Abstract
Rapid spread of antibiotic resistance throughout the kingdom bacteria is inevitably bringing humanity towards the “post-antibiotic” era. The emergence of so-called “superbugs”—pathogen strains that develop resistance to multiple conventional antibiotics—is urging researchers around the globe to work on the development or perfecting of alternative means of tackling the pathogenic bacteria infections. Although various conceptually different approaches are being considered, each comes with its advantages and drawbacks. While drug-resistant pathogens are undoubtedly represented by both Gram(+) and Gram(−) bacteria, possible target spectrum across the proposed alternative approaches of tackling them is variable. Numerous anti-virulence strategies aimed at reducing the pathogenicity of target bacteria rather than eliminating them are being considered among such alternative approaches. Sortase A (SrtA) is a membrane-associated cysteine protease that catalyzes a cell wall sorting reaction by which surface proteins, including virulence factors, are anchored to the bacterial cell wall of Gram(+) bacteria. Although SrtA inhibition seems perspective among the Gram-positive pathogen-targeted antivirulence strategies, it still remains less popular than other alternatives. A decrease in virulence due to inactivation of SrtA activity has been extensively studied in Staphylococcus aureus, but it has also been demonstrated in other Gram(+) species. In this manuscript, results of past studies on the discovery of novel SrtA inhibitory compounds and evaluation of their potency were summarized and commented on. Here, we discussed the rationale behind the inhibition of SrtA, raised some concerns on the comparability of the results from different studies, and touched upon the possible resistance mechanisms as a response to implementation of such therapy in practice. The goal of this article is to encourage further studies of SrtA inhibitory compounds.
1. Introduction
The rapid spread of antibiotic resistance throughout the kingdom of bacteria (including, but not limited to, human pathogens that are of healthcare and economic importance) has highlighted the need for alternative means of bacterial disease treatment and reinvigorated the interest in studies that are targeted towards the development of alternative approaches to their containment [1]. Although the search for novel antibiotics and registration of drugs for effective treatment of multidrug-resistant strain infections is likely to continue, the occurrence of resistance due to the natural course of evolution under the selective pressure is inevitable. The question that arises is: Can we keep up the pace of discovery and registration of novel antibiotics [2], with each successive one proving to be effective only for a while, with the rate of antibiotic-resistance emergence which is a complex phenomenon that is still considered incompletely known [3]?
While multidrug-resistant bacterial pathogens are undoubtedly found in both Gram-positive and Gram-negative groups (e.g., two Gram-positive and four Gram-negative highly virulent antibiotic-resistant species are listed in the ESKAPE list [4]), herein we largely focused on the Gram-positive species, as the presence of sortases, the focal point of this paper, is far from being ubiquitous and has been seldom observed in Gram-negative bacterium.
2. The Emergence of Antibiotic-Resistant Gram-Positive Pathogens of Healthcare Importance
It was previously noted that numerous major bacterial pathogens that still continue to pose a serious threat to humanity today have emerged in the course of the last 50 years [5]. Some of the most prominent among these are the Gram-positive drug-resistant causative agents of various hospital-acquired (also known as “nosocomial” or “healthcare-associated”) infections. These include (however are not restricted to): The notoriously known multidrug-resistant (MDRSA) and methicillin-resistant (MRSA) Staphylococcus aureus strains [6]; Staphylococcus epidermidis—for long thought to be a mere opportunistic microorganism, but recently shown to be implicated in medical device-related infections, keratitis, and bacteremia [7]; Clostridium difficile, which is considered one of the most frequent causes of hospital-acquired gastrointestinal tract infections [8,9]; different enterococci, some of which (e.g., Enterococcus faecalis and E. faecium) can be responsible for bacteremia and endocarditis in addition to urinary tract, intra-abdominal, pelvic, and soft tissue infections [10]. Whereas some other (non-nosocomial) relevant Gram-positive pathogens worth a mention are Streptococcus mutans—associated with oral diseases and infective endocarditis [11,12]; Streptococcus pneumoniae—capable of causing pneumonia, meningitis, sepsis, bacteremia, and otitis [13]; Listeria monocytogenes—a significant cause of foodborne listeriosis outbreaks with a fatality rate of up to 30% [14,15]; causative agent of zoonotic anthrax—Bacillus anthracis, for which antibiotic resistance may not yet be an urgent matter, but potentially relevant [16,17].
While the antibiotic susceptibility spectra among the different strains of the aforementioned species are highly different and have been described elsewhere (e.g., [7,14,15,17] etc.), all of them have already been documented to harbor resistance to at least one or more antimicrobials in different classes in addition to the high potential for development of further drug resistance [18,19], which signifies the importance of research on the alternative means to combat them.
3. Alternative Options
It is, however, a widely known fact that antibiotics are by no means a sole option to treat bacterial pathogens. Although probably the most widely-used and, arguably, the most effective means for treatment of bacterial infections to date, there are also some alternative approaches that were described even prior to seminal discovery of penicillin by Alexander Fleming in 1928 [20]. But, historically, these were slowly dimmed by the wide employment of antibiotics only to resurface recently, and undergo further perfecting along with the various other state-of-art approaches, each with their own advantages and disadvantages in comparison to antibiotics [21].
Further in this article we shall attempt to briefly introduce some of the chosen prospective approaches that are being considered possible alternatives to antibiotics and in the recent years have gathered substantial attention from the research community (Figure 1).
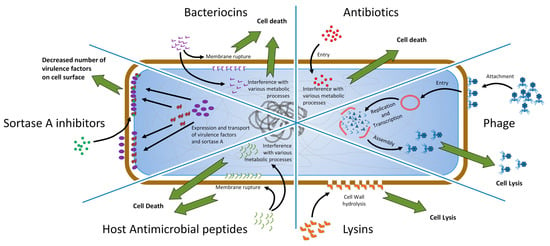
Figure 1.
Conceptual sexpartite diagram comparing simplified modes of action for antibiotics and some of the alternative approaches to combat the pathogenic bacteria. The diagram is divided into six parts, each corresponding to the given approach for combating the Gram-positive pathogenic bacteria. Brown rounded rectangle represents peptidoglycan layer and together with the contents represents the Gram-positive bacterial cell. Perimeter of the blue rounded rectangle represents the plasma membrane. Black arrows represent interactions and point at the desired spatial location/locations of the agents for interplay with their bacterial targets. Green arrows pointing away from the cell represent the desired outcome of an interplay between a given agent and its target. Colored figures unique to each of the diagram parts represent agents that can be viewed as an alternative to antibiotics (for example red circles represent molecules of antibiotics). In the case of sortase A (SrtA) inhibitors (leftmost part of the diagram): Green circles—inhibitory compounds, purple ellipses—virulence factors, amorphous brown figures—SrtA enzyme.
A detailed description of these selected methods, as well as other possible alternatives (e.g., vaccines, antibodies, immune stimulation), is not considered herein because these have been recently summarized and reviewed in detail by other authors elsewhere [1,21,22,23,24,25,26].
