1. Introduction
Malaria remains a life-threatening disease, causing millions of cases worldwide with around 405,000 deaths annually [
1]. There are five different species of the
Plasmodium parasite causing human malaria, namely
Plasmodium falciparum,
Plasmodium vivax,
Plasmodium ovale,
Plasmodium malariae and
Plasmodium knowlesi. The most severe form is caused by
P. falciparum, as it often leads to death and can be fatal not long after the first symptoms [
2]. The increase and spread of multidrug-resistant
P. falciparum have become major challenges to malaria treatment and are correlated with increases in morbidity and mortality in many malaria-endemic countries [
1]. In response to this harsh predicament, artemisinin-based combination therapies (ACTs) have been recommended and widely established in malaria-endemic regions. Unfortunately, the recent Global Malaria Programme report pointed to artemisinin and ACT resistance causing significant delays in parasite clearance [
3]. The development of novel antimalarial drugs and more effective therapy is urgently needed.
The
Plasmodium mitochondrion contains the smallest eukaryotic mtDNA (6-kb), encoding only three genes for proteins and highly fragmented rRNA genes [
4]. The differences in the mitochondria of malaria parasites from those of their vertebrate hosts allow for drug selectivity and potential targets for antimalarial drug development. Atovaquone is well known for malaria treatment, acts on the malaria mitochondrion, and has a very low frequency of side effects [
5]. Even though the parasite has developed resistance to atovaquone, it maintains high efficiency when used in combination with proguanil for prophylaxis against malaria in endemic areas [
6].
In the past ten years, it has been shown that several peptides can act on malarial parasites. The small linear angiotensin I and II-derived peptides exhibit antiplasmodial activity against the blood stage of both avian
P. gallinaceum and
P. falciparum [
7,
8]. Symplostatin 4 (Sym4), a marine-derived peptide, kills
P. falciparum at nanomolar concentrations, generating a swollen food vacuole and inhibiting the initial stage of hemoglobin degradation [
9]. Moreover, several host defense or antimicrobial peptides have been reported to disturb malaria homeostasis by disrupting parasite cellular membranes or interfering with key processes of parasite metabolism, and surprisingly kill the parasite [
10,
11]. One example is dermaseptin and its truncated derivatives, which exert anti-
P. falciparum activity in the micromolar range by permeabilization of the cell membrane [
12,
13]. These antiplasmodial peptides, both natural and synthetic molecules, vary in size, charge, amino acid composition, hydrophobicity and secondary structure, and some of them appear to act selectively on infected erythrocytes and/or intraerythrocytic parasite membranes [
11]. This implies that membrane-active peptides, such as cell-penetrating peptides (CPPs) or mitochondria-penetrating peptides (MPPs), which can translocate into cells without membrane damage, may be novel compounds for development as antimalarial agents.
Mitochondria-penetrating peptides or MPPs are a class of peptides with the ability to enter human cells and specifically target the mitochondria. The combination of two major characteristics, being cationic and lipophilic, drives the permeation of MPPs through the hydrophobic mitochondrial membrane [
14]. The mechanism of MPPs depends on their concentration. The peptide binds to the membrane’s outer leaflet at low concentrations while crossing the hydrophobic bilayer and diffuses into both leaflets at high concentrations. Moreover, MPPs can cross to the inner leaflet without disturbing the lamellarity and transmembrane, which offer adequate energy for peptides to cross the hydrophobic core [
15]. The characteristics and actions of MPPs lead to antimicrobial or antimalarial activities. One such short peptide, (L-cyclohexyl alanin-D-arginine)
3 or (F
xr)
3, exhibited efficient and promising cell penetration and mitochondrial localization [
14]. The objective of this study was to determine the antimalarial activity of (F
xr)
3 and elucidate its localization in
P. falciparum. Our findings suggest that (F
xr)
3 might be developed as a new antimalarial agent for use alone or in combination with current antimalarial agents.
