
Elexacaftor–Tezacaftor–Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Patients Characterization
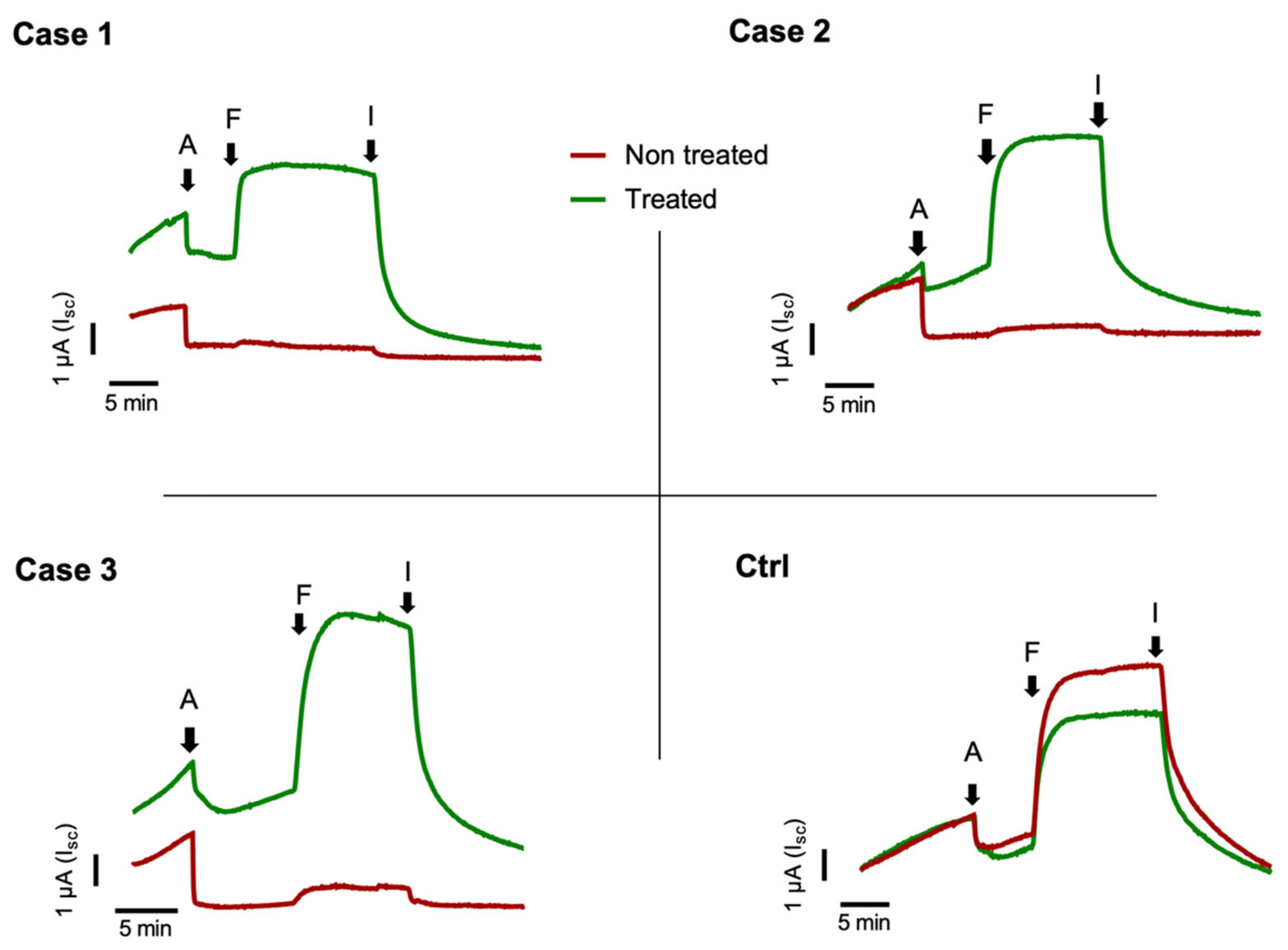
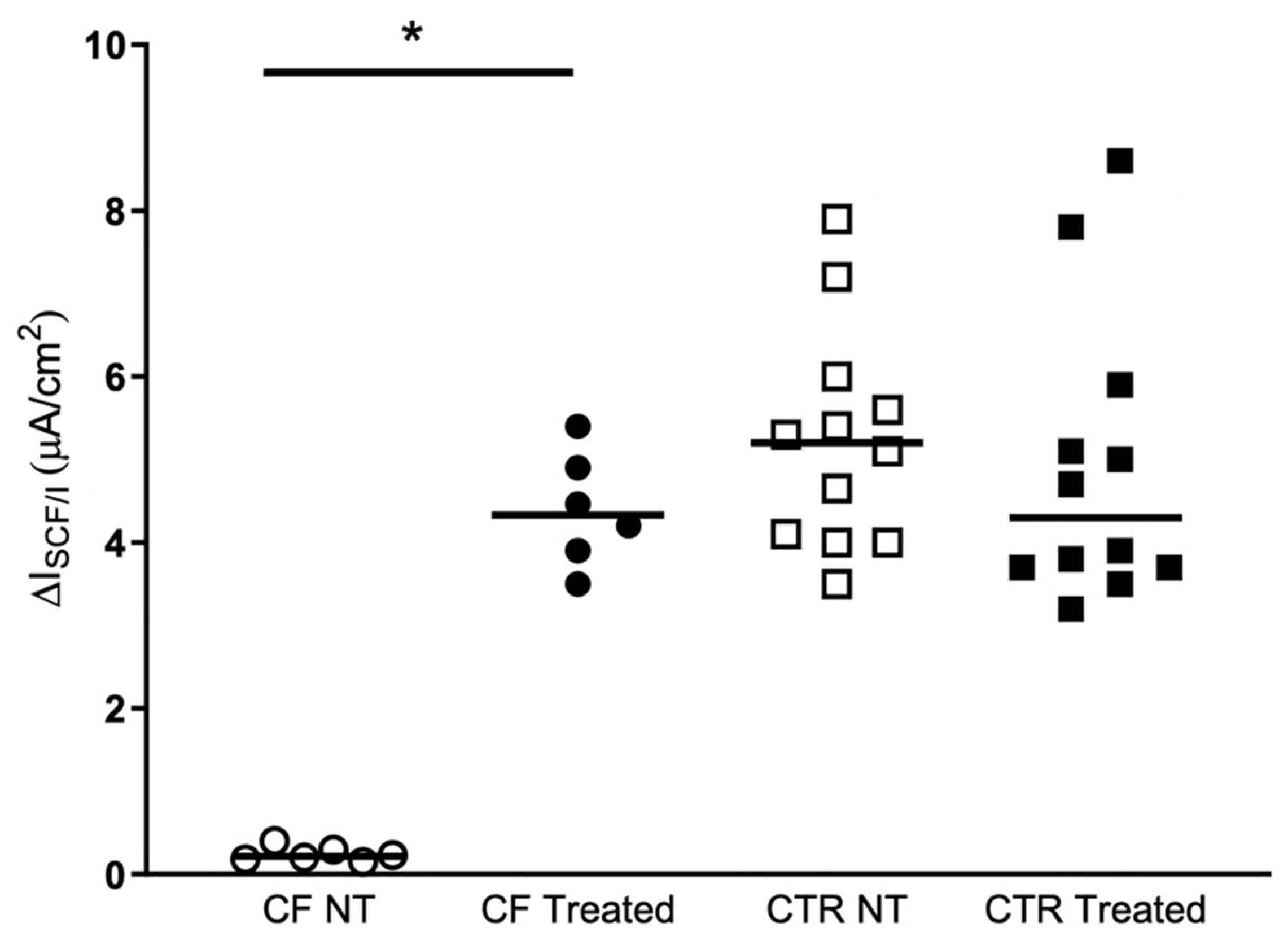
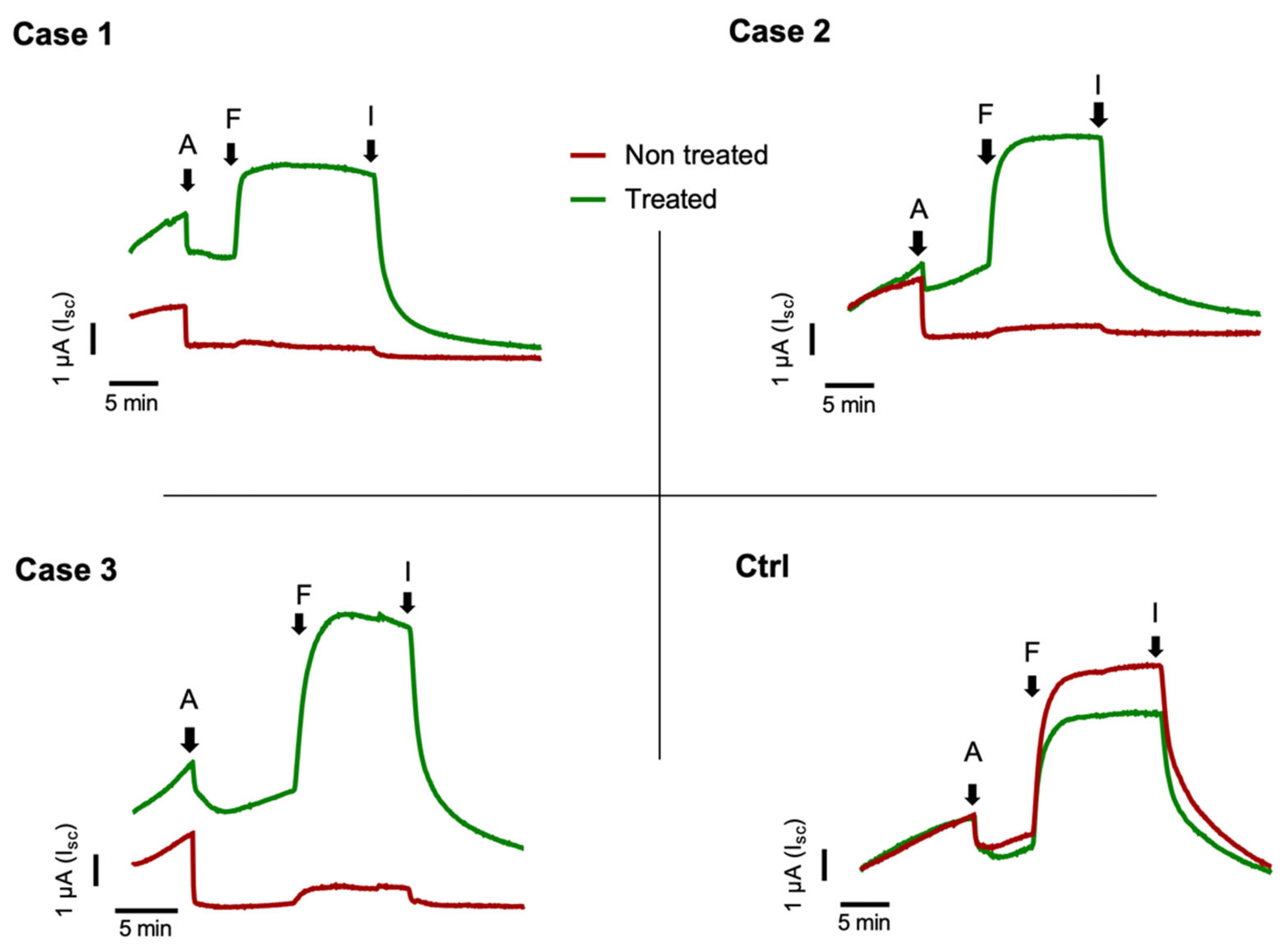
2.2. Nasal Brushing and Short-Circuit Current Recordings
3. Discussion
4. Materials and Methods
4.1. Patients Characterization
4.2. Nasal Brushing
4.3. Short-Circuit Current Recordings
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castanos, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.C.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Robertson, S.; Green, Y.; Cooke, J.; et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): An open-label, single-arm study. Lancet Respir. Med. 2016, 4, 107–115. [Google Scholar] [CrossRef]
- Ren, C.L.; Morgan, R.L.; Oermann, C.; Resnick, H.E.; Brady, C.; Campbell, A.; DeNagel, R.; Guill, M.; Hoag, J.; Lipton, A.; et al. Cystic Fibrosis Foundation Pulmonary Guidelines. Use of Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2018, 15, 271–280. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordani, B.; Amato, A.; Majo, F.; Ferrari, G.; Quattrucci, S.; Minicucci, L.; Padoan, R.; Floridia, G.; Salvatore, D.; Carnovale, V.; et al. Italian Cystic Fibrosis Registry (ICFR). Report 2015–2016. Epidemiol. Prev. 2019, 43, 1–36. [Google Scholar] [CrossRef]
