
Streptococcus sputorum, a Novel Member of Streptococcus with Multidrug Resistance, Exhibits Cytotoxicity

Abstract
1. Introduction
2. Results and Discussion
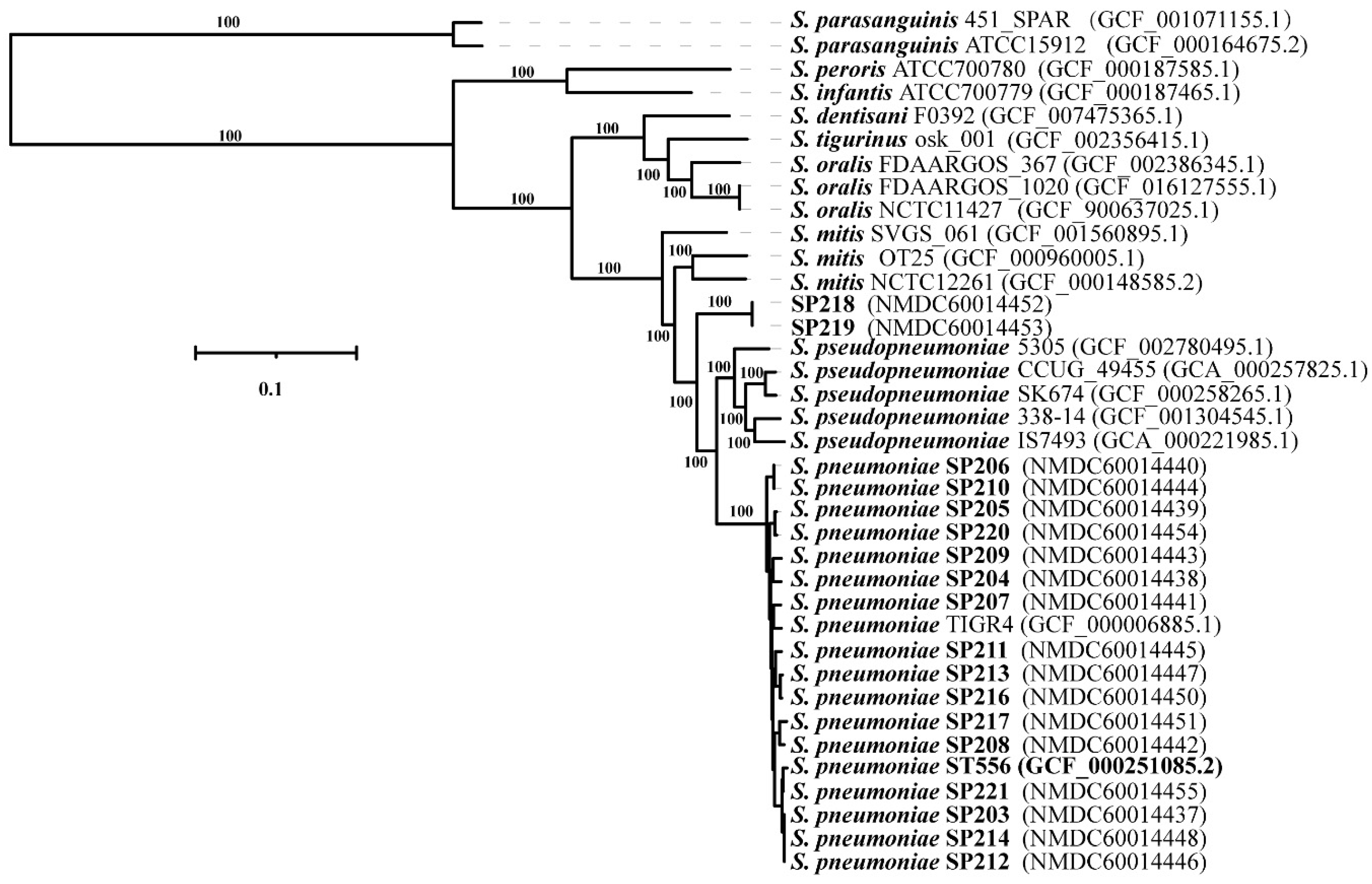
2.1. Phylogenetic Analysis of the Isolates
2.2. Morphological and Physiological Characterization of the New Species
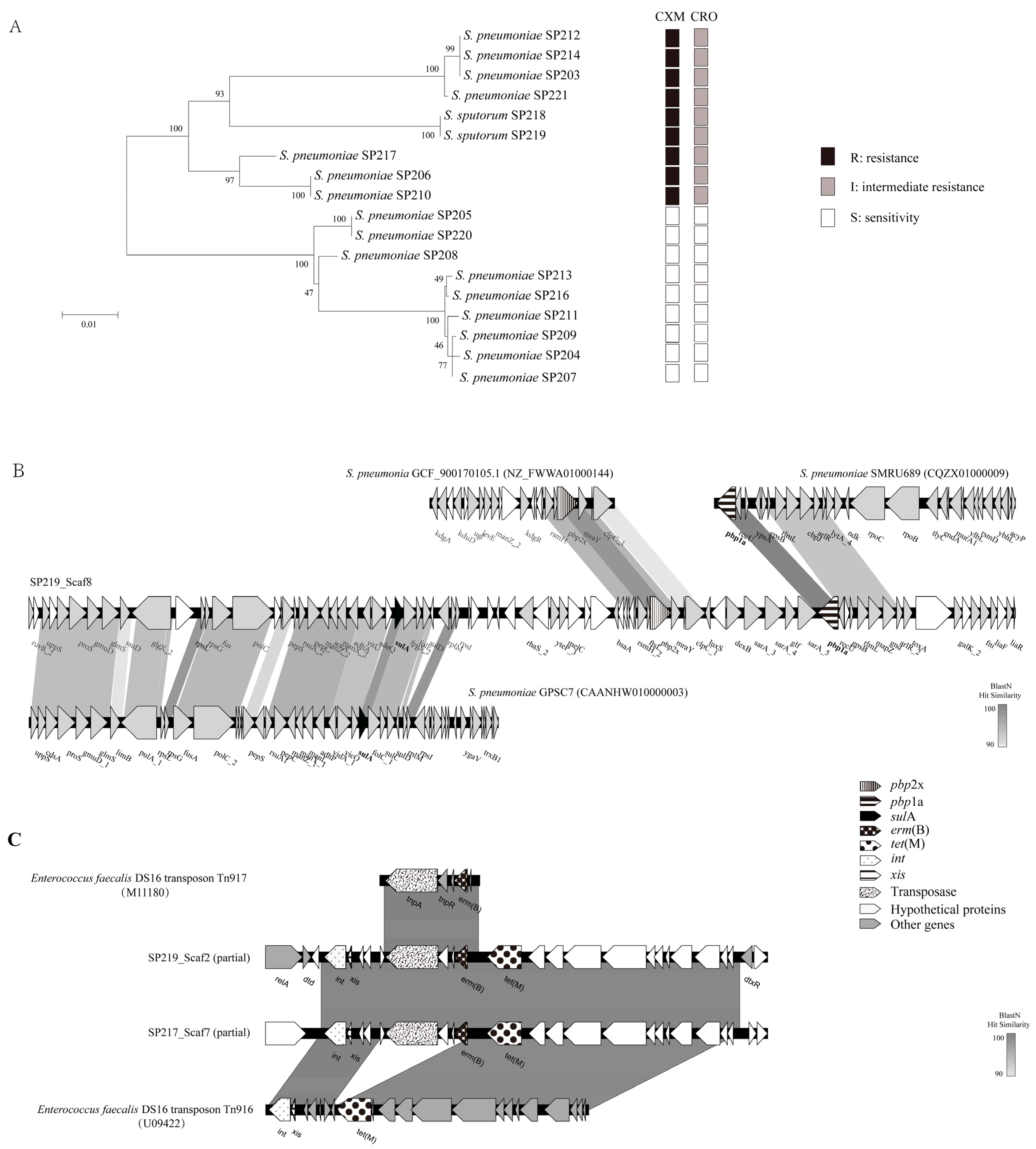
2.3. Antibiotic Resistance via HGT
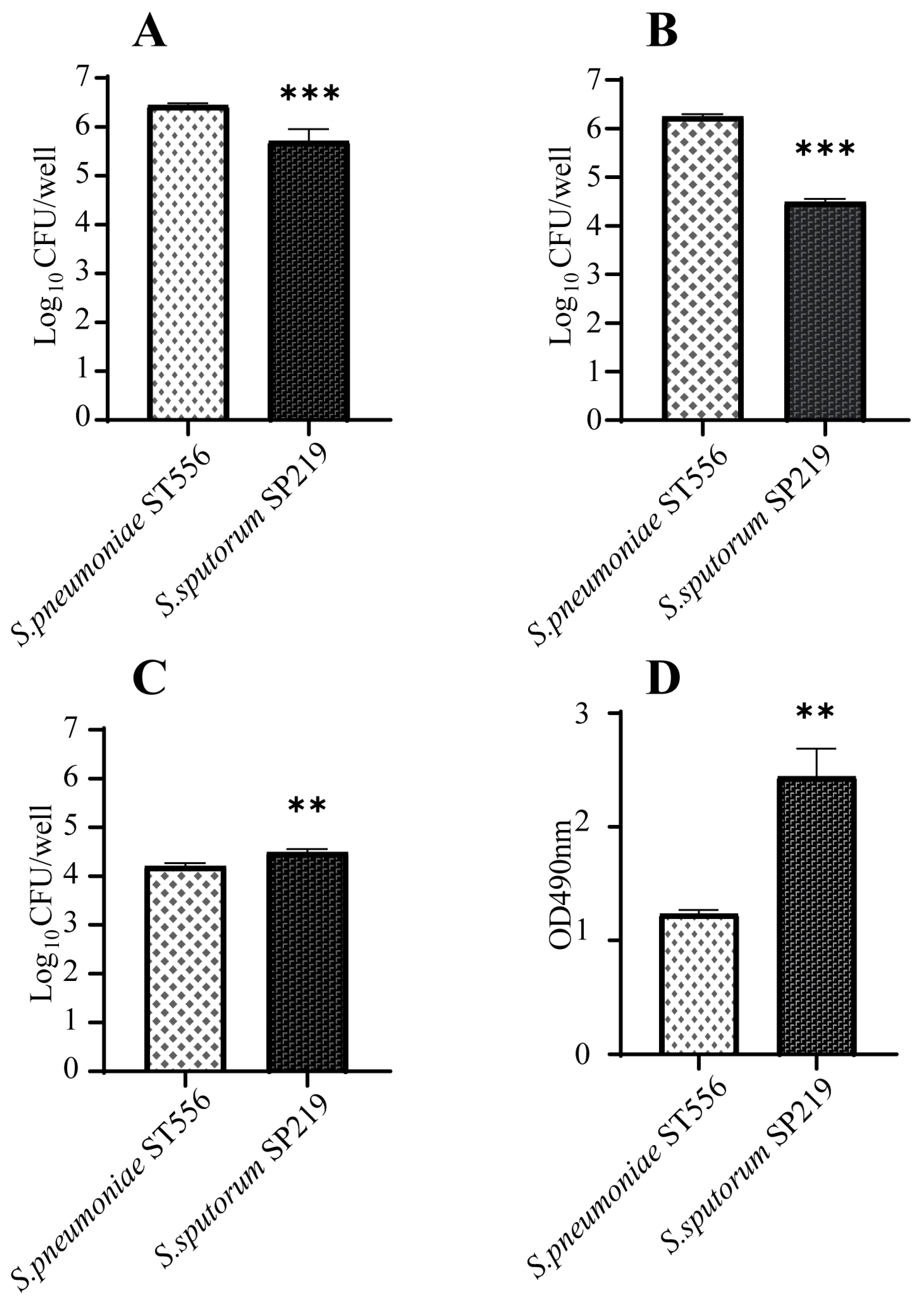
2.4. Virulence Genes and Cytotoxicity of the New Species
3. Conclusions
4. Materials and Methods
4.1. Bacterial Strains, MIC Determination, and Morphological and Physiological Characterization of the Novel Strain
4.2. Whole-Genome Sequencing and Genome Analysis
4.3. Cell Adhesion and Invasion Assays
4.4. LDH Release Test
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Nucleotide Sequence Accession Numbers
References
- Murdoch, D.R.; Corey, G.R.; Hoen, B.; Miró, J.M.; Fowler, V.G., Jr.; Bayer, A.S.; Karchmer, A.W.; Olaison, L.; Pappas, P.A.; Moreillon, P.; et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: The International Collaboration on Endocarditis-Prospective Cohort Study. Arch. Intern Med. 2009, 169, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Fuursted, K.; Littauer, P.J.; Greve, T.; Scholz, C.F. Septicemia with Streptococcus pseudopneumoniae: Report of three cases with an apparent hepatic or bile duct association. Infect. Dis. 2016, 48, 636–639. [Google Scholar] [CrossRef]
- Jensen, A.; Scholz, C.F.P.; Kilian, M. Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int. J. Syst. Evol. Microbiol. 2016, 66, 4803–4820. [Google Scholar] [CrossRef]