3.1. Phage Therapy
Bacteriophages (“bacteria eaters”)—viruses of bacteria, the most abundant biological entities in the biosphere—were already being viewed as a tool for treatment of bacterial infections shortly after their independent discovery by Frederick Twort [27] and Felix d’Herelle [28] in the beginning of the 20th century [29]. The usage of strictly lytic bacteriophages in the therapy, although somewhat abandoned during the antibiotic era, has proven to be effective against various human bacterial diseases caused by different cocci, pseudomonads, coliforms, and other pathogens [30]. Some of the benefits of phage therapy, as outlined by Loc–Carrillo and Abedon [31], include: Bactericidal mode of action; “auto-dosing” upon infection of host bacteria population; low inherent toxicity, minimal disruption of normal bacterial flora, narrower potential for inducing resistance, lack of cross-resistance with antibiotics, application versatility, and capability of biofilm clearance. Arguably, the main disadvantage of phage therapy, however, is strongly linked to one of their greatest benefits—the narrow host range of phages, which might span just a few strains of a particular bacterial species. This suggests the need to formulate cocktails of multiple carefully characterized phages with a good proven bacterial killing potential, covering, ideally, different host receptors to rule out the phage-resistance emergence in the course of treatment. However, even then, they are not guaranteed to eliminate every strain of the target species encountered. Despite the fact that strictly lytic phages were almost exclusively used as such natural antibacterial agents for obvious reasons, efforts to use lytic-derivatives of temperate phages with a broad host range to treat bacterial infections in humans were successfully partaken as of recently [32].
3.2. Lysins
Lysins—enzymes that are produced by bacteriophages to disrupt the cell wall of their hosts during the last phases of the infection cycle to release the phage progeny into the environment—are also being considered a prospective alternative to antibiotics [33]. Although the ability of lysins to reach the peptidoglycan layer from within the phage-infected cell is usually dependent on another phage-encoded protein (holin) in the natural setting, it has been previously shown that exogenous application of lysins is a rather prospective strategy for treatment of bacterial infections. When applied to Gram-positive bacteria exogenously, lysins have an immediate access to the cell wall, as opposed to being obstructed by the membrane from within the cell, which is the case during the natural phage life cycle; thus, even the small amounts of recombinant phage lysins have demonstrated promising therapeutic potential due to their capability of rapidly lysing the target bacteria. [34]. The first study showing phage lysin effectiveness in vivo was reported in 2001. It described the disease prevention and pathogen elimination in the upper respiratory tracts of mice that were colonized by streptococci [35]. Since then, a plethora of novel lysins have been discovered [36] and numerous advances in the field have been made (recently reviewed in detail by De Maesschalck et al. [37]). The main advantages of lysin therapeutic applications over antibiotics include: Selectiveness regarding their targets, low risk of lysin-resistance emergence, potent activity that occurs within seconds after application of even highly diluted preparations, synergy between different lysins, and lack of toxicity, among others [21]. The main hurdle of “classical” lysin therapy, however, was general ineffectiveness of exogenously applied lysins against Gram-negative bacteria. Nevertheless, this issue is being addressed by the development of state-of-art endolysin-based anti-bacterials termed Artilysin®s that are active against Gram-negative pathogens [38].
3.3. Antimicrobial Peptides
Antimicrobial peptides (AMPs) are a diverse class of small molecules (generally between 10–50 amino acids long) that are naturally produced by both single-cell and multicellular organisms to either directly kill/inhibit the growth of competing/foreign microorganisms or modulate the innate immune response of the higher organism. These are being considered as yet another alternative tool of combating various pathogens (including antibiotic-resistant bacteria) [39,40]. The main mechanism of action for AMPs lies in their ability to interact with bacterial membranes/cell walls via electrostatic interactions, which results in either rupturing of the membrane or entry into the bacteria with subsequent inhibition of intracellular functions [41]. Although the first antimicrobial peptide lysozyme was discovered by Alexander Fleming as early as 1922 [42], until the 1980s there was a relatively low number of research reports on AMPs [43]. The latest antimicrobial peptide database (APD3) lists 2169 AMPs of various origins, with the vast majority being animal host defense peptides [44].
3.4. Bacteriocins
Bacteriocins are highly potent bactericidal agents representing a subclass of antimicrobial peptides produced by bacteria. In general terms these can be classified as either post-translationally modified (class I) or made up of peptides with unmodified amino acids (class II) [45]. The discovery of bacteriocins is attributed to André Gratia, who described the first bacteriocin (colicin V) produced by verotoxin-producing E. coli strain to act against other nearby E. coli in 1925 [46]. The bacteriocins are structurally diverse and, thus, exhibit different mechanisms of action. Most bacteriocins, however, act by forming pores in bacterial cell membranes, which leads to their disruption and subsequent collapse of the phospholipid bilayer that ultimately leads to death of a bacterial cell [21]. Although their main function is to enhance the competitiveness of bacteriocin-producing bacteria in their natural environment through the elimination of contestant co-inhabitant bacteria, researchers have found applications for bacteriocins in food preservation and clinical setting despite the potential for resistance-development [47,48]. In addition, some of the state-of-art studies have suggested that bacteriocin application might be even more feasible if these peptides are formulated using the state-of-art nanotechnological approaches, and these findings pave way for further enhancements of their usability [49].
3.5. Antivirulence Strategies
The rationale behind antivirulence strategies lies in the assumption that “disarmed” pathogens can do no harm; thus, the aim of such strategies is to interfere with the bacterial virulence factors that help the pathogen to either cause damage to the host or evade its immune system and persist within the host [50]. The current anti-virulence strategies tend to target the processes of bacterial quorum sensing systems and biofilm formation ability, as well as to disassemble functional membrane domains and neutralize bacterial toxins via usage of small molecule compounds that show corresponding inhibitory activity against at least one of the virulence factors of the pathogen of interest [51].
It is clearly evident that only a few cells of any pathogenic bacteria are of no immediate concern to the host; however, strength of all pathogens “lies in numbers” that they eventually reach through inevitable propagation in appropriate environmental conditions. With growth of bacterial cell population also grows the necessity for “communication” between the cells, which is essential for coordination of the whole community towards situations that are advantageous to a population as a whole. This communication was revealed to be mediated by small signal molecules that auto-induce expression of particular genes [52]. The cell density-dependent process of such communication is termed “quorum sensing” [53] and there are indications that it might occur even between different species [54]. It is thought that as much as 4–10% of bacterial genome and ≥20% of proteome could be influenced by quorum sensing, and the effects of quorum sensing range from harmless metabolic or phenotypic adaptations to modulation of pathogenicity related virulence (e.g., biofilm formation, toxin expression) [55]. Thus, interference with quorum sensing cascades that blocks bacterial virulence-associated “communication” (“quorum quenching”) was outlined as a promising antivirulence strategy [56].
Whereas inhibition of the transcription might seem a more appropriate approach for some of the bacterial virulence factors, such as protein-based toxins, thus, eliminating the cause rather than a consequence, it is also possible to target these molecules after their synthesis. Studies in this direction have led to the development of approaches that enable evasion from the destructive effects of toxins on the host through their neutralization by antibodies, toxin activity blocking by small molecule compounds [57], or even sequestration of toxins in artificial liposomes [58].