3. Discussion
Malaria mitochondrion could be an attractive and selective target for the development of antimalarial drugs, as it plays a key role in energy production and is different from those of their vertebrate hosts. Even though the outer membrane of a mitochondrion is semi-permeable, allowing free access of small and large molecules up to 5 kDa, the inner membrane maintains a very tight barrier to the transport of many small and large molecules (including drugs) to the mitochondrial matrix [
16]. Mitochondria-penetrating peptides or MPPs have been developed to accelerate the transport of molecules into the human mitochondrion [
14]. MPPs can target and penetrate the mitochondria of active eukaryotic cells by two major properties, being cationic and lipophilic. These characteristics may explain the antiplasmodial activity of our peptide. In order to exploit MPPs as antimalarial agents, they must also be able to pass through the red blood cell membrane without disruption or destruction. After passing through this membrane, they must penetrate the parasite membrane and specifically target the mitochondria. However, mitochondria of malaria exhibit some extraordinary evolutionary and functional features, such as the membrane composition, which is different from that of eukaryotic cells [
5]. Moreover, the mitochondrial characteristics of each stage of the parasite are different, so the ability of MPPs to act on parasite mitochondrion must be observed. In this study, a promising mitochondria-targeting peptide, known as (L-cyclohexyl alanine-D-arginine)
3 or (F
xr)
3, was selected for elucidating the antimalarial activity of MPPs.
(F
xr)
3 exhibited antimalarial activity against both chloroquine-sensitive (3D7) and resistant (K1) strains of
P. falciparum with, notably, more effective antiplasmodial activity toward late-stage parasites as shown by IC
50 values. This might be due to more active mitochondria in early-stage parasites. There was a report on the stage-specific presence of cristae inside
P. falciparum mitochondrion. In ring-stage parasites, the mitochondrion is elongated, acristate with low electron density. In trophozoites, the mitochondrion is significantly larger with an unchanged appearance from that of the ring stage. In mature schizonts, each daughter merozoite harbors one small, acristate, electron-lucent mitochondrion in close proximity to one four-membrane-bound apicoplast [
17]. There are discrepancies in the IC
50 results between strains K1 and 3D7. The antimalarial drug susceptibility of both strains revealed that they showed different sensitivity to antimalarial drugs, including chloroquine, piperine, mefloquine and artesunate. Strain K1 is highly resistant to chloroquine (IC
50 is 115.0 nM), while strain 3D7 is very sensitive (IC
50 of 3.9 nM). In contrast, strain K1 exhibited lower sensitivity toward other antimalarial drugs such as piperine and artesunate. The IC
50 of strains K1 and 3D7 against mefloquine were 4.22 nM and 14.6 nM, respectively. The IC
50 of strains K1 and 3D7 against artesunate were 1.06 nM and 2.48 nM, respectively [
18]. It has been noted that the lowest IC
50 of (F
xr)
3 toward chloroquine-resistant
P. falciparum, K1 strain, was only 59.16 ng/mL. The IC
50 values are within the nM range and are comparable to the IC
50 values of established drugs toward different laboratory and field-isolated strains of
P. falciparum such as chloroquine (9.8–744 nM) and artemisinin monomer (6.6–42.5 nM) and their derivatives: dihydroartemisinin or DHA (2.09–14.8 nM) and artesunic acid or ARS (0.82 nM) [
19]. In general, it can be said that (F
xr)
3 exhibits high antimalarial activities against chloroquine-resistant strains of
P. falciparum.
The action of (F
xr)
3 was confirmed by cellular localization at the mitochondria of living malaria parasites. (F
xr)
3 exhibited efficient cellular uptake and specific mitochondrial localization in human cells such as HeLa and MRC-5 cells [
14]. Late-stage parasites (trophozoite and schizont) displayed higher amounts of (F
xr)
3 localized in mitochondria than did ring stage (early-stage), and this might be due to the large mitochondrion of trophozoite and schizont compared to that of the ring stage [
17]. This was in accordance with the higher antimalarial activity of (F
xr)
3 toward parasites observed in late-stage. The antimalarial action of (F
xr)
3 might be similar to that of atovaquone (ATQ), which caused a collapse of mitochondrial membrane potential. ATQ targets cytochrome
b, which plays a role in electron transport during mitochondrial respiration [
20] and functions by inhibiting mitochondrial electron transport and depolarizing malarial mitochondria leading to cell damage and death, while showing no effect on mammalian mitochondrial membrane potential [
5]. However, ATQ is expensive and subject to relatively facile development of resistance [
21]. The target of (F
xr)
3 in the parasite’s mitochondrion is still unelucidated and needs to be identified in order to pave the way for the development of new antimalarial agents. MPPs have been widely applied for gene and drug delivery to mitochondria, especially in conjunction with anticancer drugs such as cisplatin [known as MPP-cisplatin conjugate (mtPt)], which can induce apoptosis by damaging mitochondrial DNA without damaging nuclear DNA and overcomes cisplatin resistance in cancer cells [
22].
