrieras, J.-B.; Guigon, G.; Mugnier, N.; Schicklin, S.; Kaneko, G.; Santiago-Allexant, E.; Bouchiat, C.; et al. Routine Whole-Genome Sequencing for Outbreak Investigations of Staphylococcus aureus in a National Reference Center. Front. Microbiol. 2018, 9, 511. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Molina, A.; de Toro, M.; Alexa, E.A.; Álvarez-Ordóñez, A. 1.12—Applying Genomics to Track Antimicrobial Resistance in the Food Chain. In Comprehensive Foodomics; Elsevier: Amsterdam, The Netherlands, 2020; pp. 188–211. [Google Scholar] [CrossRef]
- Nurjadi, D.; Zizmann, E.; Chanthalangsy, Q.; Heeg, K.; Boutin, S. Integrative Analysis of Whole Genome Sequencing and Phenotypic Resistance Toward Prediction of Trimethoprim-Sulfamethoxazole Resistance in Staphylococcus aureus. Front. Microbiol. 2021, 11, 607842. [Google Scholar] [CrossRef]
- Yee, R.; Bard, J.D.; Simner, P.J. The Genotype-to-Phenotype Dilemma: How Should Laboratories Approach Discordant Susceptibility Results? J. Clin. Microbiol. 2021, 59, e00138-20. [Google Scholar] [CrossRef]
- Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F.; Lindqvist, R. Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J. 2019, 17, e05898. [Google Scholar] [CrossRef]
- Challagundla, L.; Reyes, J.; Rafiqullah, I.; Sordelli, D.O.; Echaniz-Aviles, G.; Velazquez-Meza, M.E.; Castillo-Ramírez, S.; Fittipaldi, N.; Feldgarden, M.; Chapman, S.B.; et al. Phylogenomic Classification and the Evolution of Clonal Complex 5 Methicillin-Resistant Staphylococcus aureus in the Western Hemisphere. Front. Microbiol. 2018, 9, 1901. [Google Scholar] [CrossRef]
- Jamrozy, D.M.; Harris, S.R.; Mohamed, N.; Peacock, S.J.; Tan, C.Y.; Parkhill, J.; Anderson, A.S.; Holden, M.T.G. Pan-genomic perspective on the evolution of the Staphylococcus aureus USA300 epidemic. Microb. Genom. 2016, 2, e000058. [Google Scholar] [CrossRef]
- Deurenberg, R.; Vink, C.; Kalenic, S.; Friedrich, A.; Bruggeman, C.; Stobberingh, E. The molecular evolution of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2007, 13, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.; Ledo, C.; Lattar, S.; Llana, M.N.; Bertelli, A.M.; Di Gregorio, S.; Sordelli, D.O.; Gómez, M.I.; Mollerach, M.E. High virulence of methicillin resistant Staphylococcus aureus ST30-SCCmecIVc-spat019, the dominant community-associated clone in Argentina. Int. J. Med. Microbiol. 2017, 307, 191–199. [Google Scholar] [CrossRef]
- Mairi, A.; Touati, A.; Lavigne, J.-P. Methicillin-Resistant Staphylococcus aureus ST80 Clone: A Systematic Review. Toxins 2020, 12, 119. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, M.; Lin, Q.; Ye, Y.; Fan, H.; Wen, K.; Zeng, J.; Huang, D.; Mo, W.; Lei, Y.; et al. Identification of Methicillin-Resistant Staphylococcus Aureus from Methicillin-Sensitive Staphylococcus aureus and Molecular Characterization in Quanzhou, China. Front. Cell Dev. Biol. 2021, 9, 629681. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Ip, M.; Sun, M.; Sun, J.; Huang, G.; Wang, C.; Deng, L.; Zheng, Y.; Fu, Z.; et al. Molecular and Clinical Characteristics of Clonal Complex 59 Methicillin-Resistant Staphylococcus aureus Infections in Mainland China. PLoS ONE 2013, 8, e70602. [Google Scholar] [CrossRef]
- Lv, G.; Jiang, R.; Zhang, H.; Wang, L.; Li, L.; Gao, W.; Zhang, H.; Pei, Y.; Wei, X.; Dong, H.; et al. Molecular Characteristics of Staphylococcus aureus from Food Samples and Food Poisoning Outbreaks in Shijiazhuang, China. Front. Microbiol. 2021, 12, 1436. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Genet. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Song, J.-H.; Hsueh, P.-R.; Chung, D.R.; Ko, K.S.; Kang, C.-I.; Peck, K.R.; Yeom, J.-S.; Kim, S.-W.; Chang, H.-H.; Kim, Y.-S.; et al. Spread of methicillin-resistant Staphylococcus aureus between the community and the hospitals in Asian countries: An ANSORP study. J. Antimicrob. Chemother. 2011, 66, 1061–1069. [Google Scholar] [CrossRef]