Recently discovered bacterial functional membrane microdomains (FMMs), which resemble lipid rafts of eukaryotic cells in both structure and function, were immediately proposed as yet another novel antivirulence strategy target due to the involvement of FMMs/FMM-associated proteins (e.g., flotillins) in biofilm formation, attachment, virulence, and signaling [59,60,61].
The ability of some pathogens to form biofilms, which are bacterial communities enclosed in structure formed by thereof extracellular matrix components, helps embedded bacterial communities to evade host defensive responses, mitigate environmental stresses, and provide protection from antibiotics [62]. Pathogenic biofilm formation on the host tissues facilitates the onset of a chronic bacterial disease that is considerably more difficult to treat than acute infections caused by pathogens in the planctonic (“free”) state [63]. Thus, the prevention of biofilm formation is also recognized as a prospective antivirulence strategy that strives to limit bacterial adhesion to surfaces or affect the extracellular matrix component production, and sometimes even destroy the extracellular matrix post factum, when biofilm has already been formed [51].
One of the particularly promising antivirulence strategies that is aimed at Gram-positive bacteria is the inhibition of bacterial cysteine protease—sortase (especially SrtA)—activity by small molecule compounds [64].
4. Sortase A (SrtA)
Sortases are cysteine transpeptidases that play a pivotal role in shaping the architecture of microorganisms by mediating the covalent protein attachment to their cell wall [65]. While sortase enzymes are found ubiquitously in Gram-positive bacteria, where their functions have been largely elucidated, genes encoding proteins belonging to the sortase superfamily have also been documented in a fraction of genomes from Gram-negative and archaeal species, although their functions in these organisms have not yet been completely understood [66]. Despite the fact that up to eight different classes of sortases (A, B, C, D1, D2, E, F, and “Marine”) are already being recognized as of now, the “canonical” Staphylococcus aureus SrtA still remains the most extensively studied enzyme of this group [66,67,68,69,70].
SrtA is a membrane-associated cysteine protease that catalyzes a cell wall sorting reaction by which surface proteins, including virulence factors, are anchored to the bacterial cell wall. The steps involved in the SrtA-assisted protein anchoring to the cell wall of Gram-positive bacteria have been uncovered and, in the case of Staphylococcus aureus, are as follows [71]: (1) Proteins with the N-terminal secretion signal peptide are transported to the cell surface via the secretory (Sec) pathway; proteins that are destined to be anchored to the cell-wall contain an additional LPXTG (leucine–proline–any residue–threonine–glycine) motif followed by a hydrophobic region and a tail of charged residues within their C-terminus; (2) the LPXTG motif is recognized by the membrane-associated SrtA enzyme; (3) cleavage between T and G of a LPXTG motif is introduced by two-step transpeptidation reaction and threonine is covalently attached to the cell wall amino group of a pentaglycine [72].
It has previously been shown that the C-terminal sorting signal with a highly-conserved LPXTG motif is prevalent within cell wall-anchored surface proteins of Gram-positive bacteria [73] and the elucidation of Gram-positive bacteria surface protein roles in interactions with the host, which also includes virulence, is a topic that has gathered substantial research attention in the past. These studies have led to hypotheses that it might be possible to reduce pathogen virulence by interfering with the display of the aforementioned proteins on the surface of the cells [74,75,76]. However, it was not before the seminal study of Mazmanian and colleagues [77], which demonstrated that the SrtA enzyme is an absolute necessity for surface protein anchoring to the cell wall envelope and consequent pathogenesis of S. aureus infections, that the sortase inhibitor research era really begun. The primary goal of the subsequent studies was to find a way to disrupt the pathogenesis of bacteria without affecting microbial viability, thus, treating infections caused by Gram-positive pathogens. In addition to S. aureus, a decrease in virulence due to inactivation of SrtA activity has also been demonstrated in Listeria monocytogenes [78], Streptococcus pneumoniae [79], S. suis [80], and S. mutans [81].
5. SrtA Inhibitors
In comparison to the conventional antibiotics and other anti-virulence strategies, the main advantage of SrtA inhibitor usage as a potential Gram-positive pathogen infection treatment lies in the relative harmlessness of possible resistance emergence to the host [82]. We hypothesize that, if the inhibitor is properly designed and targeted at the substrate binding site, the most likely outcome of the inhibitor-induced selective pressure shall be either in the formation of a mutated SrtA gene that produces enzyme with an altered inhibitor binding site, which most probably shall also alter the enzymatic core structure, decreasing enzyme activity and, along with it, overall virulence of the pathogen, or increase the production of sortases to cope with the decrease in enzymatic activity, which shall result in an increased metabolic burden and decreased pathogen proliferation rate. In either way, acquisition of resistance shall come with the cost that decreases the overall pathogenicity of the target bacteria. Alternatively, pathogens might also start to produce a novel protein that either enzymatically alters the inhibitor, thus abolishing its inhibitory properties, or binds it to effectively remove the inhibitor from the surrounding environment. However, we believe that such a scenario is unlikely, as it would require the coincidence of far too many favorable factors. This, in our opinion, provides a prospective of the fail-safer therapy concept ultimately resulting in the greater success rate regardless of the resistance emergence—the pathogen is either being “disarmed” by the designated SrtA inhibitor treatment or is “significantly hampered” in the course of evolution under the selective pressure imposed by such treatment. This situation, however, would differ significantly from the previously described if the selected inhibitor were to work as an allosteric modulator that could bind to the enzyme at other sites than the active center. In this case it is highly probable that the exerted selective pressure shall result in a development of metabolically or enzymatically unburdened resistance through the introduction of only the allosteric binding site altering mutation that would not change the overall molecular structure of the enzyme molecule. Therefore, we believe that the development of sortase inhibitors should be exclusively directed towards the identification of molecules binding to the enzymatic core. An alternative approach for treatment, of course, might also be an employment of cocktails of sortase inhibitors that couple to different allosteric binding sites, as it is highly unlikely that several inhibitory effect inactivating mutations would occur at the same time. However, such an approach would require a careful characterization of each individual compound and their interactions with the sortase, as well as elucidation of possible side effects on the health of the host, prior to being employed for the treatment of bacterial infections in a therapeutical setting, while implementation of such extensive thorough studies undoubtedly requires lengthy allocation of significant financial and labor resources.
Conceptually different strategies for the initial discovery of novel potential SrtA inhibitors have previously been adopted in the field, including: Screening of natural products [83], small compound library high throughput screening [84], virtual screening in silico [85], and fragment based lead discovery [86]. Although initial hits can be generated by each of the approaches individually, these approaches complement each other well, and, thus, combining them in a single study could yield better results. While working on this article and exploring the repertoire of uncovered sortase inhibitor leads (extensively summarized in Supplementary Material Table S1 [84,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118]), we observed that despite the great advances in this field, there was also a great ambiguity in the acquired results.