- Kitten, T.; Munro, C.L.; Zollar, N.Q.; Lee, S.P.; Patel, R.D. Oral streptococcal bacteremia in hospitalized patients: Taxonomic identification and clinical characterization. J. Clin. Microbiol. 2012, 50, 1039–1042. [Google Scholar] [CrossRef][Green Version]
- Shelburne, S.A.; Sahasrabhojane, P.; Saldana, M.; Yao, H.; Su, X.; Horstmann, N.; Thompson, E.; Flores, A.R. Streptococcus mitis strains causing severe clinical disease in cancer patients. Emerg. Infect. Dis. 2014, 20, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Nemoto, R.; Kodana, M.; Tarumoto, N.; Sakai, J.; Kawamura, T.; Ikebuchi, K.; Mitsutake, K.; Murakami, T.; Maesaki, S.; et al. Rapid and Accurate Species Identification of Mitis Group Streptococci Using the MinION Nanopore Sequencer. Front. Cell Infect. Microbiol. 2020, 10, 11. [Google Scholar] [CrossRef]
- Arbique, J.C.; Poyart, C.; Trieu-Cuot, P.; Quesne, G.; Carvalho Mda, G.; Steigerwalt, A.G.; Morey, R.E.; Jackson, D.; Davidson, R.J.; Facklam, R.R. Accuracy of phenotypic and genotypic testing for identification of Streptococcus pneumoniae and description of Streptococcus pseudopneumoniae sp. nov. J. Clin. Microbiol. 2004, 42, 4686–4696. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, A.; Mueller, N.J.; Tarr, P.E.; Eich, G.; Schulthess, B.; Bahlmann, A.S.; Keller, P.M.; Bloemberg, G.V. Streptococcus tigurinus, a novel member of the Streptococcus mitis group, causes invasive infections. J. Clin. Microbiol. 2012, 50, 2969–2973. [Google Scholar] [CrossRef]
- Camelo-Castillo, A.; Benítez-Páez, A.; Belda-Ferre, P.; Cabrera-Rubio, R.; Mira, A. Streptococcus dentisani sp. nov., a novel member of the mitis group. Int. J. Syst. Evol. Microbiol. 2014, 64, 60–65. [Google Scholar] [CrossRef]