(F
xr)
3 could be an alternative choice for development as an antimalarial drug attacking mitochondria or used in combination with ATQ. Antagonism between ATQ and antimalarial drugs (quinolines and artesunate) has been previously reported [
23]. Additionally, atovaquone-proguanil or malarone is prescribed as a fixed-dose prophylactic agent for travelers in malaria-endemic areas and used for treating uncomplicated malaria [
21]. The combination study of ATQ and (F
xr)
3 will be further elucidated to identify a potential benefit of antimitochondrial effects. By comparing the death rate of parasites after different time intervals, (F
xr)
3 was found to kill
P. falciparum strain K1 more rapidly than did ATQ with significant differences at 24 and 48 h. The combination of ATQ and (F
xr)
3, focused on the same target, could rapidly kill parasites throughout their eukaryotic life cycle. There are reports of substantial genetic differentiation among mitochondrial genomes of
P. vivax collected from different regions [
24] and the difficulty in vitro culturing due to the low amounts and mixed stages of the parasite. Therefore, knowledge of the parasite’s biology was far less than that of
P. falciparum. However, the usefulness of (F
xr)
3 as an anti-vivax agent still needs to be explored, as drugs with the same target (such as ATQ) can kill
P. vivax with an IC
50 value of 30 nM [
25].
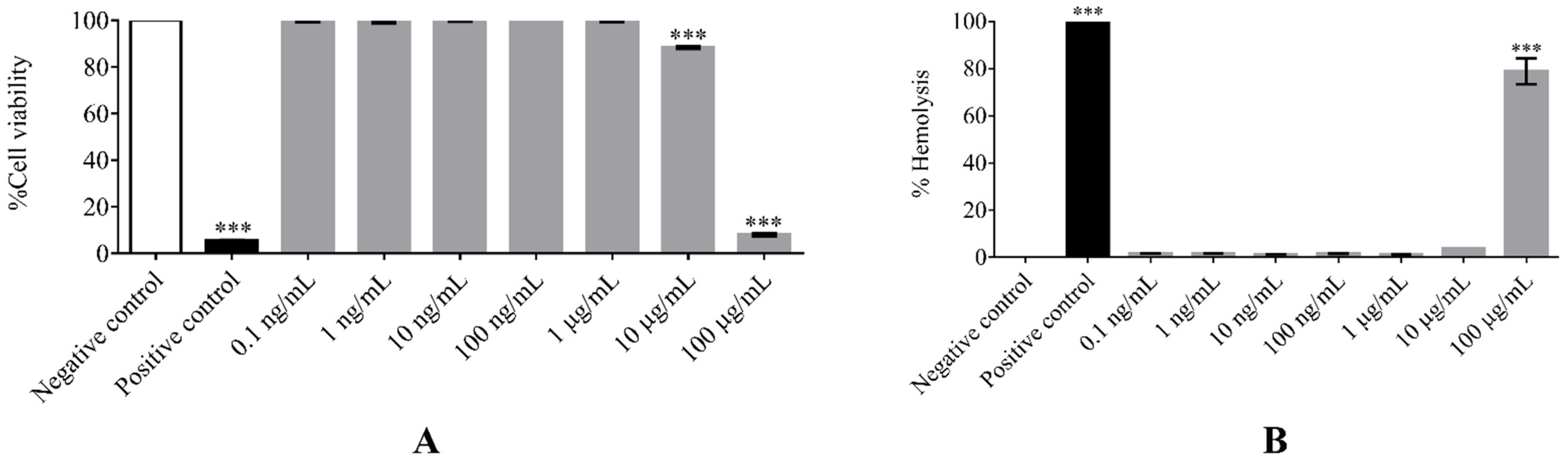
As evaluated by cytotoxicity assays, (F
xr)
3 showed very low toxicity toward normal mouse fibroblast L929 and human peripheral white blood cells at concentrations ranging from 1 ng/mL to 10 µg/mL. Moreover, it did not exhibit any toxicity toward red blood cells. This might be due to the cationic and lyophilic characteristics of (F
xr)
3 facilitating cellular uptake and permeation of the hydrophobic mitochondrial membrane without cytotoxic activity [
14]. Even though (F
xr)
3 at the highest concentration tested (100 µg/mL) exhibited cytotoxicity and hemolytic activity, this concentration was around one thousand times higher than that of antimalarial activity. Several studies found that MPPs have low toxicity both in vitro and in vivo [
14,
26,
27], suggesting the applicability of MPPs as delivery vectors and/or bioactive compounds. Besides toxicity, the sensitivity of peptides to protease degradation is a critical concern. Hemolysis is a hallmark of malaria infection and can release proteases. Moreover, there are several proteases released from parasites/lysing host cells and present in plasma/serum. Fortunately, the sequence of (F
xr)
3, which is composed of L-cyclohexylalanine and an artificial arginine residue, is not cleaved by endogenous protease [
28].