- Kuntová, L.; Pantůček, R.; Rájová, J.; Růžičková, V.; Petráš, P.; Mašlaňová, I.; Doškař, J. Characteristics and distribution of plasmids in a clonally diverse set of methicillin-resistant Staphylococcus aureus strains. Arch. Microbiol. 2012, 194, 607–614. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.J.; Lindsay, J.A. The distribution of plasmids that carry virulence and resistance genes in Staphylococcus aureus is lineage associated. BMC Microbiol. 2012, 12, 104. Available online: http://www.biomedcentral.com/1471-2180/12/104 (accessed on 1 November 2021). [CrossRef]
- Jamrozy, D.; Misra, R.; Xu, Z.; Ter-Stepanyan, M.M.; Kocharyan, K.S.; Cave, R.; Hambardzumyan, A.D.; Mkrtchyan, H.V. Novel Methicillin-Resistant Staphylococcus aureus CC8 Clone Identified in a Hospital Setting in Armenia. Front. Microbiol. 2019, 10, 1592. [Google Scholar] [CrossRef]
- McDougal, L.K.; Fosheim, G.E.; Nicholson, A.; Bulens, S.N.; Limbago, B.M.; Shearer, J.E.S.; Summers, A.O.; Patel, J.B. Emergence of Resistance among USA300 Methicillin-Resistant Staphylococcus aureus Isolates Causing Invasive Disease in the United States. Antimicrob. Agents Chemother. 2010, 54, 3804–3811. [Google Scholar] [CrossRef] [PubMed]
- Strommenger, B.; Bartels, M.D.; Kurt, K.; Layer, F.; Rohde, S.M.; Boye, K.; Westh, H.; Witte, W.; De Lencastre, H.; Nübel, U. Evolution of methicillin-resistant Staphylococcus aureus towards increasing resistance. J. Antimicrob. Chemother. 2014, 69, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Feßler, A.; Kadlec, K.; Wang, Y.; Zhang, W.-J.; Wu, C.; Shen, J.; Schwarz, S. Small Antimicrobial Resistance Plasmids in Livestock-Associated Methicillin-Resistant Staphylococcus aureus CC398. Front. Microbiol. 2018, 9, 2063. [Google Scholar] [CrossRef]
- Jamrozy, D.; Coll, F.; Mather, A.E.; Harris, S.R.; Harrison, E.M.; MacGowan, A.; Karas, A.; Elston, T.; Török, M.E.; Parkhill, J.; et al. Evolution of mobile genetic element composition in an epidemic methicillin-resistant Staphylococcus aureus: Temporal changes correlated with frequent loss and gain events. BMC Genom. 2017, 18, 684. [Google Scholar] [CrossRef] [PubMed]
- Lozano, C.; García-Migura, L.; Aspiroz, C.; Zarazaga, M.; Torres, C.; Aarestrup, F.M. Expansion of a Plasmid Classification System for Gram-Positive Bacteria and Determination of the Diversity of Plasmids in Staphylococcus aureus Strains of Human, Animal, and Food Origins. Appl. Environ. Microbiol. 2012, 78, 5948–5955. [Google Scholar] [CrossRef]
- Liang, Y.; Tu, C.; Tan, C.; El-Sayed Ahmed, M.A.E.-G.; Dai, M.; Xia, Y.; Liu, Y.; Zhong, L.-L.; Shen, C.; Chen, G.; et al. Antimicrobial resistance, virulence genes profiling and molecular relatedness of methicillin-resistant Staphylococcus aureus strains isolated from hospitalized patients in Guangdong Province, China. Infect. Drug Resist. 2019, 12, 447–459. [Google Scholar] [CrossRef]
- Benito, D.; Lozano, C.; Rezusta, A.; Ferrer, I.; Vasquez, M.A.; Ceballos, S.; Zarazaga, M.; Revillo, M.J.; Torres, C. Characterization of tetracycline and methicillin resistant Staphylococcus aureus strains in a Spanish hospital: Is livestock-contact a risk factor in infections caused by MRSA CC398? Int. J. Med. Microbiol. 2014, 304, 1226–1232. [Google Scholar] [CrossRef]