- Gao, X.Y.; Zhi, X.Y.; Li, H.W.; Klenk, H.P.; Li, W.J. Comparative genomics of the bacterial genus Streptococcus illuminates evolutionary implications of species groups. PLoS ONE 2014, 9, e101229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ju, Y.; Tang, N.; Li, Y.; Zhang, G.; Song, Y.; Fang, H.; Yang, L.; Feng, J. Systematic analysis of supervised machine learning as an effective approach to predicate β-lactam resistance phenotype in Streptococcus pneumoniae. Brief. Bioinform. 2020, 21, 1347–1355. [Google Scholar] [CrossRef]
- Kilian, M.; Riley, D.R.; Jensen, A.; Brüggemann, H.; Tettelin, H. Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. mBio 2014, 5, e01490-14. [Google Scholar] [CrossRef] [PubMed]
- Chi, F.; Nolte, O.; Bergmann, C.; Ip, M.; Hakenbeck, R. Crossing the barrier: Evolution and spread of a major class of mosaic pbp2x in Streptococcus pneumoniae, S. mitis and S. oralis. Int. J. Med. Microbiol. 2007, 297, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Sauerbier, J.; Maurer, P.; Rieger, M.; Hakenbeck, R. Streptococcus pneumoniae R6 interspecies transformation: Genetic analysis of penicillin resistance determinants and genome-wide recombination events. Mol. Microbiol. 2012, 86, 692–706. [Google Scholar] [CrossRef]
- Brenciani, A.; Bacciaglia, A.; Vecchi, M.; Vitali, L.A.; Varaldo, P.E.; Giovanetti, E. Genetic elements carrying erm(B) in Streptococcus pyogenes and association with tet(M) tetracycline resistance gene. Antimicrob. Agents Chemother. 2007, 51, 1209–1216. [Google Scholar] [CrossRef]