Our results demonstrated that (Fxr)3 can pass through the red blood cell membrane without disruption or destruction, and subsequently kill the blood stage of P. falciparum. (Fxr)3 showed more potent antimalarial activity toward late-stage (trophozoite and schizont) parasites, consistent with high intensity of (Fxr)3 localized in the parasites’ mitochondria observed by confocal microscopy. Moreover, (Fxr)3 exhibited high antimalarial activity against the chloroquine-resistant K1 strain of P. falciparum and showed very low toxicity toward normal mouse fibroblast L929 cells, human peripheral white blood cells (PBMCs) and red blood cells. To our knowledge, there is no study on the sensitivity and specificity of (Fxr)3 toward the malaria parasite, P. falciparum. This is the first report on the antimalarial activity of (Fxr)3. (Fxr)3 or other MPPs might be developed as new antimalarial agents or used in combination with current antimalarial agents targeting the mitochondrion, such as atovaquone.
4. Materials and Methods
4.1. Culture of Parasite
Plasmodium falciparum strains 3D7 (CQ-sensitive clone), K1 (CQ-resistance clone) and two field-isolated strains (J1 and MR4) were used in this study. They were kindly provided by the Department of Parasitology, Phramongkutklao College of Medicine, Thailand. All isolate strains were continuously cultured using standard methods with modifications [
29]. For parasite culture, human blood group O
+ RBCs were used, and white blood cells were removed by washing with RPMI 1640. Complete culture medium was prepared by adding 10 mL of 0.5% albumax II (Invitrogen, Waltham, MA, USA) to each 200 mL aliquot of stock RPMI 1640 medium (Gibco URL, Waltham, MA, USA) containing 2.05 mM L-glutamine, 25 mM HEPES, 21 mM sodium bicarbonate and gentamycin (5 g/mL). A 5% parasitemia and 5% hematocrit were maintained under 5% CO
2, 3% O
2 and 92% N2 at 37 °C. Five percent D-sorbitol (Sigma, St. Louis, MO, USA) was used for parasite synchronization by adding 10 mL of 5% D-sorbitol per 1 mL packed red cells and incubating at 37 °C for 30 min, and after that washing with RPMI1640 at 2000×
g for 5 min.
4.2. Mitochondria-Penetrating Peptide
A short peptide (L-cyclohexyl alanine-D-arginine)
3 or (F
xr)
3, composed of alternating hydrophobic cyclohexylalanine (F
x) and D-arginine (r), was used as a mitochondria-penetrating peptide (MPP) in this study [
14]. It was kindly provided by Professor Dr. Shana O. Kelley, University of Toronto, Canada. Synthesis of the peptide was performed by using solid-phase synthesis on Rink amide resin (0.6–0.7mmol/g, 100,200 mesh) (NovaBiochem, Darmstadt, Germany). The peptide was purified by reversed-phase high-pressure liquid chromatography, and the purity was greater than or equal to 95%. Liquid chromatography-mass spectrometry (LC-MS) coupled with electron-spray ionization was used to confirm the identity of the conjugate.
4.3. Cytotoxicity Determination
The cytotoxic activity of (Fxr)3 was assessed with the L929 mouse skin fibroblast cell line. Cells were cultured in RPMI 1640 medium with 10% fetal bovine serum, 2 mM of L-glutamine, 100 U/mL of penicillin and 100 mg/mL of streptomycin at 37 °C. Cells were incubated at 37 °C in a humidified incubator with 5% CO2. Confluent cells were treated with 0.5% trypsin solution for detachment, diluted with 10% serum-containing medium. The cells were then plated in 96-well culture plates at 30,000 cells/cm2. Culture plates were incubated for 24 h at 37 °C in a humidified incubator with 5% CO2 prior to stimulation of the cells with the compound. After that, L929 was incubated with ten-fold serial dilutions of (Fxr)3 with concentrations ranging from 0.1 ng/mL-100 μg/mL. Viability of the cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium or MTT assay. After removing the medium, cells were incubated with MTT solution (5 mg/mL in PBS) for 4 h and the resulting formazan was solubilized with DMSO (100 μL). Formazan absorbance was measured at 550 nm using an automated microplate reader. Cell viability was expressed as a percentage of the control culture value. Untreated cells and cells treated with 20% DMSO were used as negative and positive controls, respectively. All experiments were carried out in triplicate.