- WHO. Human Genetics Programme; The Molecular Genetic Epidemiology of Cystic Fibrosis. 2002. Available online: http://apps.who.int/iris/bitstream/handle/10665/68702/WHO_HGN_CF_WG_04.02.pdf;jsessionid=4D8CCCEF242626F889F39FCB96CC50DE?sequence=1 (accessed on 7 July 2021).
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Campagna, G.; Amato, A.; Majo, F.; Ferrari, G.; Quattrucci, S.; Padoan, R.; Floridia, G.; Salvatore, D.; Carnovale, V.; Puppo Fornaro, G.; et al. Italian Cystic Fibrosis Registry (ICFR). Report 2017–2018. Epidemiol. Prev. 2021, 45, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martinez-Bartolome, S.; Lavallee-Adam, M.; Balch, W.E.; Yates, J.R., 3rd. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Tomati, V.; Pesce, E.; Caci, E.; Sondo, E.; Scudieri, P.; Marini, M.; Amato, F.; Castaldo, G.; Ravazzolo, R.; Galietta, L.J.V.; et al. High-throughput screening identifies FAU protein as a regulator of mutant cystic fibrosis transmembrane conductance regulator channel. J. Biol. Chem. 2018, 293, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Castellani, C.; Cuppens, H.; Macek, M., Jr.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Terlizzi, V.; Mergni, G.; Buzzetti, R.; Centrone, C.; Zavataro, L.; Braggion, C. Cystic fibrosis screen positive inconclusive diagnosis (CFSPID): Experience in Tuscany, Italy. J. Cyst. Fibros. 2019, 18, 484–490. [Google Scholar] [CrossRef]