- Brueggemann, A.B.; Coffman, S.L.; Rhomberg, P.; Huynh, H.; Almer, L.; Nilius, A.; Flamm, R.; Doern, G.V. Fluoroquinolone resistance in Streptococcus pneumoniae in United States since 1994–1995. Antimicrob. Agents Chemother. 2002, 46, 680–688. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Keith, E.R.; Podmore, R.G.; Anderson, T.P.; Murdoch, D.R. Characteristics of Streptococcus pseudopneumoniae isolated from purulent sputum samples. J. Clin. Microbiol. 2006, 44, 923–927. [Google Scholar] [CrossRef]
- Kilian, M.; Tettelin, H. Identification of Virulence-Associated Properties by Comparative Genome Analysis of Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, Three, S. oralis Subspecies, and S. infantis. mBio 2019, 10, e01985-19. [Google Scholar] [CrossRef] [PubMed]
- Slotved, H.C.; Facklam, R.R.; Fuursted, K. Assessment of a novel bile solubility test and MALDI-TOF for the differentiation of Streptococcus pneumoniae from other mitis group streptococci. Sci. Rep. 2017, 7, 7167. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Meier-Kolthof, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Novick, S.; Shagan, M.; Blau, K.; Lifshitz, S.; Givon-Lavi, N.; Grossman, N.; Bodner, L.; Dagan, R.; Mizrachi Nebenzahl, Y. Adhesion and invasion of Streptococcus pneumoniae to primary and secondary respiratory epithelial cells. Mol. Med. Rep. 2017, 15, 65–74. [Google Scholar] [CrossRef]
- Lim, Y.K.; Park, S.N.; Shin, J.H.; Chang, Y.H.; Shin, Y.; Paek, J.; Kim, H.; Kook, J.K. Streptococcus chosunense sp. nov., Isolated from Human Postoperative Maxillary Cyst. Curr. Microbiol. 2019, 76, 1193–1198. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| National Microbiology Data Center Accession Number | Strain Number | Serotype | AMX | CXM | CRO | CPM | ERY | AZM | CLI | CIP | LVX | MXF | TC | TMP | SMZ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMDC60014437 | SP203 | 271 | 2 | 4 | 1 | 1 | 256 | 256 | 512 | 1 | 1 | 0.125 | 16 | 16 | 128 |