- Lozano, C.; Rezusta, A.; Gómez, P.; Gómez-Sanz, E.; Báez, N.; Martin-Saco, G.; Zarazaga, M.; Torres, C. High prevalence of spa types associated with the clonal lineage CC398 among tetracycline-resistant methicillin-resistant Staphylococcus aureus strains in a Spanish hospital. J. Antimicrob. Chemother. 2012, 67, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Nurjadi, D.; Olalekan, A.O.; Layer, F.; Shittu, A.O.; Alabi, A.; Ghebremedhin, B.; Schaumburg, F.; Hofmann-Eifler, J.; Van Genderen, P.J.J.; Caumes, E.; et al. Emergence of trimethoprim resistance gene dfrG in Staphylococcus aureus causing human infection and colonization in sub-Saharan Africa and its import to Europe. J. Antimicrob. Chemother. 2014, 69, 2361–2368. [Google Scholar] [CrossRef]
- Resistancemap 2021. Available online: https://resistancemap.cddep.org/ (accessed on 15 November 2021).
- Resistancemap—Antibiotic Use 2021. Available online: https://resistancemap.cddep.org/AntibioticUse.php (accessed on 15 November 2021).
- Resistancemap—Methodology 2021. Available online: https://resistancemap.cddep.org/MethodologyAU.php (accessed on 15 November 2021).
- Deiters, C.; Günnewig, V.; Friedrich, A.W.; Mellmann, A.; Köck, R. Are cases of Methicillin-resistant Staphylococcus aureus clonal complex (CC) 398 among humans still livestock-associated? Int. J. Med. Microbiol. 2015, 305, 110–113. [Google Scholar] [CrossRef] [PubMed]
- El Dessouky, Y.; Mouftah, S.; Elhadidy, M. Genomic Overview into the Evolving Epidemiology of Methicillin-Resistant Staphylococcus aureus. Mansoura Vet. Med. J. 2020, 21, 125–131. [Google Scholar] [CrossRef]
- Chen, C.-J.; Huang, Y.-C. New epidemiology of Staphylococcus aureus infection in Asia. Clin. Microbiol. Infect. 2014, 20, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-H.; Greenwood-Quaintance, K.E.; Uhl, J.R.; Cunningham, S.A.; Chia, N.; Jeraldo, P.; Sampathkumar, P.; Nelson, H.; Patel, R. Molecular epidemiology of Staphylococcus aureus bacteremia in a single large Minnesota medical center in 2015 as assessed using MLST, core genome MLST and spa typing. PLoS ONE 2017, 12, e0179003. [Google Scholar] [CrossRef]
- Li, H.; Andersen, P.S.; Stegger, M.; Sieber, R.N.; Ingmer, H.; Staubrand, N.; Dalsgaard, A.; Leisner, J.J. Antimicrobial Resistance and Virulence Gene Profiles of Methicillin-Resistant and -Susceptible Staphylococcus aureus from Food Products in Denmark. Front. Microbiol. 2019, 10, 2681. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Coombs, G.; Shore, A.C.; Coleman, D.C.; Akpaka, P.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A Field Guide to Pandemic, Epidemic and Sporadic Clones of Methicillin-Resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- Kanjilal, S.; Sater, M.R.A.; Thayer, M.; Lagoudas, G.K.; Kim, S.; Blainey, P.C.; Grad, Y.H. Trends in Antibiotic Susceptibility in Staphylococcus aureus in Boston, Massachusetts, from 2000 to 2014. J. Clin. Microbiol. 2018, 56, e01160-17. [Google Scholar] [CrossRef]
- Jones, M.; Jernigan, J.A.; Evans, M.E.; Roselle, G.A.; Hatfield, K.M.; Samore, M.H. Vital Signs: Trends in Staphylococcus aureus infections in veterans affairs medical centers—United States, 2005–2017. Morb. Mortal. Wkly. Rep. 2019, 68, 220–224. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antibiotic Resistance Threats in the United States, 2019; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019. [CrossRef]
- CDDEP. Infectious Diseases in the South-East Asia Region. Available online: https://cddep.org/wp-content/uploads/2021/02/Infectious-diseases-in-the-south-east-asia-region.pdf (accessed on 15 November 2021).
