Novel Molecular Markers Linked to Pseudomonas aeruginosa Epidemic High-Risk Clones



Abstract
1. Introduction
2. Results
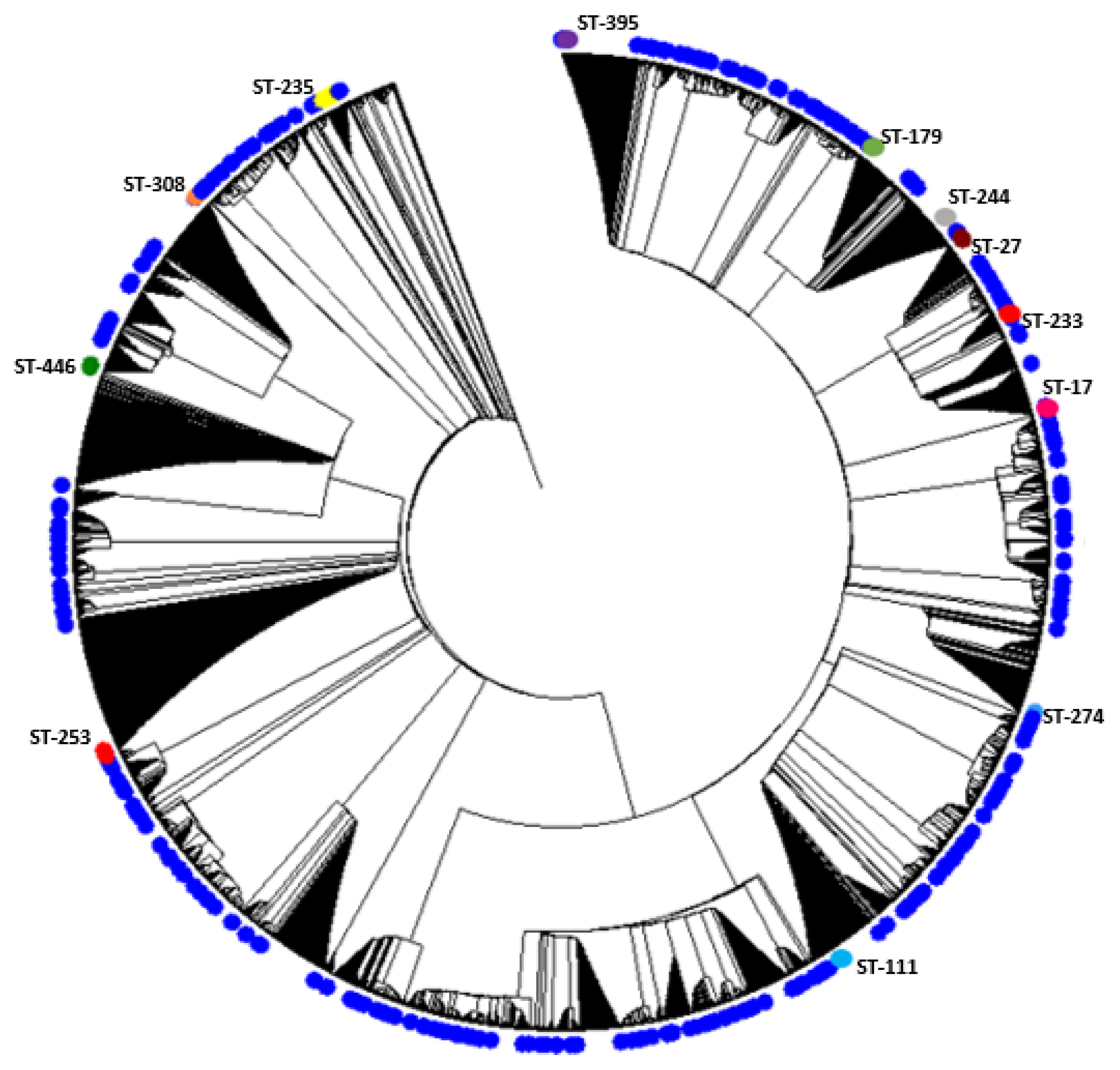
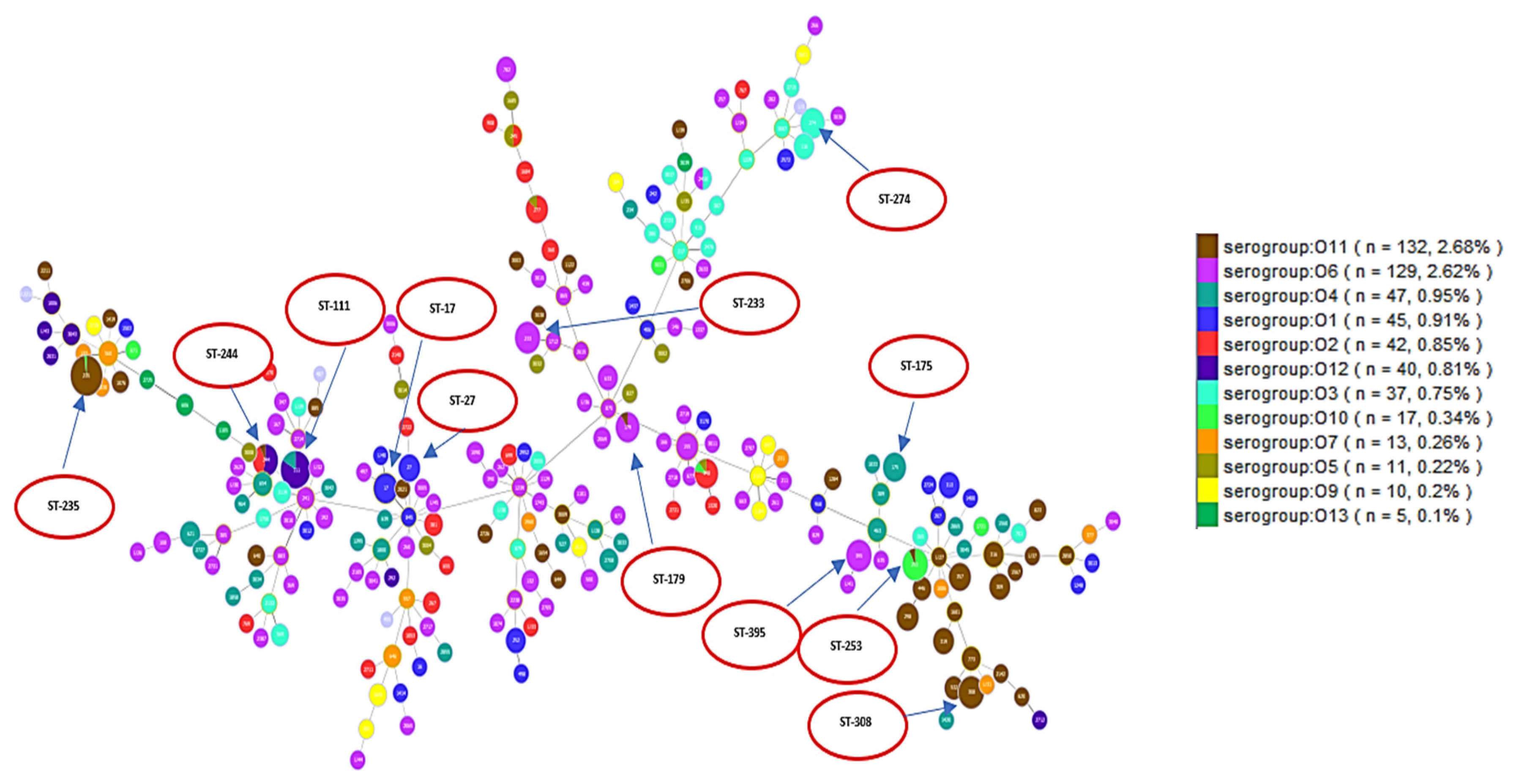
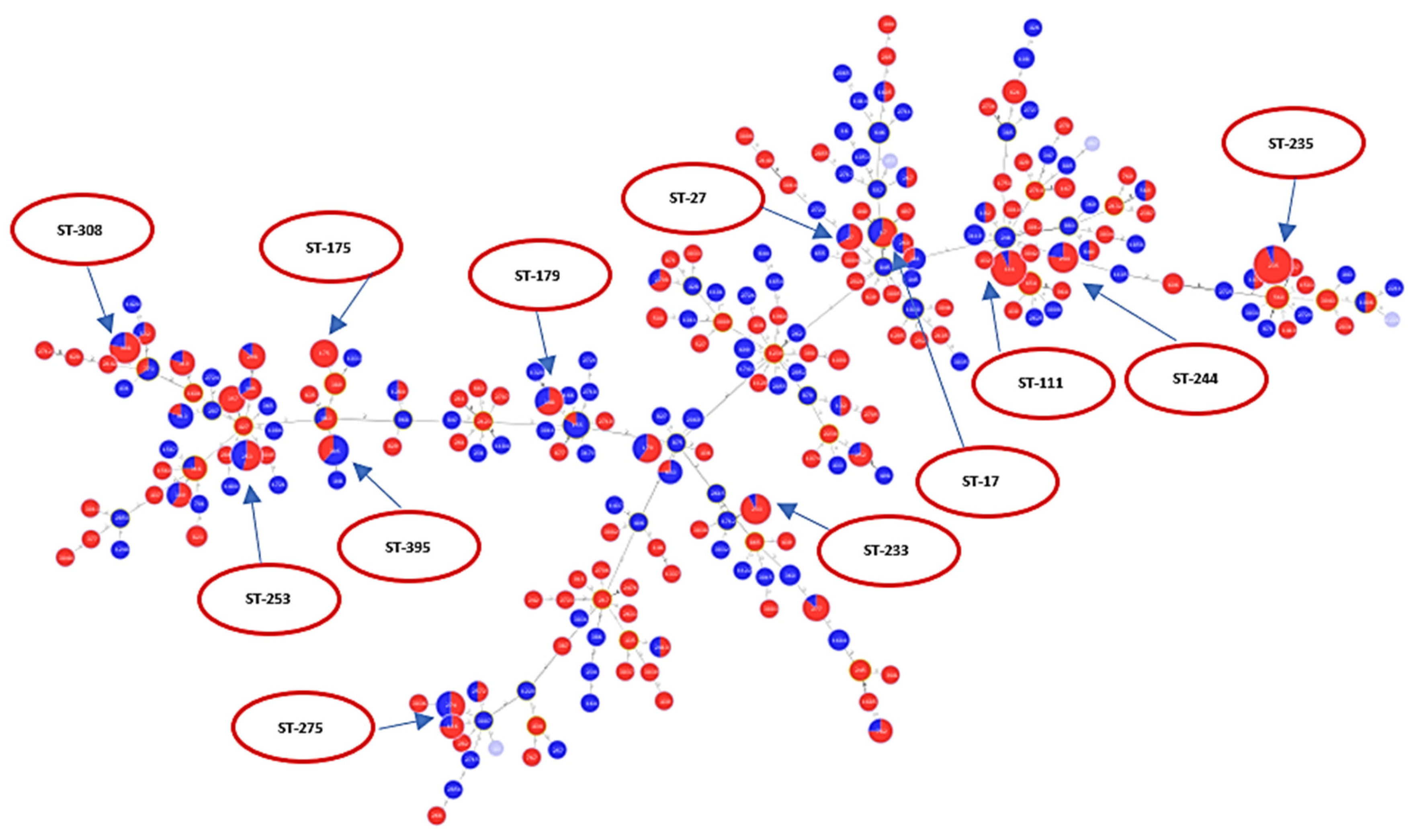
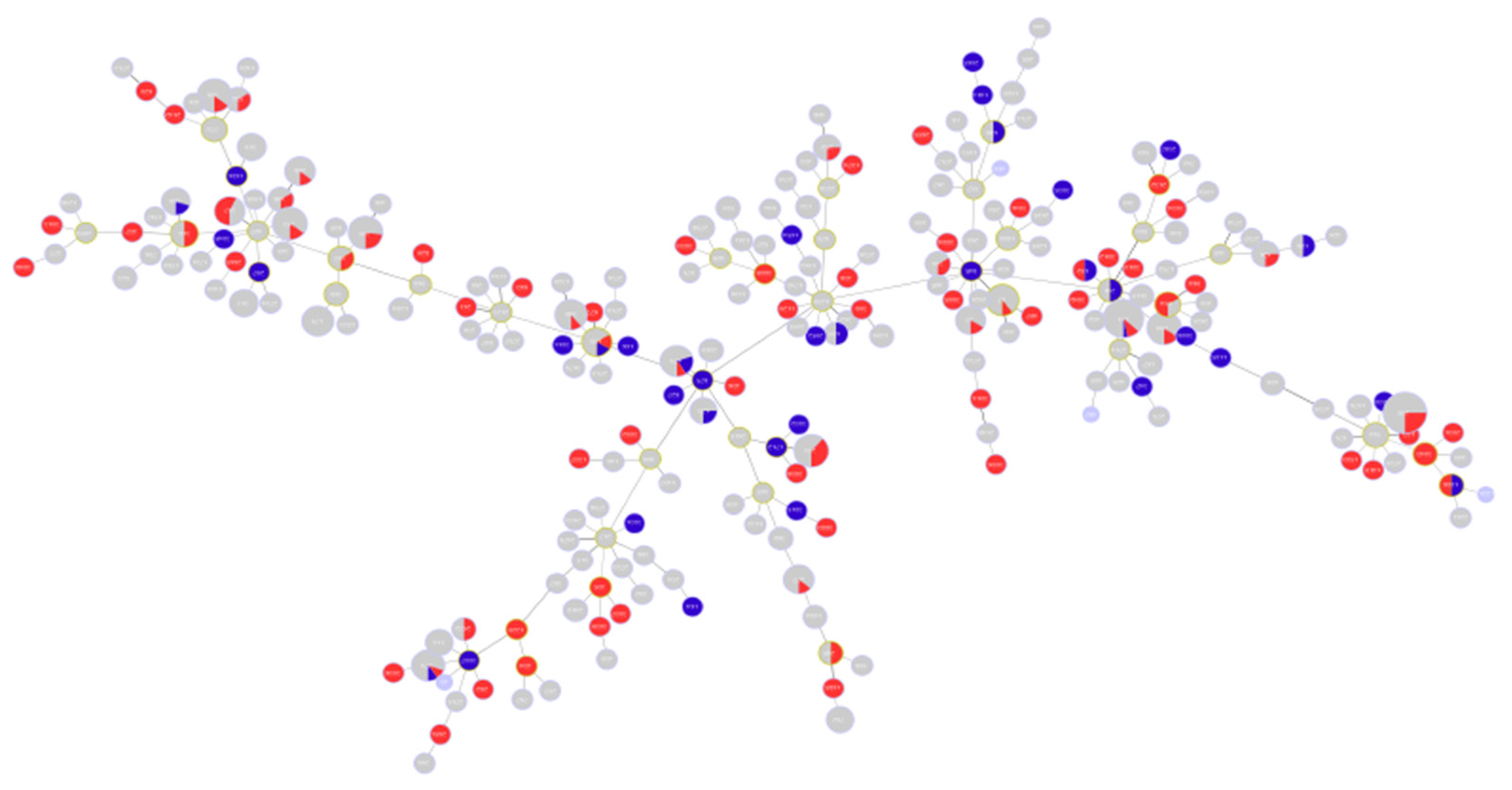
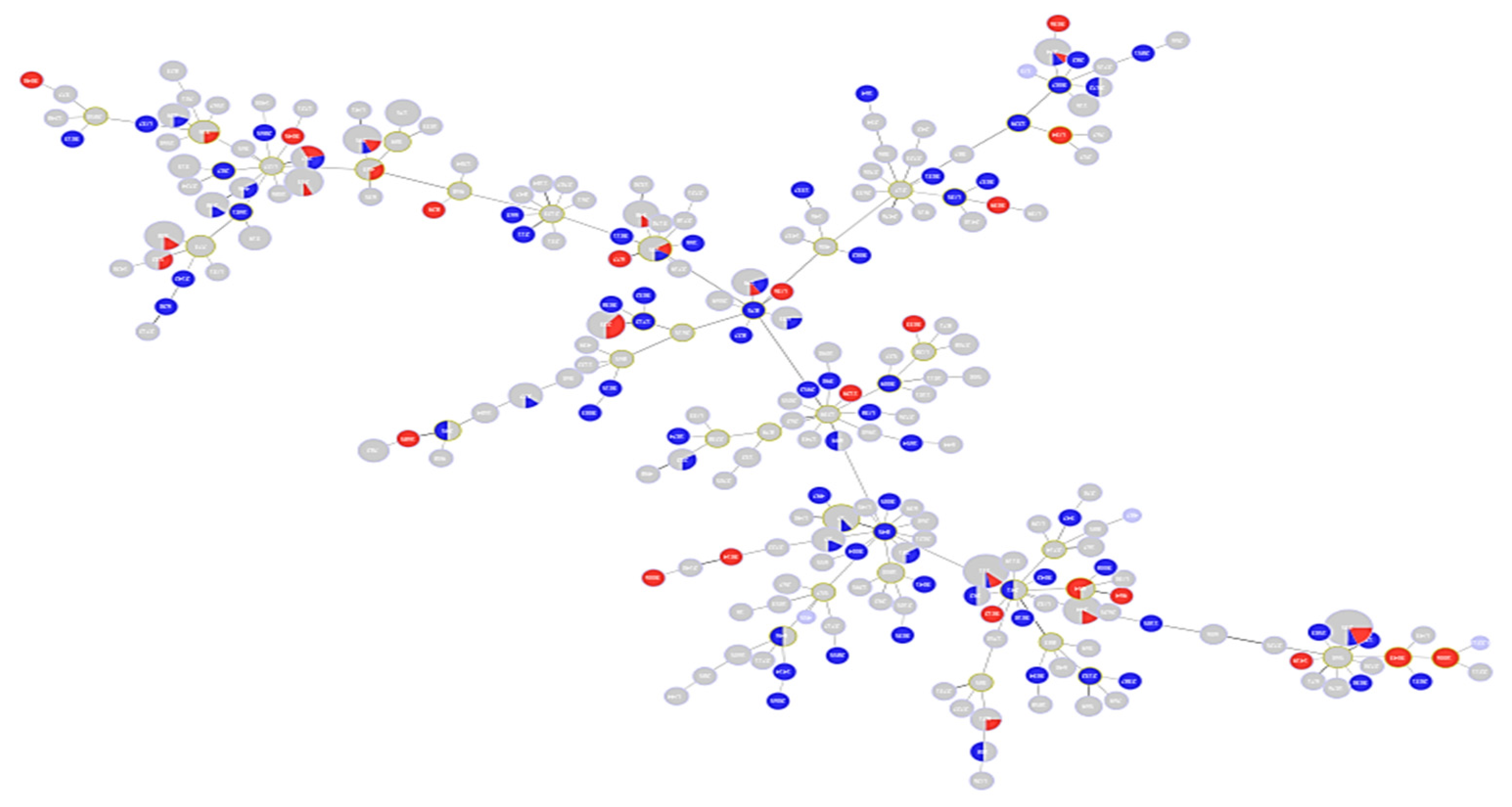