| NMDC60014438 | SP204 | unannotated | 0.031 | 0.008 | 0.008 | 0.031 | 256 | 256 | 512 | 0.25 | 0.5 | 0.125 | 64 | 1 | 512 |
| NMDC60014439 | SP205 | unannotated | 0.008 | 0.016 | 0.031 | 0.062 | 256 | 256 | 512 | 1 | 1 | 0.125 | 64 | 128 | 1024 |
| NMDC60014440 | SP206 | 81 | 1 | 4 | 0.5 | 1 | 256 | 256 | 512 | 1 | 1 | 0.125 | 64 | 64 | 512 |
| NMDC60014441 | SP207 | 505 | 0.016 | 0.016 | 0.008 | 0.031 | 256 | 256 | 512 | 1 | 1 | 0.125 | 32 | 2 | 32 |
| NMDC60014442 | SP208 | 342 | 0.031 | 0.062 | 0.031 | 0.062 | 256 | 128 | 256 | 0.25 | 0.5 | 0.062 | 32 | 16 | 32 |
| NMDC60014443 | SP209 | unannotated | 0.016 | 0.016 | 0.008 | 0.031 | 256 | 256 | 512 | 1 | 1 | 0.125 | 64 | 128 | 128 |
| NMDC60014444 | SP210 | 81 | 1 | 4 | 0.5 | 1 | 256 | 256 | 512 | 1 | 1 | 0.125 | 32 | 64 | 128 |
| NMDC60014445 | SP211 | unannotated | 0.031 | 0.016 | 0.016 | 0.031 | 128 | 256 | 512 | 1 | 1 | 0.125 | 32 | 64 | 128 |
| NMDC60014446 | SP212 | 271 | 1 | 4 | 0.5 | 1 | 256 | 256 | 512 | 0.25 | 0.5 | 0.125 | 16 | 64 | >1024 |
| NMDC60014447 | SP213 | 7751 | 0.008 | 0.016 | 0.008 | 0.062 | 256 | 256 | 512 | 1 | 1 | 0.25 | 16 | 2 | 32 |
| NMDC60014448 | SP214 | 271 | 1 | 4 | 1 | 1 | 256 | 256 | 512 | 2 | 1 | 0.25 | 16 | 32 | 128 |
| NMDC60014450 | SP216 | 6946 | 0.016 | 0.031 | 0.016 | 0.062 | 128 | 256 | 512 | 1 | 1 | 0.125 | 16 | 1 | 128 |
| NMDC60014451 | SP217 | 3173 | 1 | 4 | 0.5 | 1 | 128 | 256 | 512 | 8 | 8 | 2 | 32 | 8 | >1024 |
| NMDC60014454 | SP220 | 10085 | 0.031 | 0.25 | 0.031 | 0.031 | 256 | 256 | 512 | 0.5 | 0.5 | 0.125 | 16 | 16 | 32 |