4.4. Peripheral White Blood Cell Toxicity Assay
Blood group O+ human PBMCs were isolated from fresh heparinized blood by Lymphoprep (r = 1.077 g/mL; Axis-Shield, Oslo, Norway). Lymphocytes were isolated from buffy coats by Lymphoprep followed by staining with Trypan Blue and number counting to determine cell survival (live and dead cells) with 96% live cell. Human lymphocytes were conserved in RPMI 1640 medium until use, no longer than two weeks. Cells were plated on 96-well plates, 1000 cells per well, and co-incubated with ten-fold serial dilution of (Fxr)3 with concentrations ranging from 0.1 ng/mL–100 μg/mL, at 37 °C for 24 h. Supernatants were collected after incubation, and the viability of the cells was determined by MTT assay as described above.
4.5. Hemolytic Assay
The human red blood cells (hRBCs) (2% vol/vol) were incubated with ten-fold serial dilutions of (Fxr)3 with concentrations ranging from 0.1 ng/mL–100 μg/mL at 37 °C for 24 h. Supernatants were collected after centrifugation at 2000× g for 10 min, and the absorbance was determined by spectrophotometry at 540 nm (Labomed Inc, Los Angeles, CA, USA). The values were converted to % hemolysis by comparing to the control (100% hydrolysis) using distilled water (DW).
4.6. In Vitro Drug Sensitivity Assay
Sensitivities of
P. falciparum to (F
xr)
3 and atovaquone (ATQ) were investigated based on the incorporation of [
3H] hypoxanthine into parasite nucleic acids or radioisotopic technique [
30]. The level of radioactivity uptake was used as the index of parasite growth. All strains were maintained in continuous culture at 5% parasite, 5% hematocrit using type O
+ human erythrocytes in RPMI 1640 with 0.5% Albumax (Invitrogen), 25 mM HEPES (Sigma), under a 3% O
2, 4% CO
2 and 93% N2 gas mixture. Parasites (1% hematocrit) were incubated with ten-fold serial dilutions of (F
xr)
3 or ATQ in the microtiter plate at 37 °C. At the end of the incubation period, the plates were removed from the chamber, and a final concentration of 50 µCi [
3H] hypoxanthine (NEN, Boston, MA, USA) was added to each well. The plates were gently agitated to ensure adequate mixing of the parasite/drug solutions with the radiolabeled hypoxanthine and thereafter placed back into the incubation chamber and gassed for 5 min prior to incubation at 37 °C for 24 h. After incubation, the assay plates were harvested onto filtermats (Wallac A printed Filltermats, Turku, Finland), using a Tomtec March III M semi-automatic harvester. The filtermats were subsequently removed from the harvester and dried at 60 °C in an oven prior to scintillation counting. Each dry filtermat was placed inside a plastic sample bag, and 1 mL of a Beta-plate Scint (Wallac, Turku, Finland) was added on top of it. The bag was then sealed before being heated in a 1495-021 Microsealer (Wallac, Finland). Each filtermat was placed in a cassette, and the radioactivity measured using a 1450 Micro-Beta Trilux liquid scintillation and luminescence counter (Wallac, Turku, Finland).
4.7. (Fxr)3 Localization
The late stage of P. falciparum strain K1 was incubated with (Fxr)3 linked with TAMRA or TM-(Fxr)3 at concentration of the IC50 for 4 h. Hoechst 3342 (Sigma, Ronkonkoma, NY, USA), at a final concentration of 10 μg/mL, was added into the cultures in order to determine the live parasite cells. Mitotracker green (Cell Signalling, Danvers, MA, USA) was added to achieve a final concentration of 400 nM and incubated for 30 min at 37 °C. Then, the cells were washed two times with PBS pH 7.4. Images were taken with confocal laser scanning (ZEISS LSM 700). The excitation wavelength for visualization for Hoechst 33,342 was 355 nm, and emissions were collected at 461 nM. The excitation wavelength for visualization of the mitotracker green was 490 nm, and emissions were collected at 516 nM. The excitation wavelength for visualization TAMRA or (Fxr)3 was 555 nm, and emissions were collected at 580 nM.
4.8. Data Analysis
All experiments were performed in triplicate, and the results are presented as mean ± standard deviation (SD). The data were analyzed by one-way analysis of variance (ANOVA) with GraphPad Prism version 5; p-value < 0.05 was considered statistically significant.


 and
and 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}