The presentation of the determined compound-specific parameters differed significantly as IC50 values were presented both as molar and mass concentrations, with the latter being less informative due to the great differences in tested compound molecular weight. Even more, the minimum inhibitory concentration (MIC) test has not been performed for many of the reported leads; thus, their bacterial toxicity and possible drawbacks of their applicability in anti-virulence therapies remains unexplored. An additional aspect that seems to hamper the effective cross-study comparison of various compounds is related to the basic enzyme kinetics, because, as clearly demonstrated by the data that we acquired in one of our latest studies (Supplementary Material Table S1 and Figure 2) where, while searching for suitable substrate, we tested the same SrtA-compound combination with two different substrates, and acquired IC50 values were dependent on a combination of both the selected substrate and tested compound. Thus, this parameter is usable for compound comparison only if the overall setting of the experiments is identical. This situation clearly demonstrates that standardization of experimental procedures and results’ presentation is needed in this field to improve cross-study comparison capabilities and achieve a greater whole SrtA research community success rate.
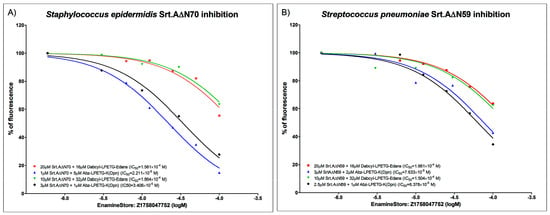
Figure 2.
Enzyme inhibition curves for (A) recombinant Δ70 Staphylococcus epidermidis SrtA, and (B) recombinant Δ59 Streptococcus pneumoniae SrtA. N-terminal parts containing membrane anchor domain were cleaved to improve solubility and purification of enzyme. FRET-based inhibition assays were carried out employing two different substrates: Abz-LPETG-K (Dnp), Dabcyl-LPETG-Edans, one lead compound, Z1758047752, which was acquired from Enamine Ltd. (https://www.enaminestore.com/). Measurements were performed in triplicates and repeated twice. Results demonstrate that acquired IC50 values are substrate dependent.
It is also worth noting that only few of the studies on the identification of SrtA inhibitory compounds that were reviewed in this article have actually taken their studies further and performed the actual efficacy evaluation for their most potent compounds beyond mere documentation of their inhibitory effects in enzymatic models (e.g., no MIC tests or in vivo models), which suggests that there is a need for follow-up in-depth studies of these compounds to evaluate the plausibility of their development into an actual therapeutic agents of a new kind. This necessity is further highlighted by the fact that, to the best of our knowledge, none of the thus far identified SrtA inhibitors has yet been advanced to the clinical trials, while numerous products from some other classes of possible antibiotic alternatives (e.g., phage therapy, lysins) are already seeking medical approval by moving through the different phases of clinical trials.
Therefore, we believe that, in the near future, assessment of the previously described SrtA inhibitor efficacy should be conducted in the natural setting using a standardized approach, with the acquired results made available to the scientific community as soon as possible, so that aggregation of the pro and cons evidence would either enable the consolidation of the SrtA inhibitor position among the potential antibiotic alternatives, thus drawing more attention from the funding bodies and attracting additional researchers to the field, or prove the inefficiency of this approach, thus enabling the redirection of resources to the further development of approaches that would prove to be more reasonable.
6. Materials and Methods
6.1. SrtA Expression and Purification
The presumed catalytic core of the SrtA gene from Staphylococcus epidermidis and Streptococcus pneumoniae microorganisms that encode enzymatically active transpeptidase domains were PCR amplified and inserted in the vector for bacterial expression [119,120]. The DNA sequence encoding for N-terminally truncated Staphylococcus epidermidis SrtA (Se-SrtA∆N70) was PCR-amplified from genomic DNA of S. epidermidis YC-1 strain using 5′-TATACATATGGGTTATATAGAAGTTCCAGATG-3′ and 5′-TAATCTCGAGTTAGTTAATTTGTGTAGCTATG-3′ primers. The DNA sequence encoding for N-terminally truncated Streptococcus pneumoniae SrtA (Sp-SrtA∆N59) was PCR-amplified from genomic DNA of the S. pneumoniae D39 strain using 5′-ATTACATATGGAAGAAAATCAGGATACAGAAG-3′ and 5′-TATACTCGAGTTAATAAAATTGTTTATATGGT-3′ primers. The PCR-amplified DNA sequences were cloned into the pET28b (Novagen, Madison, WI, USA) vector through NdeI and XhoI restriction sites for expression as His-tagged proteins in E. coli. The plasmid DNAs were isolated using the Plasmid Miniprep Kit (Thermo Fisher Scientific, Waltham, MA, USA), verified by restriction analysis, and approved by sequencing using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, MA, USA).
The recombinant sortases were expressed in E. coli BL21 (DE3) cells using a standard protocol and purified as previously described [121]. In short, transformed bacteria were cultured in Luria–Bertani medium at 37 °C until an optic density at 600 nm (OD600) of 0.6–0.7 was reached. Recombinant protein synthesis was induced at 37 °C, 200 rpm by adding isopropylthiogalactoside to a final concentration of 1 mM and continued for 3 h. The cells were then sedimented by centrifugation and resuspended in phosphate-buffered saline buffer pH 7.5 with 1 mM dithiothreitol, 0.1% Triton X100, and protease inhibitor cocktail added prior to sonication. The recombinant soluble sortases were further purified under native conditions.
Both histidine-tagged recombinant SrtA proteins were purified by affinity chromatography using the Ni-nitrilotriacetic acid agarose (Qiagen, Hilden, Germany) pre-equilibrated with buffer A (10 mM imidazole, 1 mM DTT, 50 mM NaH2PO4, and 300 mM NaCl, pH 8.0). Protein binding was performed for 1 h at room temperature on the rotator and 20 mM imidazole buffer A pH 8.0 was then used during the wash procedure to remove unbound proteins. The proteins were eluted by adding 300 mM imidazole buffer A to the column and then additionally purified on Superdex75 120 mL packed on XK16/70 gel filtration column (GE Healthcare, Chicago, IL, USA) by using 1 mM DTT, 50 mM Tris-HCl, and 150 mM NaCl pH 7.5 end buffer. Purified proteins were concentrated to 10 mg/mL by centrifugation at 4000 rpm at 4 °C in Amicon Ultra centrifugal filter units with a 3 kDa cut off (Millipore, Burlington, VT, USA). Further clarification of protein was achieved by centrifugation at 16,400 rpm for 10 min at 4 °C, and 20% glycerol was added to the buffer for prolonged protein storage on ice.
6.2. FRET Enzymatic Assay and IC50 Determination
The tested compounds were commercial products purchased from Enamine Ltd. The half maximal inhibitory concentration (IC50) values for compounds were determined by monitoring the increase in fluorescence intensity upon cleavage of the Dabcyl-LPETG-Edans FRET peptide (excitation/emission wavelength of 360/485 nm) or Abz-LPETG-K (Dnp) FRET peptide (Ex/Em 320/420 nm), which were used as the substrates for both sortases [108]. In brief, the test compounds of various concentrations (3–100 μM) were added to 1–20 μM of each of the recombinant sortase in 20 mM HEPES, 5 mM CaCl2, 0.05% Tween-20 buffer pH 7.5 [122]. Subsequently, the peptide Dabcyl-LPETG-Edans at a final concentration 16–32 μM (depending on each sortase enzymatic activity) was added to the reaction. Similarly, test compounds were added to the 1–20 μM recombinant sortases with subsequent peptide Abz-LPETG-K (Dnp)-NH2 addition at a final concentration 1 or 5 μM (depending on each sortase enzymatic activity). Fluorescence was recorded for 15 h within an interval of 30 min at 37 °C temperature using a Tecan F200 microplate reader. IC50 values were calculated using GraphPad Prism Software [123].