2.1. Description of Population Structure and Diversity in the Studied Set of Sequences
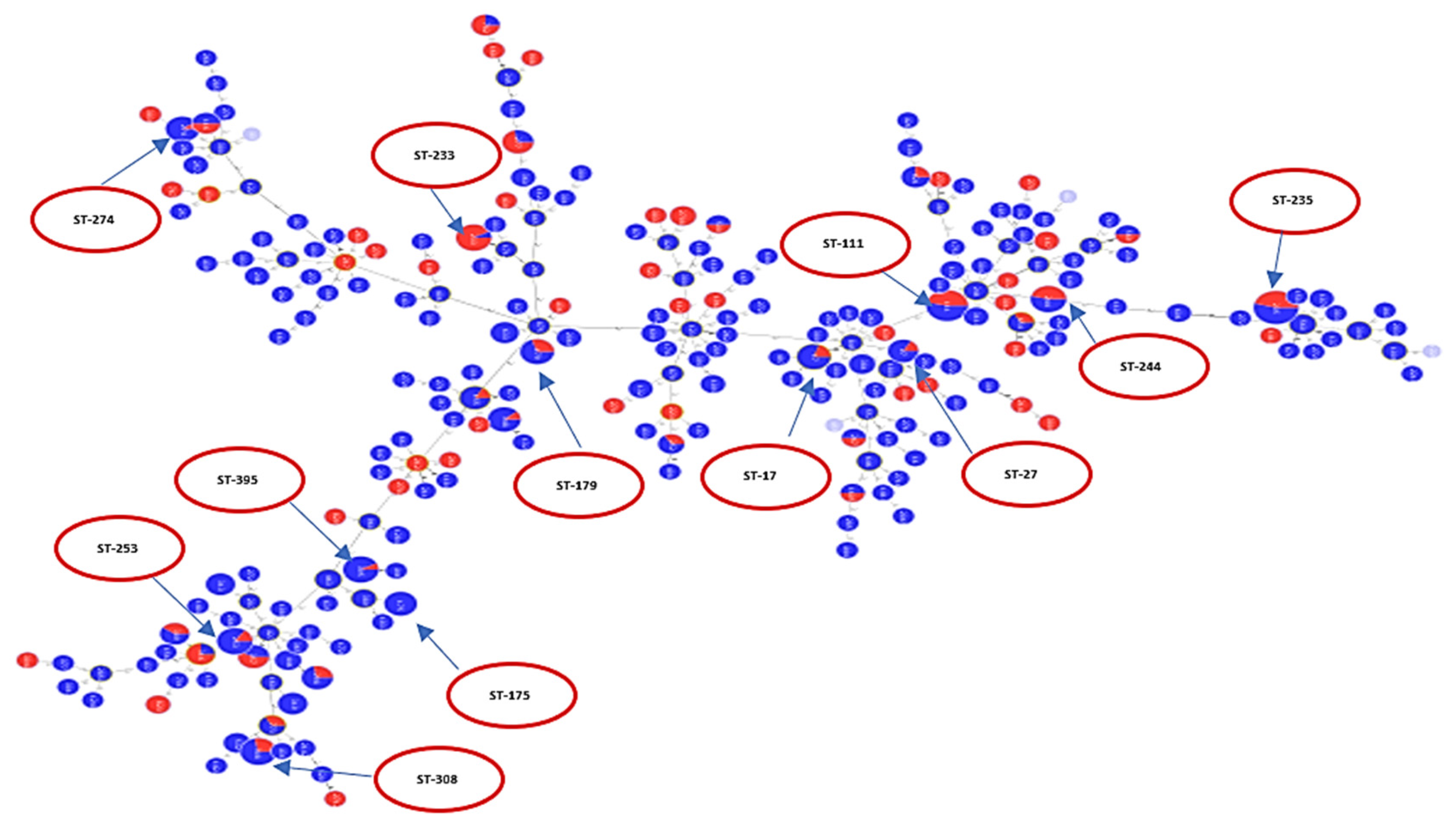
2.2. Analysis of Different Multilocus Sequence Typing (MLST) Profiles
2.3. Resistance Profile of P. aeruginosa Epidemic High-Risk Clones
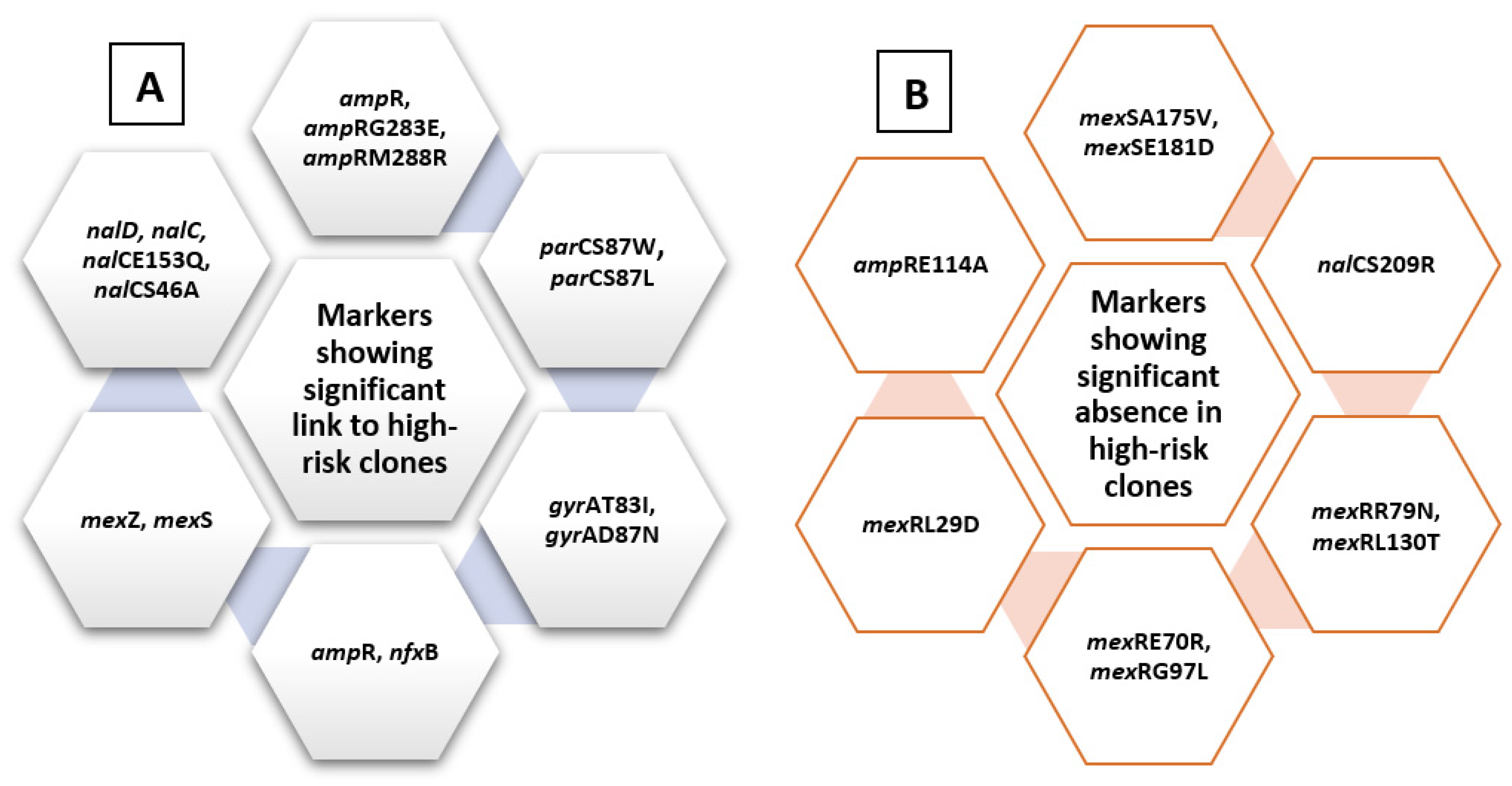
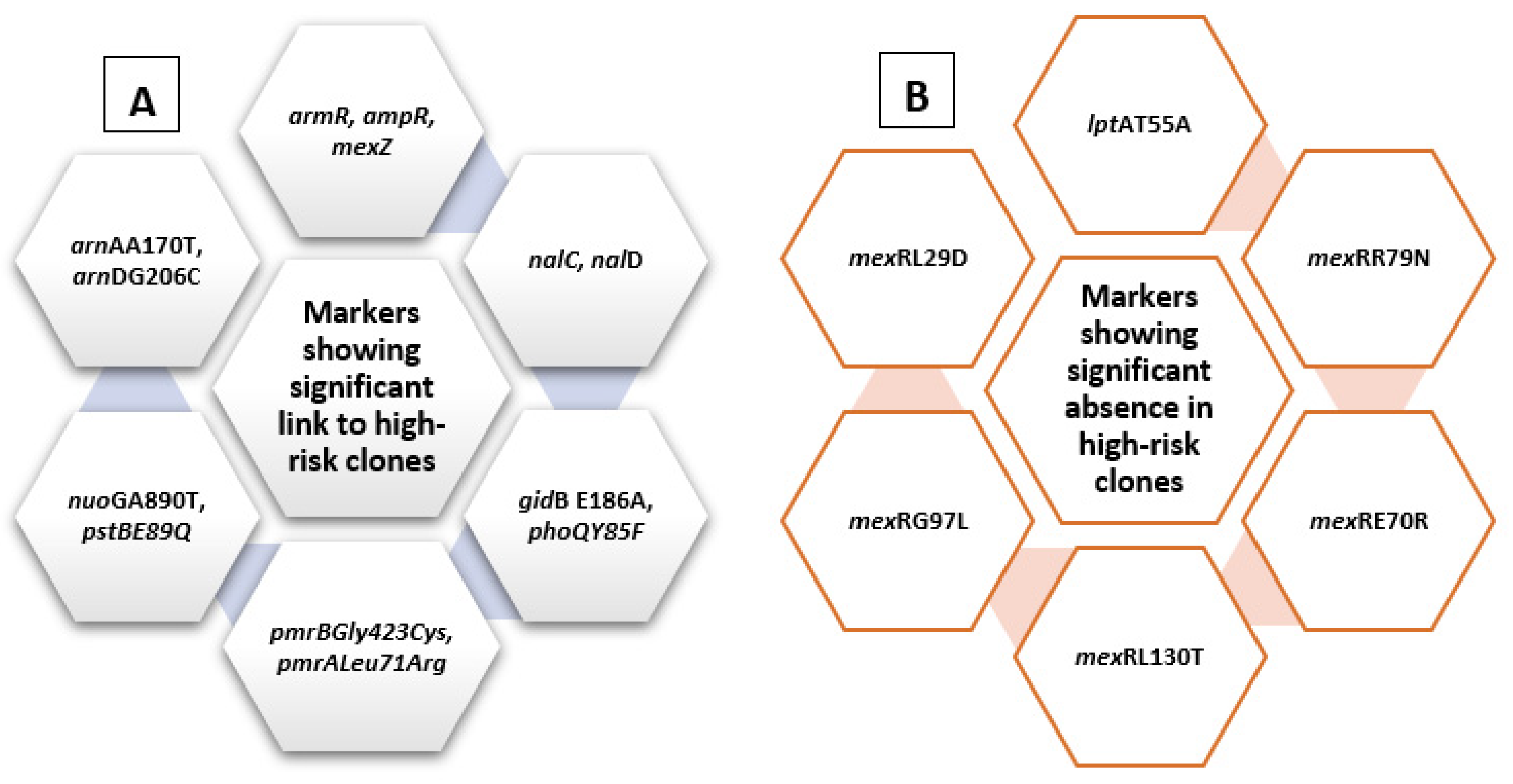
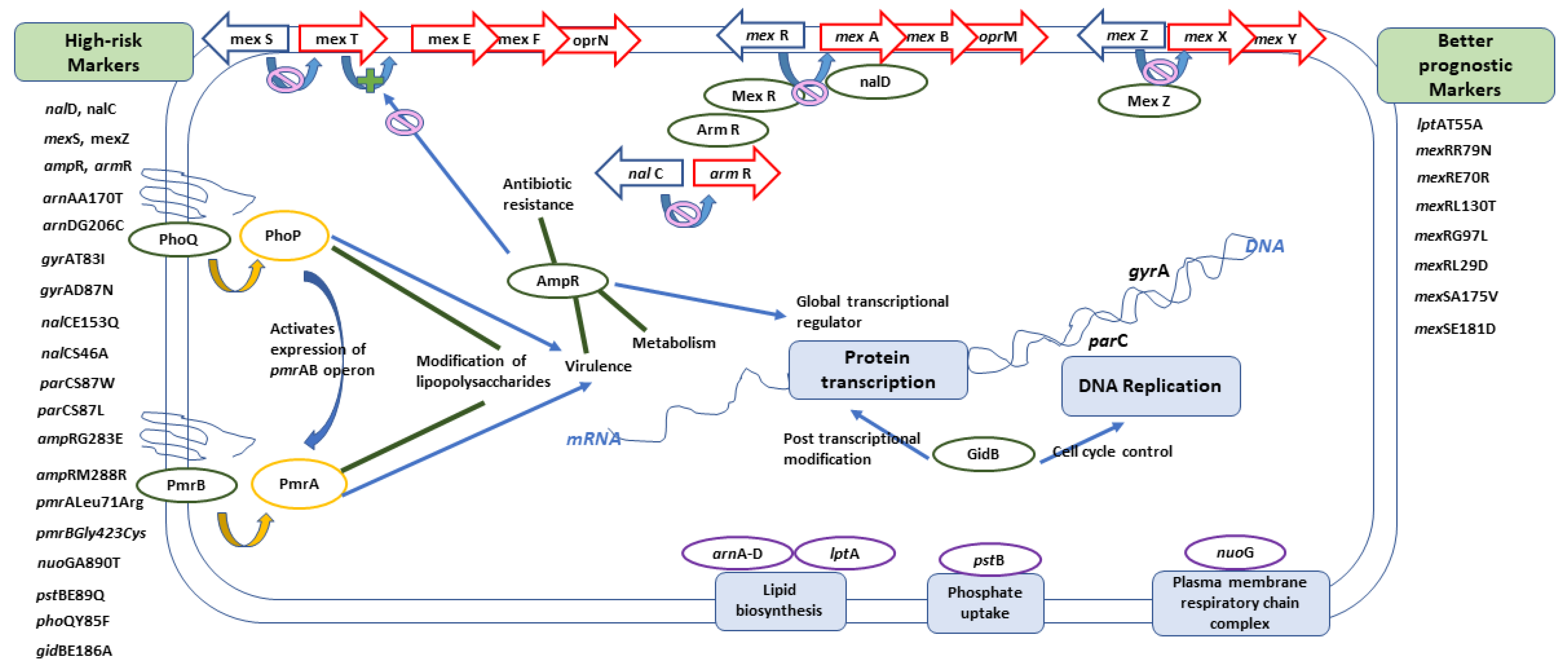
2.4. Analysis of Molecular Markers in Relation to High-Risk Clones
2.4.1. Levofloxacin-Related Molecular Markers
2.4.2. Ciprofloxacin-Related Molecular Markers
2.4.3. Amikacin-Related Molecular Markers
2.4.4. Gentamycin-Related Molecular Markers
3. Discussion
4. Materials and Methods
4.1. Multilocus Sequence Typing (MLST) and Serotypes (O-type) Analysis
4.2. Phylogenetic Analysis
4.3. Population Structure and Diversity Analysis
4.4. Resistance Genes and Markers Correlations
“Pseudomonas aeruginosa” [title/abstract] AND “aminoglycosides resistance”[title/abstract]
“Pseudomonas aeruginosa” [title/abstract] AND “Quinolone resistance”[title/abstract].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Centre for Disease Prevention and Control. Antimicrobial Resistance Surveillance in Europe 2013; Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net); ECDC: Stockholm, Sweden, 2014. [Google Scholar]
- CDC. Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019; pp. 1–150. Available online: http://dx.doi.org/10.15620/cdc:82532 (accessed on 30 November 2020).