- Ministry of Health Malaysia. Malaysian Action Plan on Antimicrobial Resistance (MyAP-AMR) 2017–2021. Available online: https://www.moh.gov.my/moh/resources/Penerbitan/Garis%20Panduan/Garis%20panduan%20Umum%20(Awam)/National_Action_Plan_-_FINAL_29_june.pdf (accessed on 15 November 2021).
- Norwegian Ministeries. National Strategy against Antibiotic Resistance 2015–2020. Available online: https://www.regjeringen.no/contentassets/5eaf66ac392143b3b2054aed90b85210/antibiotic-resistance-engelsk-lavopploslig-versjon-for-nett-10-09-15.pdf (accessed on 15 November 2021).
- Swedish Institute for Infectious Disease Control. SWEDRES 2001 a Report on Swedish Antibiotic Utilisation and Resistance in Human Medicine. Available online: https://www.folkhalsomyndigheten.se/contentassets/a21b86df576844cab59537f4d793ed87/swedres-2001.pdf (accessed on 15 November 2021).
- Tadesse, B.T.; Ashley, E.A.; Ongarello, S.; Havumaki, J.; Wijegoonewardena, M.; González, I.J.; Dittrich, S. Antimicrobial resistance in Africa: A systematic review. BMC Infect. Dis. 2017, 17, 616. [Google Scholar] [CrossRef] [PubMed]
- Thailand Ministry of Public Health. Thailand’s National Strategic Plan on Antimicrobial Resistance 2017–2021. Available online: https://rr-asia.oie.int/wp-content/uploads/2020/03/thailand_thailands-national-strategic-plan-on-amr-2017-2021.pdf (accessed on 15 November 2021).
- The Government of Japan. National Action Plan on Antimicrobial Resistance (AMR). Available online: https://www.mhlw.go.jp/file/06-Seisakujouhou-10900000-Kenkoukyoku/0000138942.pdf (accessed on 15 November 2021).
- WHO. WHO Report on Surveillance of Antibiotic Consumption. Available online: https://www.who.int/medicines/areas/rational_use/who-amr-amc-report-20181109.pdf (accessed on 15 November 2021).
- NCBI. NCBI 2021. Available online: https://ftp.ncbi.nlm.nih.gov/genomes/GENOMES_REPORTS/ (accessed on 12 November 2021).
- PATRIC. PATRIC 2021. Available online: https://ftp.patricbrc.org/ (accessed on 12 November 2021).
- PUBMLST. PUBMLST 2021. Available online: https://pubmlst.org (accessed on 12 November 2021).
- Cobo-Díaz, J.F.; del Río, P.G.; Álvarez-Ordóñez, A. Whole Resistome Analysis in Campylobacter jejuni and C. coli Genomes Available in Public Repositories. Front. Microbiol. 2021, 12, 662144. [Google Scholar] [CrossRef] [PubMed]
- StarAMR. Staramr 2021. Available online: https://github.com/phac-nml/staramr (accessed on 12 November 2021).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- PubMLST S. aureus Scheme. 2021. Available online: https://github.com/tseemann/mlst/blob/master/db/pubmlst/saureus/saureus.txt (accessed on 17 December 2021).