7. Conclusions
To conclude, while SrtA activity inhibition by small molecule compounds that leads to a decrease of Gram-positive pathogen virulence seems to be a reasonable approach to combat drug-resistant pathogens and many of such compounds are displaying promising potency, the SrtA research community still has a long way to go before a definite conclusion on the feasibility of this strategy can be reached. We believe that employment of standardized procedures (both in vitro and in vivo, with an employment of appropriate controls to elucidate the non-specific effects) for testing of identified leads and reporting of the acquired results in a standardized manner that would rapid cross study comparison (MIC tests, ki value calculations, etc.) should be a top priority of the field. Collectively defining the recommended workflows and/or guidelines is absolutely possible and would prove to be extremely beneficial for a relatively small SrtA research community, possibly leading to a faster accumulation of knowledge in the field while ensuring comparability of the results.
Supplementary Materials
The following are available online at https://www.mdpi.com/2079-6382/10/2/164/s1, Table S1: Examples of the potent SrtA inhibitors and their sources. Source column is color-coded as follows: Green entries—compounds initially described as SrtA inhibitors derived from natural sources, blue—from small molecule screening, orange—from in silico screening. IC50 columns represent the half maximal inhibitory concentration (μM and μg/mL). MIC columns represent the minimum inhibitory concentration (μM and μg/mL). Asterisk (*) after the value in either of the IC50 or MIC columns indicates that the value was absent in the referenced study and was derived herein. Double asterisk (**) in “SrtA for IC50 determination” and “Strain for MIC determination” column indicates that the given compound was tested on more than one variant of SrtA or more than one bacterial strain in the referenced original study; values of these columns then, respectively, represent the SrtA for which the IC50 value is given and bacterial strain for which the MIC value is given. “Title of the compound from the original study” indicates how the compound is referred to by the original authors in the referenced experimental paper.
Author Contributions
Conceptualization, N.Z., V.K., Z.R., A.L., and D.F.; methodology, V.K., Z.R., and A.L.; validation, V.K., Z.R., and A.L.; formal analysis, V.K., Z.R., and A.L.; investigation, N.Z., A.L., and D.F.; resources, A.L.; data curation, N.Z., A.L., and D.F.; writing—original draft preparation, N.Z. and D.F.; writing—review and editing, NZ., V.K., Z.R., A.L., and D.F.; visualization, N.Z. and D.F.; supervision, A.L. and D.F.; project administration, A.L.; funding acquisition, A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the European Regional Development Fund (agreement no. 1.1.1.1/16/A/107).
Data Availability Statement
The data presented in this study are available in Supplementary Material Table S1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- David, M.Z.; Dryden, M.; Gottlieb, T.; Tattevin, P.; Gould, I.M. Recently approved antibacterials for methicillin-resistant Staphylococcus aureus (MRSA) and other Gram-positive pathogens: The shock of the new. Int. J. Antimicrob. Agents 2017, 50, 303–307. [Google Scholar] [CrossRef]
- Waldetoft, K.W.; Brown, S.P. Alternative therapeutics for self-limiting infections—An indirect approach to the antibiotic resistance challenge. PLoS Biol. 2017, 15, 1–10. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Vouga, M.; Greub, G. Emerging bacterial pathogens: The past and beyond. Clin. Microbiol. Infect. 2016, 22, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Namvar, A.E.; Bastarahang, S.; Abbasi, N.; Ghehi, G.S.; Farhadbakhtiarian, S.; Arezi, P.; Hosseini, M.; Baravati, S.Z.; Jokar, Z.; Chermahin, S.G. Clinical characteristics of Staphylococcus epidermidis: A systematic review. GMS Hyg. Infect. Control 2014, 9. [Google Scholar] [CrossRef]
- Peng, Z.; Jin, D.; Kim, H.B.; Stratton, C.W.; Wu, B.; Tang, Y.W.; Suna, X. Update on antimicrobial resistance in Clostridium difficile: Resistance mechanisms and antimicrobial susceptibility testing. J. Clin. Microbiol. 2017, 55, 1998–2008. [Google Scholar] [CrossRef]
- Spigaglia, P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther. Adv. Infect. Dis. 2016, 3, 23–42. [Google Scholar] [CrossRef]
- Agudelo Higuita, N.I.; Huycke, M.M. Enterococcal disease, epidemiology, and implications for treatment. In Enterococci—From Commensals to Leading Causes to Drug Resistant Infection; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014; pp. 1–35. [Google Scholar]
- Karikalan, S.; Mohankumar, A. Antibiogram of streptococcus mutans isolated from dental caries patients. Int. J. Med. Health Res. 2016, 2, 79–83. [Google Scholar]
- Salehi, B.; Kregiel, D.; Mahady, G.; Sharifi-Rad, J.; Martins, N.; Rodrigues, C.F. Management of Streptococcus mutans-Candida spp. oral biofilms’ infections: Paving the way for effective clinical interventions. J. Clin. Med. 2020, 9, 517. [Google Scholar] [CrossRef]
- Cherazard, R.; Epstein, M.; Doan, T.L.; Salim, T.; Bharti, S.; Smith, M.A. Antimicrobial resistant Streptococcus pneumoniae: Prevalence, mechanisms, and clinical implications. Am. J. Ther. 2017, 24, e361–e369. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.L.; Gorris, L.G.M.; Hayman, M.M.; Jackson, T.C.; Whiting, R.C. A review of Listeria monocytogenes: An update on outbreaks, virulence, dose-response, ecology, and risk assessments. Food Control 2017, 75, 1–13. [Google Scholar] [CrossRef]
- Olaimat, A.N.; Al-Holy, M.A.; Shahbaz, H.M.; Al-Nabulsi, A.A.; Abu Ghoush, M.H.; Osaili, T.M.; Ayyash, M.M.; Holley, R.A. Emergence of antibiotic resistance in Listeria monocytogenes isolated from food products: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1277–1292. [Google Scholar] [CrossRef]
- Athamna, A.; Athamna, M.; Abu-Rashed, N.; Medlej, B.; Bast, D.J.; Rubinstein, E. Selection of Bacillus anthracis isolates resistant to antibiotics. J. Antimicrob. Chemother. 2004, 54, 424–428. [Google Scholar] [CrossRef]
- Cavallo, J.D.; Ramisse, F.; Girardet, M.; Vaissaire, J.; Mock, M.; Hernandez, E. Antibiotic susceptibilities of 96 isolates of Bacillus anthracis isolated in France between 1994 and 2000. Antimicrob. Agents Chemother. 2002, 46, 2307–2309. [Google Scholar] [CrossRef]