- Kos, V.N.; Déraspe, M.; McLaughlin, R.E.; Whiteaker, J.D.; Roy, P.H.; Alm, R.A.; Corbeil, J.; Gardner, H. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 2015, 59, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Willyard, C. The drug-resistant bacteria that pose the greatest health threats. Nature 2017, 543, 15. [Google Scholar] [CrossRef] [PubMed]
- Foxman, B.; Zhang, L.; Koopman, J.S.; Manning, S.D.; Marrs, C.F. Choosing an appropriate bacterial typing technique for epidemiologic studies. Epidemiol. Perspect. Innov. 2005, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, B.; Valot, B.; Abdelbary, M.M.H.; Prod’hom, G.; Greub, G.; Senn, L.; Blanc, D.S. Combining Standard Molecular Typing and Whole Genome Sequencing to Investigate Pseudomonas aeruginosa Epidemiology in Intensive Care Units. Front. Public Health 2020, 8, 3. [Google Scholar] [CrossRef]
- Spratt, B.G. Exploring the Concept of Clonality in Bacteria BT. In Genomics, Proteomics, and Clinical Bacteriology: Methods and Reviews; Woodford, N., Johnson, A.P., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 323–352. ISBN 978-1-59259-763-5. [Google Scholar]
- Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Emond-Rheault, J.-G.; Tucker, N.P.; Levesque, R.C. Genomics of antibiotic-resistance prediction in Pseudomonas aeruginosa. Ann. N. Y. Acad. Sci. 2019, 1435, 5–17. [Google Scholar] [CrossRef]
- Oliver, A.; Mulet, X.; López-Causapé, C.; Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 2015, 21–22. [Google Scholar] [CrossRef]
- Fernández-Olmos, A.; García-Castillo, M.; María Alba, J.; Morosini, M.I.; Lamas, A.; Romero, B.; Galán, J.C.; Del Campo, R.; Cantón, R. Population structure and Antimicrobial susceptibility of both nonpersistent and persistent Pseudomonas aeruginosa isolates recovered from cystic fibrosis patients. J. Clin. Microbiol. 2013, 51. [Google Scholar] [CrossRef]
- Van Mansfeld, R.; Willems, R.; Brimicombe, R.; Heijerman, H.; Van Berkhout, F.T.; Wolfs, T.; Van Der Ent, C.; Bonten, M. Pseudomonas aeruginosa genotype prevalence in Dutch Cystic Fibrosis patients and age dependency of colonization by various P. aeruginosa sequence types. J. Clin. Microbiol. 2009, 47. [Google Scholar] [CrossRef]
- Caballero, J.D.D.; del Campo, R.; Cobo, M.; Chinchón, G.; López-Causapé, C.; Antonio, O.; Tato, M.; Cantón, R. 59 Population structure and antimicrobial susceptibility of Pseudomonas aeruginosa from a national survey involving 24 cystic fibrosis units in Spain. J. Cyst. Fibros. 2014, 13, S61. [Google Scholar] [CrossRef][Green Version]
- Garcia-Castillo, M.; Morosini, M.I.; Rodriguez-Baños, M.; Máiz, L.; Lamas, A.; Baquero, F.; Cantón, R.; del Campo, R. 128 Population structure of Pseudomonas aeruginosa from cystic fibrosis patients. J. Cyst. Fibros. 2011, 10, S33. [Google Scholar] [CrossRef]
- Woodford, N.; Turton, J.F.; Livermore, D.M. Multiresistant Gram-negative bacteria: The role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 736–755. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; Cabot, G.; Gomez-Zorrilla, S.; Zamorano, L.; Ocampo-Sosa, A.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; et al. Influence of Virulence Genotype and Resistance Profile in the Mortality of Pseudomonas aeruginosa Bloodstream Infections. Clin. Infect. Dis. 2015, 60, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Mogayzel, P.J.; Naureckas, E.T.; Robinson, K.A.; Brady, C.; Guill, M.; Lahiri, T.; Lubsch, L.; Matsui, J.; Oermann, C.M.; Ratjen, F.; et al. Cystic Fibrosis Foundation Pulmonary Guideline. Pharmacologic Approaches to Prevention and Eradication of Initial Pseudomonas aeruginosa Infection. Ann. Am. Thorac. Soc. 2014, 11, 1640–1650. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, H.; Jiang, Q.; Wang, Q.; Li, S.; Huang, Y. Bronchoscope-related Pseudomonas aeruginosa pseudo-outbreak attributed to contaminated rinse water. Am. J. Infect. Control 2020, 48, 26–32. [Google Scholar] [CrossRef]
- Moloney, E.M.; Deasy, E.C.; Swan, J.S.; Brennan, G.I.; O’Donnell, M.J.; Coleman, D.C. Whole-genome sequencing identifies highly related Pseudomonas aeruginosa strains in multiple washbasin U-bends at several locations in one hospital: Evidence for trafficking of potential pathogens via wastewater pipes. J. Hosp. Infect. 2019, 4–11. [Google Scholar] [CrossRef]
- Correa, A.; Del Campo, R.; Perenguez, M.; Blanco, V.M.; Rodríguez-Baños, M.; Perez, F.; Maya, J.J.; Rojas, L.; Cantón, R.; Arias, C.A.; et al. Dissemination of high-risk clones of extensively drug-resistant pseudomonas aeruginosa in Colombia. Antimicrob. Agents Chemother. 2015, 59, 2421–2425. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Curran, B.; Jonas, D.; Grundmann, H.; Pitt, T.; Dowson, C.G. Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 2004, 42, 5644–5649. [Google Scholar] [CrossRef]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carriço, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Haubold, B.; Hudson, R.R. LIAN 3.0: Detecting linkage disequilibrium in multilocus data. Bioinformatics 2000, 16, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Severiano, A.; Pinto, F.R.; Ramirez, M.; Carriço, J.A. Adjusted Wallace coefficient as a measure of congruence between typing methods. J. Clin. Microbiol. 2011, 49, 3997–4000. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Coque, T.M.; de la Cruz, F. Ecology and evolution as targets: The need for novel eco-evo drugs and strategies to fight antibiotic resistance. Antimicrob. Agents Chemother. 2011, 55, 3649–3660. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; Ocampo-Sosa, A.A.; Domínguez, M.A.; Gago, J.F.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; Peña, C.; Martínez-Martínez, L.; et al. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob. Agents Chemother. 2012, 56, 6349–6357. [Google Scholar] [CrossRef] [PubMed]