- Terlizzi, V.; Castaldo, G.; Salvatore, D.; Lucarelli, M.; Raia, V.; Angioni, A.; Carnovale, V.; Cirilli, N.; Casciaro, R.; Colombo, C.; et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J. Med. Genet. 2017, 54, 224–235. [Google Scholar] [CrossRef]
- Terlizzi, V.; Di Lullo, A.M.; Comegna, M.; Centrone, C.; Pelo, E.; Castaldo, G.; Raia, V.; Braggion, C. S737F is a new CFTR mutation typical of patients originally from the Tuscany region in Italy. Ital. J. Pediatr. 2018, 44, 2. [Google Scholar] [CrossRef] [Green Version]
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748. [Google Scholar] [CrossRef]
- Terlizzi, V.; Amato, F.; Castellani, C.; Ferrari, B.; Galietta, L.J.V.; Castaldo, G.; Taccetti, G. Ex vivo model predicted in vivo efficacy of CFTR modulator therapy in a child with rare genotype. Mol. Genet. Genom. Med. 2021, 9, e1656. [Google Scholar] [CrossRef]
- Di Lullo, A.M.; Scorza, M.; Amato, F.; Comegna, M.; Raia, V.; Maiuri, L.; Ilardi, G.; Cantone, E.; Castaldo, G.; Iengo, M. An “ex vivo model” contributing to the diagnosis and evaluation of new drugs in cystic fibrosis. Acta Otorhinolaryngol. Ital. 2017, 37, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Amato, F.; Elce, A.; Monti, M.; Iannone, C.; Pucci, P.; Seia, M.; Angioni, A.; Zarrilli, F.; Castaldo, G.; et al. Molecular and functional analysis of the large 5’ promoter region of CFTR gene revealed pathogenic mutations in CF and CFTR-related disorders. J. Mol. Diagn. 2013, 15, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, F.; Seia, M.; Giordano, S.; Elce, A.; Zarrilli, F.; Castaldo, G.; Tomaiuolo, R. Gene mutation in microRNA target sites of CFTR gene: A novel pathogenetic mechanism in cystic fibrosis? PLoS ONE 2013, 8, e60448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianotti, A.; Delpiano, L.; Caci, E. In vitro Methods for the Development and Analysis of Human Primary Airway Epithelia. Front. Pharmacol. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Patient | Age | SCC (mEq/L) | Fecal Elastase (µg/g) | Last FEV1 (Liter, %) | Chronic Infection | CFTR-RD (Y/N) | OLTT (Y/N) |
|---|---|---|---|---|---|---|---|
| 1 * | 48 | 104 | <100 | 0.65; 26 | PA, BG | Y | Y |
| 2 * | 40 | 82 | >500 | 1.25; 38 | PA | N | Y |
| 3 | 59 | 114 | >500 | 0.59; 28 | PA | N | Y |
| Type of Cells | No-Treated | Treated | p Value a |
|---|---|---|---|
| CF HNEC (n = 6) | 0.2 (0.2–0.3) | 4.3 (3.8–5.0) | 0.031 |