- Munita, J.M.; Bayer, A.S.; Arias, C.A. Evolving resistance among gram-positive pathogens. Clin. Infect. Dis. 2015, 61, S48–S57. [Google Scholar] [CrossRef]
- Koulenti, D.; Xu, E.; Mok, Y.S.I.; Song, A.; Karageorgopoulos, D.E.; Armaganidis, A.; Lipman, J.; Tsiodras, S. Novel antibiotics for multidrug-resistant gram-positive microorganisms. Microorganisms 2019, 7, 270. [Google Scholar] [CrossRef]
- Tan, S.Y.; Tatsumura, Y. Alexander Fleming (1881–1955): Discoverer of penicillin. Singap. Med. J. 2015, 56, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Moutinho, C.G.; Pinto, F.C.; del Fiol, F.S.; Jozala, A.; Chaud, M.V.; Vila, M.M.D.C.; Teixeira, J.A.; Balcão, V.M. Alternatives to overcoming bacterial resistances: State-of-the-art. Microbiol. Res. 2016, 191, 51–80. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to conventional antibiotics in the era of antimicrobial resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.; Low, A.S.; McHugh, R.E.; Muwonge, A.; Stevens, M.P.; Corbishley, A.; Gally, D.L. Alternatives to antibiotics in a one health context and the role genomics can play in reducing antimicrobial use. Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Payne, D.J.; Rappuoli, R.; de Gregorio, E. Technologies to address antimicrobial resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12887–12895. [Google Scholar] [CrossRef]
- Wang, C.-H.; Hsieh, Y.-H.; Powers, Z.M.; Kao, C.-Y. Defeating antibiotic-resistant bacteria: Exploring alternative therapies for a post-antibiotic era. Int. J. Mol. Sci. 2020, 21, 1061. [Google Scholar] [CrossRef]
- Hauser, A.R.; Mecsas, J.; Moir, D.T. Beyond antibiotics: New therapeutic approaches for bacterial infections. Clin. Infect. Dis. 2016, 63, 89–95. [Google Scholar] [CrossRef]
- Twort, F.W. An investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef]
- D’Herelle, F. Sur un microbe invisible antagoniste des bacilles dysenteriques [An invisible microbe that is antagonistic to the dysentery bacillus]. Comptes Rendus 1917, 165, 373–375. [Google Scholar]
- Chanishvili, N. Phage Therapy-History from Twort and d’Herelle Through Soviet Experience to Current Approaches, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 83, ISBN 9780123944382. [Google Scholar]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Fischetti, V.A.; Nelson, D.; Schuch, R. Reinventing phage therapy: Are the parts greater than the sum? Nat. Biotechnol. 2006, 24, 1508–1511. [Google Scholar] [CrossRef]
- Fenton, M.; Ross, P.; Mcauliffe, O.; O’Mahony, J.; Coffey, A. Recombinant bacteriophage lysins as antibacterials. Bioeng. Bugs 2010, 1, 9–16. [Google Scholar] [CrossRef]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, I.; Coutinho, F.H.; Rodriguez-Valera, F. Thousands of novel endolysins discovered in uncultured phage genomes. Front. Microbiol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- De Maesschalck, V.; Gutiérrez, D.; Paeshuyse, J.; Lavigne, R.; Briers, Y. Advanced engineering of third-generation lysins and formulation strategies for clinical applications. Crit. Rev. Microbiol. 2020, 46, 548–564. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Rodríguez-Rubio, L.; Lavigne, R.; Briers, Y. From endolysins to Artilysin®s: Novel enzyme-based approaches to kill drug-resistant bacteria. Biochem. Soc. Trans. 2016, 44, 123–128. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Fleming, A. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1922, 93, 306–317. [Google Scholar] [CrossRef]
- Wang, G. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol. Biol. 2015, 1268, 43–66. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Martínez, B.; Rodríguez, A.; Suárez, E. Antimicrobial peptides produced by bacteria: The bacteriocins. In New Weapons to Control Bacterial Growth; Villa, T.G., Vinas, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 15–38. ISBN 978-3-319-28368-5. [Google Scholar]
- Gratia, A. Sur un remarquable exemple d’antagonisme entre deux souches de coilbacille. C. R. Seances Soc. Biol. Fil. 1925, 93, 1040–1041. [Google Scholar]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins-a viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Dreyer, L.; Smith, C.; van Staden, A.D. A review: The fate of bacteriocins in the human gastro-intestinal tract: Do they cross the gut–blood barrier? Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Fahim, H.A.; Khairalla, A.S.; El-Gendy, A.O. Nanotechnology: A valuable strategy to improve bacteriocin formulations. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Martínez, O.F.; Cardoso, M.H.; Ribeiro, S.M.; Franco, O.L. Recent advances in anti-virulence therapeutic strategies with a focus on dismantling bacterial membrane microdomains, toxin neutralization, quorum-sensing interference and biofilm inhibition. Front. Cell. Infect. Microbiol. 2019, 9, 1–24. [Google Scholar] [CrossRef]
- Nealson, K.H.; Platt, T.; Hastings, J.W. Cellular control of the synthesis and activity of the bacterial luminescent system. J. Bacteriol. 1970, 104, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density- responsive transcriptional regulators. J. Bacteriol. 1994, 176, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Bassler, B.L.; Greenberg, E.P.; Stevens, A.M. Cross-species induction of luminescence in the quorum-sensing bacterium Vibrio harveyi. J. Bacteriol. 1997, 179, 4043–4045. [Google Scholar] [CrossRef]
- Deep, A.; Chaudhary, U.; Gupta, V. Quorum sensing and bacterial pathogenicity: From molecules to disease. J. Lab. Physicians 2011, 3, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Rémy, B.; Mion, S.; Plener, L.; Elias, M.; Chabrière, E.; Daudé, D. Interference in bacterial quorum sensing: A biopharmaceutical perspective. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Kong, C.; Neoh, H.M.; Nathan, S. Targeting Staphylococcus aureus toxins: A potential form of anti-virulence therapy. Toxins 2016, 8, 72. [Google Scholar] [CrossRef]
- Fang, R.H.; Luk, B.T.; Hu, C.M.J.; Zhang, L. Engineered nanoparticles mimicking cell membranes for toxin neutralization. Adv. Drug Deliv. Rev. 2015, 90, 69–80. [Google Scholar] [CrossRef] [PubMed]
- López, D.; Kolter, R. Functional microdomains in bacterial membranes. Genes Dev. 2010, 24, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane microdomain disassembly inhibits MRSA antibiotic resistance. Cell 2017, 171, 1354–1367. [Google Scholar] [CrossRef]