- Kugelberg, E.; Löfmark, S.; Wretlind, B.; Andersson, D.I. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2005, 55, 22–30. [Google Scholar] [CrossRef]
- Chowdhury, P.R.; Scott, M.; Worden, P.; Huntington, P.; Hudson, B.; Karagiannis, T.; Charles, I.G.; Djordjevic, S.P. Genomic islands 1 and 2 play key roles in the evolution of extensively drug-resistant ST235 isolates of Pseudomonas aeruginosa. Open Biol. 2016, 6. [Google Scholar] [CrossRef]
- Roy Chowdhury, P.; Scott, M.J.; Djordjevic, S.P. Genomic islands 1 and 2 carry multiple antibiotic resistance genes in Pseudomonas aeruginosa ST235, ST253, ST111 and ST175 and are globally dispersed. J. Antimicrob. Chemother. 2017, 72, 620–622. [Google Scholar] [CrossRef]
- Treepong, P.; Kos, V.N.; Guyeux, C.; Blanc, D.S.; Bertrand, X.; Valot, B.; Hocquet, D. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin. Microbiol. Infect. 2018, 24, 258–266. [Google Scholar] [CrossRef]
- Pelegrin, A.C.; Saharman, Y.R.; Griffon, A.; Palmieri, M.; Mirande, C.; Karuniawati, A.; Sedono, R.; Aditianingsih, D.; Goessens, W.H.F.; van Belkum, A.; et al. High-risk international clones of carbapenem-nonsusceptible pseudomonas aeruginosa endemic to Indonesian intensive care units: Impact of a multifaceted infection control intervention analyzed at the genomic level. MBio 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Horna, G.; Amaro, C.; Palacios, A.; Guerra, H.; Ruiz, J. High frequency of the exoU+/exoS+ genotype associated with multidrug-resistant “high-risk clones” of Pseudomonas aeruginosa clinical isolates from Peruvian hospitals. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, E.L.A.; Kwasnicka, A.; Ochs, M.M.; Hancock, R.E.W. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol. Microbiol. 1999, 34, 305–316. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E.W. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, S.M.; Ernst, R.K.; Miller, S.I. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 2004, 186, 575–579. [Google Scholar] [CrossRef]
- Vatansever, C.; Menekse, S.; Dogan, O.; Gucer, L.S.; Ozer, B.; Ergonul, O.; Can, F. Co-existence of OXA-48 and NDM-1 in colistin resistant Pseudomonas aeruginosa ST235. Emerg. Microbes Infect. 2020, 9, 152–154. [Google Scholar] [CrossRef]
- Schniederjans, M.; Koska, M.; Häussler, S. Transcriptional and Mutational Profiling of an Aminoglycoside-Resistant Pseudomonas aeruginosa Small-Colony Variant. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Han, M.-L.; Zhu, Y.; Creek, D.J.; Lin, Y.-W.; Gutu, A.D.; Hertzog, P.; Purcell, T.; Shen, H.-H.; Moskowitz, S.M.; Velkov, T.; et al. Comparative Metabolomics and Transcriptomics Reveal Multiple Pathways Associated with Polymyxin Killing in Pseudomonas aeruginosa. mSystems 2019, 4, 1–18. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Thrane, S.W.; Taylor, V.L.; Lund, O.; Lam, J.S.; Jelsbak, L. Application of Whole-Genome Sequencing Data for O-Specific Antigen Analysis and In Silico Serotyping of Pseudomonas aeruginosa Isolates. J. Clin. Microbiol. 2016, 54, 1782–1788. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Smith, N.H.; O’Rourke, M.; Spratt, B.G. How clonal are bacteria? Proc. Natl. Acad. Sci. USA 1993, 90, 4384–4388. [Google Scholar] [CrossRef] [PubMed]
- Lenski, R.E. Assessing the genetic structure of microbial populations. Proc. Natl. Acad. Sci. USA 1993, 90, 4334–4336. [Google Scholar] [CrossRef]
- Carriço, J.A.; Silva-Costa, C.; Melo-Cristino, J.; Pinto, F.R.; de Lencastre, H.; Almeida, J.S.; Ramirez, M. Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 2006, 44, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef]
- O’Brien, S.; Williams, D.; Fothergill, J.L.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. High virulence sub-populations in Pseudomonas aeruginosa long-term cystic fibrosis airway infections. BMC Microbiol. 2017, 17, 30. [Google Scholar] [CrossRef]
- Mena, A.; Smith, E.E.; Burns, J.L.; Speert, D.P.; Moskowitz, S.M.; Perez, J.L.; Oliver, A. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 2008, 190, 7910–7917. [Google Scholar] [CrossRef]
- Oliver, A.; Cantón, R.; Campo, P.; Baquero, F.; Blázquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1253. [Google Scholar] [CrossRef]
- Mulet, X.; Cabot, G.; Ocampo-Sosa, A.A.; Domínguez, M.A.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; Peña, C.; et al. Biological markers of Pseudomonas aeruginosa epidemic high-risk clones. Antimicrob. Agents Chemother. 2013, 57, 5527–5535. [Google Scholar] [CrossRef]