| NMDC60014455 | SP221 | 9104 | 0.5 | 2 | 0.5 | 1 | 16 | 2 | 0.125 | 0.25 | 0.5 | 0.125 | 32 | 1 | 512 |
| NMDC60014452 | SP218 | unannotated | 1 | 2 | 0.25 | 1 | 64 | 128 | 0.25 | 64 | 8 | 4 | 32 | 64 | >1024 |
| NMDC60014453 | SP219 | unannotated | 1 | 2 | 0.25 | 1 | 64 | 128 | 0.25 | 64 | 8 | 4 | 32 | 64 | >1024 |
| Total number of insensitive isolates | 0 | 9 | 9 | 0 | 18 | 18 | 15 | 4 | 3 | 3 | 18 | 18 | 18 | ||
| Rate of insensitive isolates | 0 | 50% | 50% | 0 | 100% | 100% | 83.33% | 22.22% | 16.67% | 16.67% | 100% | 100% | 100% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zeng, Y.; Wei, M.; Cui, L.; Song, Y.; Zhang, G.; Li, Y.; Feng, J. Streptococcus sputorum, a Novel Member of Streptococcus with Multidrug Resistance, Exhibits Cytotoxicity. Antibiotics 2021, 10, 1532. https://doi.org/10.3390/antibiotics10121532
Wang C, Zeng Y, Wei M, Cui L, Song Y, Zhang G, Li Y, Feng J. Streptococcus sputorum, a Novel Member of Streptococcus with Multidrug Resistance, Exhibits Cytotoxicity. Antibiotics. 2021; 10(12):1532. https://doi.org/10.3390/antibiotics10121532
Chicago/Turabian StyleWang, Chao, Yuan Zeng, Mengyu Wei, Lanqing Cui, Yuqin Song, Gang Zhang, Yun Li, and Jie Feng. 2021. "Streptococcus sputorum, a Novel Member of Streptococcus with Multidrug Resistance, Exhibits Cytotoxicity" Antibiotics 10, no. 12: 1532. https://doi.org/10.3390/antibiotics10121532
APA StyleWang, C., Zeng, Y., Wei, M., Cui, L., Song, Y., Zhang, G., Li, Y., & Feng, J. (2021). Streptococcus sputorum, a Novel Member of Streptococcus with Multidrug Resistance, Exhibits Cytotoxicity. Antibiotics, 10(12), 1532. https://doi.org/10.3390/antibiotics10121532