- Koch, G.; Wermser, C.; Acosta, I.C.; Kricks, L.; Stengel, S.T.; Yepes, A.; Lopez, D. Attenuating Staphylococcus aureus virulence by targeting flotillin protein scaffold activity. Cell Chem. Biol. 2017, 24, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS Suppl. 2013, 1–51. [Google Scholar] [CrossRef]
- Ha, M.W.; Yi, S.W.; Paek, S.M. Design and synthesis of small molecules as potent staphylococcus aureus sortase a inhibitors. Antibiotics 2020, 9, 706. [Google Scholar] [CrossRef]
- Hendrickx, A.P.A.; Budzik, J.M.; Oh, S.Y.; Schneewind, O. Architects at the bacterial surface-sortases and the assembly of pili with isopeptide bonds. Nat. Rev. Microbiol. 2011, 9, 166–176. [Google Scholar] [CrossRef]
- Malik, A.; Kim, S.B. A comprehensive in silico analysis of sortase superfamily. J. Microbiol. 2019, 57, 431–443. [Google Scholar] [CrossRef]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V.L. Crystal structures of Staphylococcus aureus Sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Ton-That, H.; Schneewind, O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001, 40, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Paterson, G.K.; Mitchell, T.J. The biology of gram-positive sortase enzymes. Trends Microbiol. 2004, 12, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Schneewind, O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef]
- Foster, T.J.; McDevitt, D. Surface-associated proteins of Staphylococcus aureus: Their possible roles in virulence. FEMS Microbiol. Lett. 1994, 118, 199–205. [Google Scholar] [CrossRef]
- Foster, T.J.; Höök, M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998, 6, 484–488. [Google Scholar] [CrossRef]
- Patel, A.H.; Nowlan, P.; Weavers, E.D.; Foster, T. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 1987, 55, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef]
- Bierne, H.; Mazmanian, S.K.; Trost, M.; Pucciarelli, M.G.; Liu, G.; Dehoux, P.; Jänsch, L.; Garcia-del Portillo, F.; Schneewind, O.; Cossart, P. Inactivation of the SrtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol. Microbiol. 2002, 43, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Kharat, A.S.; Tomasz, A. Inactivation of the SrtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect. Immun. 2003, 71, 2758–2765. [Google Scholar] [CrossRef]
- Vanier, G.; Sekizaki, T.; Domínguez-Punaro, M.C.; Esgleas, M.; Osaki, M.; Takamatsu, D.; Segura, M.; Gottschalk, M. Disruption of SrtA gene in Streptococcus suis results in decreased interactions with endothelial cells and extracellular matrix proteins. Vet. Microbiol. 2008, 127, 417–424. [Google Scholar] [CrossRef]
- Hu, P.; Huang, P.; Chen, W.M. Curcumin inhibits the sortase a activity of the streptococcus mutans UA159. Appl. Biochem. Biotechnol. 2013, 171, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef]
- Nitulescu, G.; Nicorescu, I.M.; Olaru, O.T.; Ungurianu, A.; Mihai, D.P.; Zanfirescu, A.; Nitulescu, G.M.; Margina, D. Molecular docking and screening studies of new natural sortase A inhibitors. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Suree, N.; Yi, S.W.; Thieu, W.; Marohn, M.; Damoiseaux, R.; Chan, A.; Jung, M.E.; Clubb, R.T. Discovery and structure-activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg. Med. Chem. 2009, 17, 7174–7185. [Google Scholar] [CrossRef]
- Selvaraj, C.; Sivakamavalli, J.; Baskaralingam, V.; Singh, S.K. Virtual screening of LPXTG competitive SrtA inhibitors targeting signal transduction mechanism in Bacillus anthracis: A combined experimental and theoretical study. J. Recept. Signal Transduct. 2014, 34, 221–232. [Google Scholar] [CrossRef]
- Davis, B.J.; Erlanson, D.A. Learning from our mistakes: The “unknown knowns” in fragment screening. Bioorg. Med. Chem. Lett. 2013, 23, 2844–2852. [Google Scholar] [CrossRef]
- Kim, S.H.; Shin, D.S.; Oh, M.N.; Chung, S.C.; Lee, J.S.; Chang, I.M.; Oh, K.B. Inhibition of sortase, a bacterial surface protein anchoring transpeptidase, by β-sitosterol-3-O-glucopyranoside from Fritillaria verticillata. Biosci. Biotechnol. Biochem. 2003, 67, 2477–2479. [Google Scholar] [CrossRef]
- Kim, S.H.; Shin, D.S.; Oh, M.N.; Chung, S.C.; Lee, J.S.; Oh, K.B. Inhibition of the bacterial surface protein anchoring transpeptidase sortase by isoquinoline alkaloids. Biosci. Biotechnol. Biochem. 2004, 68, 421–424. [Google Scholar] [CrossRef]
- Oh, K.B.; Mar, W.; Kim, S.; Kim, J.Y.; Oh, M.N.; Kim, J.G.; Shin, D.; Sim, C.J.; Shin, J. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongosorites sp. Bioorg. Med. Chem. Lett. 2005, 15, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Chung, S.C.; Shin, J.; Lee, S.H.; Kim, T.I.; Lee, H.S.; Oh, K.B. Aaptamines as sortase A inhibitors from the tropical sponge Aaptos aaptos. Bioorg. Med. Chem. Lett. 2007, 17, 5366–5369. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.; Yang, W.Y.; Chung, S.C.; Kim, T.Y.; Oh, K.B.; Shin, J. In vitro sortase A inhibitory and antimicrobial activity of flavonoids isolated from the roots of Sophora flavescens. Arch. Pharm. Res. 2011, 34, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Kim, J.G.; Kim, M.R.; Lee, S.E.; Takeoka, G.R.; Oh, K.B.; Kim, J.H. Curcuma longa L. constituents inhibit sortase A and Staphylococcus aureus cell adhesion to fibronectin. J. Agric. Food Chem. 2005, 53, 9005–9009. [Google Scholar] [CrossRef]
- Yang, W.Y.; Won, T.H.; Ahn, C.H.; Lee, S.H.; Yang, H.C.; Shin, J.; Oh, K.B. Streptococcus mutans sortase A inhibitory metabolites from the flowers of Sophora japonica. Bioorg. Med. Chem. Lett. 2015, 25, 1394–1397. [Google Scholar] [CrossRef]
- Lee, S.; Song, I.H.; Lee, J.H.; Yang, W.Y.; Oh, K.B.; Shin, J. Sortase A inhibitory metabolites from the roots of Pulsatilla koreana. Bioorg. Med. Chem. Lett. 2014, 24, 44–48. [Google Scholar] [CrossRef]
- Kang, S.S.; Kim, J.G.; Lee, T.H.; Oh, K.B. Flavonols inhibit sortases and sortase-mediated Staphylococcus aureus clumping to fibrinogen. Biol. Pharm. Bull. 2006, 29, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.E.; Na, Z.; Jung, M.; Lee, H.S.; Sim, C.J.; Nahm, K.; Oh, K.B.; Shin, J. Discorhabdins from the Korean marine sponge Sceptrella sp. J. Nat. Prod. 2010, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Jeon, J.E.; Lee, Y.J.; Lee, H.S.; Sim, C.J.; Oh, K.B.; Shin, J. Sesterterpenes from the tropical sponge Coscinoderma sp. J. Nat. Prod. 2011, 74, 1805–1811. [Google Scholar] [CrossRef]