- Zamudio, R.; Hijazi, K.; Joshi, C.; Aitken, E.; Oggioni, M.R.; Gould, I.M. Phylogenetic analysis of resistance to ceftazidime/avibactam, ceftolozane/tazobactam and carbapenems in piperacillin/tazobactam-resistant Pseudomonas aeruginosa from cystic fibrosis patients. Int. J. Antimicrob. Agents 2019, 53, 774–780. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| High-risk clone | Number of sequences | Ciprofloxacin-resistant | Ciprofloxacin-susceptible | Unrecorded ciprofloxacin-susceptibility | Levofloxacin-resistant | Levofloxacin-susceptible | Gentamycin-resistant | Gentamycin-susceptible | Unrecorded gentamycin-susceptibility | Amikacin-resistant | Amikacin-susceptible |
| ST235 | 50 | 13 | - | 37 | 46 | 4 | 3 | 10 | 37 | 23 | 27 |
| ST111 | 30 | 3 | 1 | 26 | 28 | 2 | 3 | 1 | 26 | 14 | 16 |
| ST244 | 13 | 2 | - | 11 | 10 | 3 | 2 | - | 11 | 6 | 7 |
| ST395 | 13 | 3 | - | 10 | 5 | 8 | 2 | 1 | 10 | 1 | 12 |
| ST175 | 8 | - | - | 8 | 8 | - | - | - | 8 | - | 8 |
| ST17 | 10 | 1 | - | 9 | 6 | 4 | - | 1 | 9 | 2 | 8 |
| ST274 | 10 | 1 | 1 | 8 | 5 | 5 | 1 | 1 | 8 | 1 | 9 |
| Molecular Marker | High-Risk Clone | Chi-Square | p-Value | Phi Coefficient |
|---|---|---|---|---|
| mexSSer124Arg | ST235 | 39.6 | <0.005 | 0.274 |
| mexSSer124Arg | ST395 | 14.06 | <0.005 | 0.163 |
| nalD | ST395 | 3.97 | 0.046 | 0.274 |
| nalCG71E | ST395 | 5.7 | 0.017 | 0.103 |
| nalCA186T | ST233 | 147.6 | <0.005 | 0.529 |
| nalCS46A | ST235 | 3.06 | 0.08 | 0.076 |
| nalCS46A | ST244, ST253 | 4.44 | 0.035 | 0.092 |
| nalCE153Q | ST235 | 71.06 | <0.005 | 0.367 |
| nalID153Q | ST235 | 20.7 | <0.005 | 0.198 |
| parCS87W | ST274 | 7.9 | 0.005 | 0.122 |
| parCS87L | ST111 | 17.09 | <0.005 | 0.18 |
| parCS87L | ST233 | 32.3 | <0.005 | 0.248 |
| parCS87L | ST235 | 49 | <0.005 | 0.306 |
| parCS87L | ST244 | 9.5 | 0.005 | 0.135 |
| parCS87L | ST308 | 16.6 | <0.005 | 0.178 |
| gyrAT83I | ST111 | 25.7 | <0.005 | 0.22 |
| gyrAT83I | ST233 | 20.1 | <0.005 | 0.195 |
| gyrAT83I | ST235 | 35.42 | <0.005 | 0.259 |
| gyrAT83I | ST244 | 4.5 | 0.035 | 0.092 |
| gyrAT83I | ST308 | 12.7 | <0.005 | 0.155 |
| gyrAD87N | ST395 | 7.13 | 0.008 | 0.116 |
| ampRG283E | ST235 | 49.85 | <0.005 | 0.307 |
| ampRG283E | ST308 | 9.78 | 0.002 | 0.136 |
| ampRM288R | ST235 | 82.776 | <0.005 | 0.396 |
| ampRM288R | ST308 | 18.658 | <0.005 | 0.188 |
| Molecular Marker | High-Risk Clone | Chi-Square | p-Value | Phi Coefficient |
|---|---|---|---|---|
| phoQY85F | ST235 | 400.38 | <0.005 | 0.871 |
| nuoGA890T | ST395 | 455.82 | <0.005 | 0.929 |
| pstBE89Q | ST233 | 338.5 | <0.005 | 0.801 |
| arnAA170T | ST233 | 292.64 | <0.005 | 0.744 |
| arnDG206C | ST233 | 292.64 | <0.005 | 0.744 |
| gidBE186A | ST235 | 493.946 | <0.005 | 0.967 |
| lptAR62S | ST446 | 85.48 | <0.005 | 0.402 |
| faoAT385A | ST244 | 121.92 | <0.005 | 0.481 |
| pmrBGly423Cys | ST111 | 22.492 | <0.005 | 0.206 |
| pmrBGly423Cys | ST235 | 28.103 | <0.005 | 0.231 |
| pmrBGly423Cys | ST253 | 14.338 | <0.005 | 0.165 |
| pmrBGly423Cys | ST308 | 12.493 | <0.005 | 0.154 |
| pmrALeu71Arg | ST17 | 29.38 | <0.005 | 0.236 |
| pmrALeu71Arg | ST233 | 30.868 | <0.005 | 0.242 |
| pmrALeu71Arg | ST253 | 24.145 | <0.005 | 0.214 |
| pmrALeu71Arg | ST308 | 27.034 | <0.005 | 0.226 |
| pmrALeu71Arg | ST175 | 23.414 | <0.005 | 0.211 |
| pmrALeu71Arg | ST179 | 29.38 | <0.005 | 0.236 |
| pmrALeu71Arg | ST532 | 8.696 | 0.003 | 0.128 |
| Molecular Marker | High-Risk Clone | Chi-Square | p-Value | Phi Coefficient |
|---|---|---|---|---|
| nalDSer32Asn | ST235 | 3.955 | 0.047 | 0.169 |
| nalCE153Q | ST235 | 98.99 | <0.005 | 0.844 |
| pmrALeu71Arg | ST308 | 5.594 | 0.018 | 0.201 |
| pmrALeu71Arg | ST233 | 14.298 | <0.005 | 0.321 |
| pmrALeu71Arg | ST179 | 8.453 | 0.004 | 0.247 |
| pstBE89Q | ST233 | 84.93 | <0.005 | 0.782 |
| arnA A170T | ST233 | 74.917 | <0.005 | 0.734 |
| arnDG206C | ST233 | 84.93 | <0.005 | 0.782 |
| gidBE97Q | ST235 | 139 | <0.005 | 1 |
| gidBE186A | ST235 | 139 | <0.005 | 1 |
| Molecular Markers Previously Identified in Relation to Antibiotic Resistance | Newly Identified Molecular Markers |
|---|---|
| mexZ nalCS46A nalCS209R nalCG71E gyrAT83I gyrAD87N parCS87W parCS87L nalCE153Q nalCThr50pro mexS gene mexSA175V mexSE181D nalD gene nfxB armR parEV460G ampRD135N nalC ampR pmrBGly423Cys pmrALeu71Arg nalDser32Asn ampRA51T ampRG283E ampRM288R | phoQY85F nuoGA890T pstBE89Q lptAT55A lptAR62S faoAT385A arnAA170T arnDG206C mexRR79N mexRE70R mexRL130T mexRG97L mexRL29D fusA1D588G gidBE186A gidBQ28K gidBE97Q |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nageeb, W.; Amin, D.H.; Mohammedsaleh, Z.M.; Makharita, R.R. Novel Molecular Markers Linked to Pseudomonas aeruginosa Epidemic High-Risk Clones. Antibiotics 2021, 10, 35. https://doi.org/10.3390/antibiotics10010035
Nageeb W, Amin DH, Mohammedsaleh ZM, Makharita RR. Novel Molecular Markers Linked to Pseudomonas aeruginosa Epidemic High-Risk Clones. Antibiotics. 2021; 10(1):35. https://doi.org/10.3390/antibiotics10010035
Chicago/Turabian StyleNageeb, Wedad, Dina H. Amin, Zuhair M. Mohammedsaleh, and Rabab R. Makharita. 2021. "Novel Molecular Markers Linked to Pseudomonas aeruginosa Epidemic High-Risk Clones" Antibiotics 10, no. 1: 35. https://doi.org/10.3390/antibiotics10010035
APA StyleNageeb, W., Amin, D. H., Mohammedsaleh, Z. M., & Makharita, R. R. (2021). Novel Molecular Markers Linked to Pseudomonas aeruginosa Epidemic High-Risk Clones. Antibiotics, 10(1), 35. https://doi.org/10.3390/antibiotics10010035