| NO-CF HNEC (n = 12) | 5.2 (4.0–5.9) | 4.3 (3.7–5.7) | 0.116 |
| p value b | <0.0001 | 0.964 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comegna, M.; Terlizzi, V.; Salvatore, D.; Colangelo, C.; Di Lullo, A.M.; Zollo, I.; Taccetti, G.; Castaldo, G.; Amato, F. Elexacaftor–Tezacaftor–Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype. Antibiotics 2021, 10, 828. https://doi.org/10.3390/antibiotics10070828
Comegna M, Terlizzi V, Salvatore D, Colangelo C, Di Lullo AM, Zollo I, Taccetti G, Castaldo G, Amato F. Elexacaftor–Tezacaftor–Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype. Antibiotics. 2021; 10(7):828. https://doi.org/10.3390/antibiotics10070828
Chicago/Turabian StyleComegna, Marika, Vito Terlizzi, Donatello Salvatore, Carmela Colangelo, Antonella Miriam Di Lullo, Immacolata Zollo, Giovanni Taccetti, Giuseppe Castaldo, and Felice Amato. 2021. "Elexacaftor–Tezacaftor–Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype" Antibiotics 10, no. 7: 828. https://doi.org/10.3390/antibiotics10070828
APA StyleComegna, M., Terlizzi, V., Salvatore, D., Colangelo, C., Di Lullo, A. M., Zollo, I., Taccetti, G., Castaldo, G., & Amato, F. (2021). Elexacaftor–Tezacaftor–Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype. Antibiotics, 10(7), 828. https://doi.org/10.3390/antibiotics10070828