- Won, T.H.; Jeon, J.E.; Kim, S.H.; Lee, S.H.; Rho, B.J.; Oh, D.C.; Oh, K.B.; Shin, J. Brominated aromatic furanones and related esters from the ascidian synoicum sp. J. Nat. Prod. 2012, 75, 2055–2061. [Google Scholar] [CrossRef]
- Won, T.H.; Jeon, J.E.; Lee, S.H.; Rho, B.J.; Oh, K.B.; Shin, J. Beta-carboline alkaloids derived from the ascidian Synoicum sp. Bioorg. Med. Chem. 2012, 20, 4082–4087. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bi, C.; Cai, H.; Liu, B.; Zhong, X.; Deng, X.; Wang, T.; Xiang, H.; Niu, X.; Wang, D. The therapeutic effect of chlorogenic acid against Staphylococcus aureus infection through sortase A inhibition. Front. Microbiol. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Lee, Y.J.; Han, Y.R.; Park, W.; Nam, S.H.; Oh, K.B.; Lee, H.S. Synthetic analogs of indole-containing natural products as inhibitors of sortase A and isocitrate lyase. Bioorg. Med. Chem. Lett. 2010, 20, 6882–6885. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Y.; Jing, S.; Dong, H.; Wang, D.; Wang, T. Astilbin inhibits the activity of sortase a from Streptococcus mutans. Molecules 2019, 24, 465. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, L.; Xu, N.; Zhou, S.; Song, Y.; Yang, Q.; Liu, Y.; Yang, Y.; Ai, X. Rutin reduces the pathogenicity of Streptococcus agalactiae to tilapia by inhibiting the activity of sortase A. Aquaculture 2021, 530, 735743. [Google Scholar] [CrossRef]
- Oh, K.B.; Kim, S.H.; Lee, J.; Cho, W.J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule Sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef]
- Oh, K.B.; Nam, K.W.; Ahn, H.; Shin, J.; Kim, S.; Mar, W. Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl) acrylonitrile (DMMA) against Staphylococcus aureus infection in a murine model. Biochem. Biophys. Res. Commun. 2010, 396, 440–444. [Google Scholar] [CrossRef]
- Oh, K.B.; Oh, M.N.; Kim, J.G.; Shin, D.S.; Shin, J. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 2006, 70, 102–106. [Google Scholar] [CrossRef]
- Maresso, A.W.; Wu, R.; Kern, J.W.; Zhang, R.; Janik, D.; Missiakas, D.M.; Duban, M.-E.; Joachimiak, A.; Schneewind, O. Activation of inhibitors by sortase triggers irreversible modification of the active site. J. Biol. Chem. 2007, 282, 23129–23139. [Google Scholar] [CrossRef]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and structure-activity relationship studies of irreversible benzisothiazolinone-based inhibitors against Staphylococcus aureus sortase A transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef]
- Wehrli, P.M.; Uzelac, I.; Olsson, T.; Jacso, T.; Tietze, D.; Gottfries, J. Discovery and development of substituted thiadiazoles as inhibitors of Staphylococcus aureus Sortase A. Bioorg. Med. Chem. 2019, 27, 115043. [Google Scholar] [CrossRef]
- Maggio, B.; Raffa, D.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Schillaci, D.; Cusimano, M.G.; Leonchiks, A.; Zhulenkovs, D.; Basile, L.; et al. Discovery of a new class of sortase a transpeptidase inhibitors to tackle gram-positive pathogens: 2-(2-phenylhydrazinylidene)alkanoic acids and related derivatives. Molecules 2016, 21. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, T.; Guan, X.N.; Dong, Z.; Lan, L.; Yang, S.; Yang, C.G. Tideglusib and its analogues as inhibitors of Staphylococcus aureus SrtA. J. Med. Chem. 2020, 63, 8442–8457. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, P.; He, X.; Yuan, Z.W.; Yin, Z.Q.; Fu, H.; Lin, J.; He, C.; Liang, X.; Lv, C.; Shu, G.; et al. Erianin against staphylococcus aureus infection via inhibiting sortase A. Toxins 2018, 10, 385. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Pan, J.; Dong, J.; Zhou, X.; Niu, X.; Deng, X. Oligopeptide targeting sortase a as potential anti-infective therapy for Staphylococcus aureus. Front. Microbiol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chenna, B.C.; Shinkre, B.A.; King, J.R.; Lucius, A.L.; Narayana, S.V.L.; Velu, S.E. Identification of novel inhibitors of bacterial surface enzyme Staphylococcus aureus Sortase A. Bioorg. Med. Chem. Lett. 2008, 18, 380–385. [Google Scholar] [CrossRef]
- Thappeta, K.R.V.; Zhao, L.N.; Nge, C.E.; Crasta, S.; Leong, C.Y.; Ng, V.; Kanagasundaram, Y.; Fan, H.; Ng, S.B. In-silico identified new natural sortase a inhibitors disrupt s. Aureus biofilm formation. Int. J. Mol. Sci. 2020, 21, 8601. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Wereszczynski, J.; Amer, B.R.; Yi, S.W.; Jung, M.E.; Mccammon, J.A.; Clubb, R.T. Discovery of staphylococcus aureus sortase a inhibitors using virtual screening and the relaxed complex scheme. Chem. Biol. Drug Des. 2013, 82, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Chenna, B.C.; King, J.R.; Shinkre, B.A.; Glover, A.L.; Lucius, A.L.; Velu, S.E. Synthesis and structure activity relationship studies of novel Staphylococcus aureus Sortase A inhibitors. Eur. J. Med. Chem. 2010, 45, 3752–3761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Zhu, K.; Gong, S.; Dramsi, S.; Wang, Y.T.; Li, J.; Chen, F.; Zhang, R.; Zhou, L.; et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 2014, 111, 13517–13522. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Fouts, D.E.; Archer, G.L.; Mongodin, E.F.; DeBoy, R.T.; Ravel, J.; Paulsen, I.T.; Kolonay, J.F.; Brinkac, L.; Beanan, M.; et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005, 187, 2426–2438. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Biswas, T.; Das, S.; Marathe, U.; Sehgal, D.; Roy, R.P.; Suryanarayanarao, R. Crystallization and preliminary X-ray diffraction studies of sortase A from Streptococcus pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1195–1198. [Google Scholar] [CrossRef]
- Jaudzems, K.; Kurbatska, V.; Jekabsons, A.; Bobrovs, R.; Rudevica, Z.; Leonchiks, A. Targeting bacterial Sortase A with covalent inhibitors: 27 new starting points for structure-based hit-to-lead optimization. ACS Infect. Dis. 2020, 6, 186–194. [Google Scholar] [CrossRef]
- Chan, A.H.; Yi, S.W.; Weiner, E.M.; Amer, B.R.; Sue, C.K.; Wereszczynski, J.; Dillen, C.A.; Senese, S.; Torres, J.Z.; McCammon, J.A.; et al. NMR structure-based optimization of Staphylococcus aureus sortase A pyridazinone inhibitors. Chem. Biol. Drug Des. 2017, 90, 327–344. [Google Scholar] [CrossRef]
- Tonge, P.J. Quantifying the interactions between biomolecules: Guidelines for assay design and data analysis. ACS Infect. Dis. 2019, 5, 796–808. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).