- Bouiller, K.; Bertrand, X.; Hocquet, D.; Chirouze, C. Human Infection of Methicillin-Susceptible Staphylococcus aureus CC398: A Review. Microorganisms 2020, 8, 1737. [Google Scholar] [CrossRef]
- Gómez, P.; Ruiz-Ripa, L.; Fernández-Fernández, R.; Gharsa, H.; Ben Slama, K.; Höfle, U.; Zarazaga, M.; Holmes, M.A.; Torres, C. Genomic Analysis of Staphylococcus aureus of the Lineage CC130, Including mecC-Carrying MRSA and MSSA Isolates Recovered of Animal, Human, and Environmental Origins. Front. Microbiol. 2021, 12, 655994. [Google Scholar] [CrossRef]
- Pang, R.; Wu, S.; Zhang, F.; Huang, J.; Wu, H.; Zhang, J.; Li, Y.; Ding, Y.; Zhang, J.; Chen, M.; et al. The Genomic Context for the Evolution and Transmission of Community-Associated Staphylococcus aureus ST59 through the Food Chain. Front. Microbiol. 2020, 11, 422. [Google Scholar] [CrossRef]
- Shen, P.; Zhou, K.; Wang, Y.; Song, J.; Liu, Y.; Zhou, Y.; Xiao, Y. High prevalence of a globally disseminated hypervirulent clone, Staphylococcus aureus CC121, with reduced vancomycin susceptibility in community settings in China. J. Antimicrob. Chemother. 2019, 74, 2537–2543. [Google Scholar] [CrossRef]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
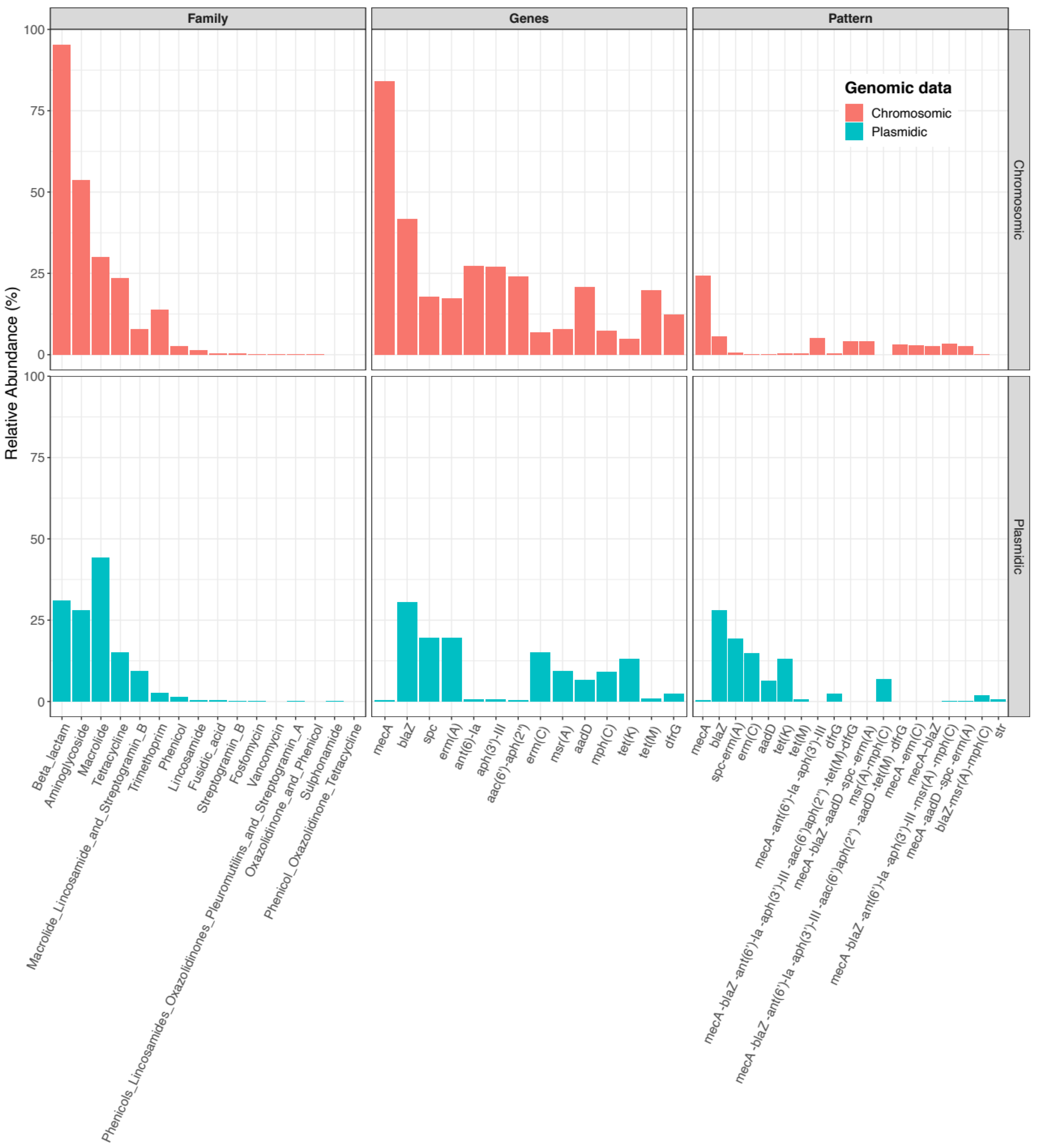
| # Pattern | # Contigs | % Contigs | % Cumulative | # ARGs | ARG Genotype—Plasmidic Contigs |
|---|---|---|---|---|---|
| 1 | 5862 | 27.91 | 27.91 | 1 | blaZ |
| 2 | 4068 | 19.37 | 47.27 | 2 | spc—erm(A) |
| 3 | 3115 | 14.83 | 62.10 | 1 | erm(C) |
| 4 | 2741 | 13.05 | 75.15 | 1 | tet(K) |
| 5 | 1420 | 6.76 | 81.91 | 2 | msr(A)—mph(C) |
| 6 | 1322 | 6.29 | 88.20 | 1 | aadD |
| 7 | 491 | 2.34 | 90.54 | 1 | dfrG |
| 8 | 392 | 1.87 | 92.41 | 3 | blaZ—msr(A)—mph(C) |
| # Pattern | # Chromosomes | % Chromosomes | % Cumulative | # ARGs | ARG Genotype—Chromosomes |
| 1 | 6002 | 24.24 | 24.24 | 1 | mecA |
| 2 | 1416 | 5.72 | 29.95 | 1 | blaZ |
| 3 | 1233 | 4.98 | 34.93 | 3 | mecA—ant(6)-Ia—aph(3’)-III |
| 4 | 1046 | 4.22 | 39.16 | 7 | mecA—blaZ—ant(6)-Ia—aph(3’)-III—aac(6’)-aph(2’’)—tet(M)—dfrG |
| 5 | 1031 | 4.16 | 43.32 | 5 | mecA—blaZ—aadD—spc—erm(A) |
| 6 | 831 | 3.36 | 46.67 | 6 | mecA—blaZ—ant(6)-Ia—aph(3’)-III—msr(A)—mph(C) |
| 7 | 764 | 3.08 | 49.76 | 8 | mecA—blaZ—ant(6)-Ia—aph(3’)-III—aac(6’)-aph(2’’)—aadD—tet(M)—dfrG |
| 8 | 726 | 2.93 | 52.69 | 2 | mecA—erm(C) |
| 9 | 676 | 2.73 | 55.42 | 4 | mecA—aadD—spc—erm(A) |
| 10 | 668 | 2.70 | 58.12 | 2 | mecA—blaZ |
| 11 | 628 | 2.54 | 60.65 | 2 | mecA—aac(6’)-aph(2’’) |
| 12 | 608 | 2.46 | 63.11 | 1 | mecC |
| 13 | 459 | 1.85 | 64.96 | 2 | mecA—aadD |
| 14 | 324 | 1.31 | 66.27 | 4 | mecA—blaZ—aac(6’)-aph(2’’)—tet(M) |
| 15 | 302 | 1.22 | 67.49 | 4 | mecA—ant(6)-Ia—aph(3’)-III—aac(6’)-aph(2’’) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennone, V.; Prieto, M.; Álvarez-Ordóñez, A.; Cobo-Diaz, J.F. Antimicrobial Resistance Genes Analysis of Publicly Available Staphylococcus aureus Genomes. Antibiotics 2022, 11, 1632. https://doi.org/10.3390/antibiotics11111632
Pennone V, Prieto M, Álvarez-Ordóñez A, Cobo-Diaz JF. Antimicrobial Resistance Genes Analysis of Publicly Available Staphylococcus aureus Genomes. Antibiotics. 2022; 11(11):1632. https://doi.org/10.3390/antibiotics11111632
Chicago/Turabian StylePennone, Vincenzo, Miguel Prieto, Avelino Álvarez-Ordóñez, and José F. Cobo-Diaz. 2022. "Antimicrobial Resistance Genes Analysis of Publicly Available Staphylococcus aureus Genomes" Antibiotics 11, no. 11: 1632. https://doi.org/10.3390/antibiotics11111632
APA StylePennone, V., Prieto, M., Álvarez-Ordóñez, A., & Cobo-Diaz, J. F. (2022). Antimicrobial Resistance Genes Analysis of Publicly Available Staphylococcus aureus Genomes. Antibiotics, 11(11), 1632. https://doi.org/10.3390/antibiotics11111632