Mining the Potential of Label-Free Biosensors for In Vitro Antipsychotic Drug Screening

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Apparatus
2.2. Electrochemical Impedance Spectroscopy (EIS) Measurements
2.3. Nanoplasmonic Spectroscopic Imaging Measurements
2.4. Synthesis of DA-BSA Conjugate and Surface Functionalization
2.5. D3 Receptor Binding Assay Procedure
3. Results
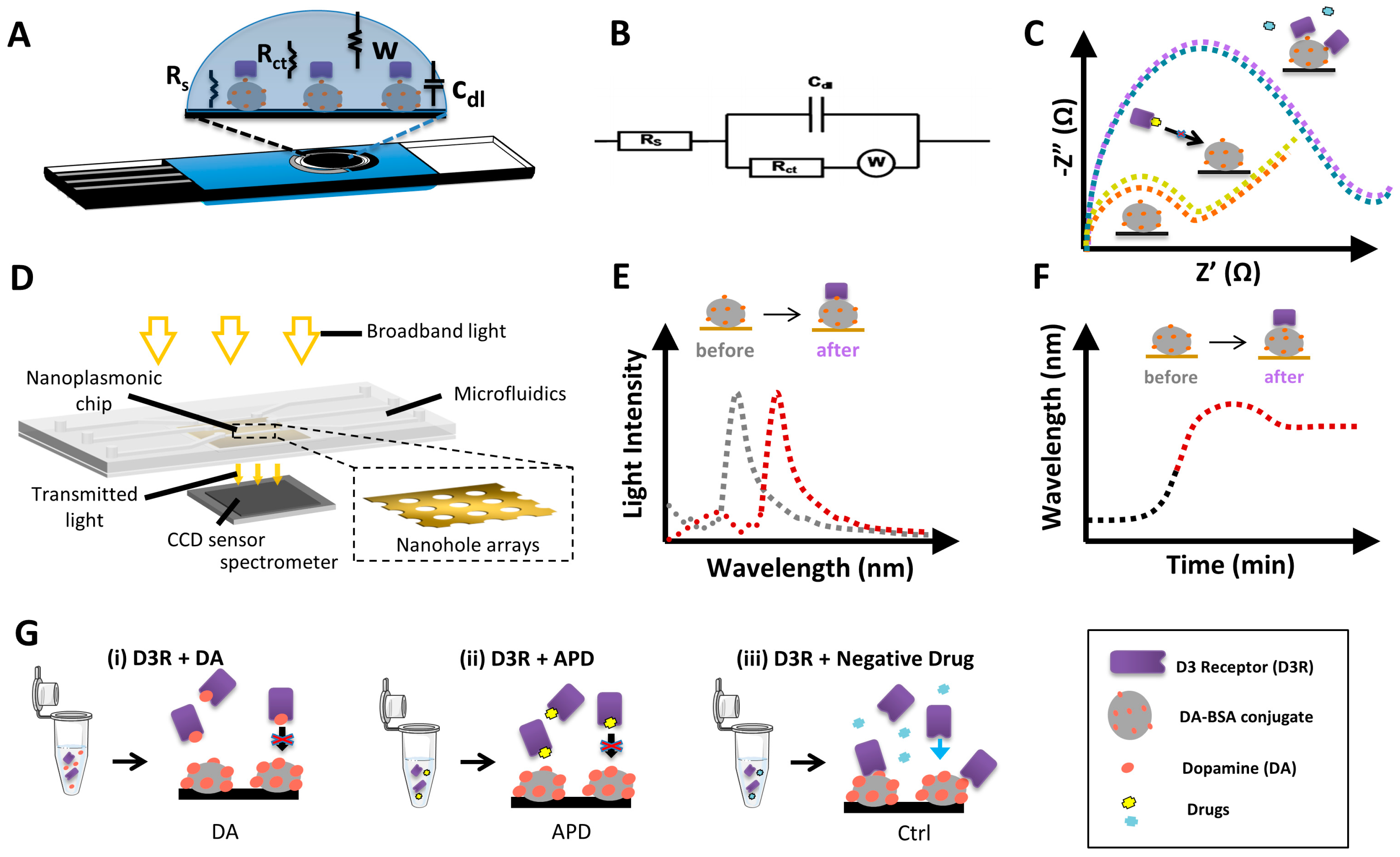
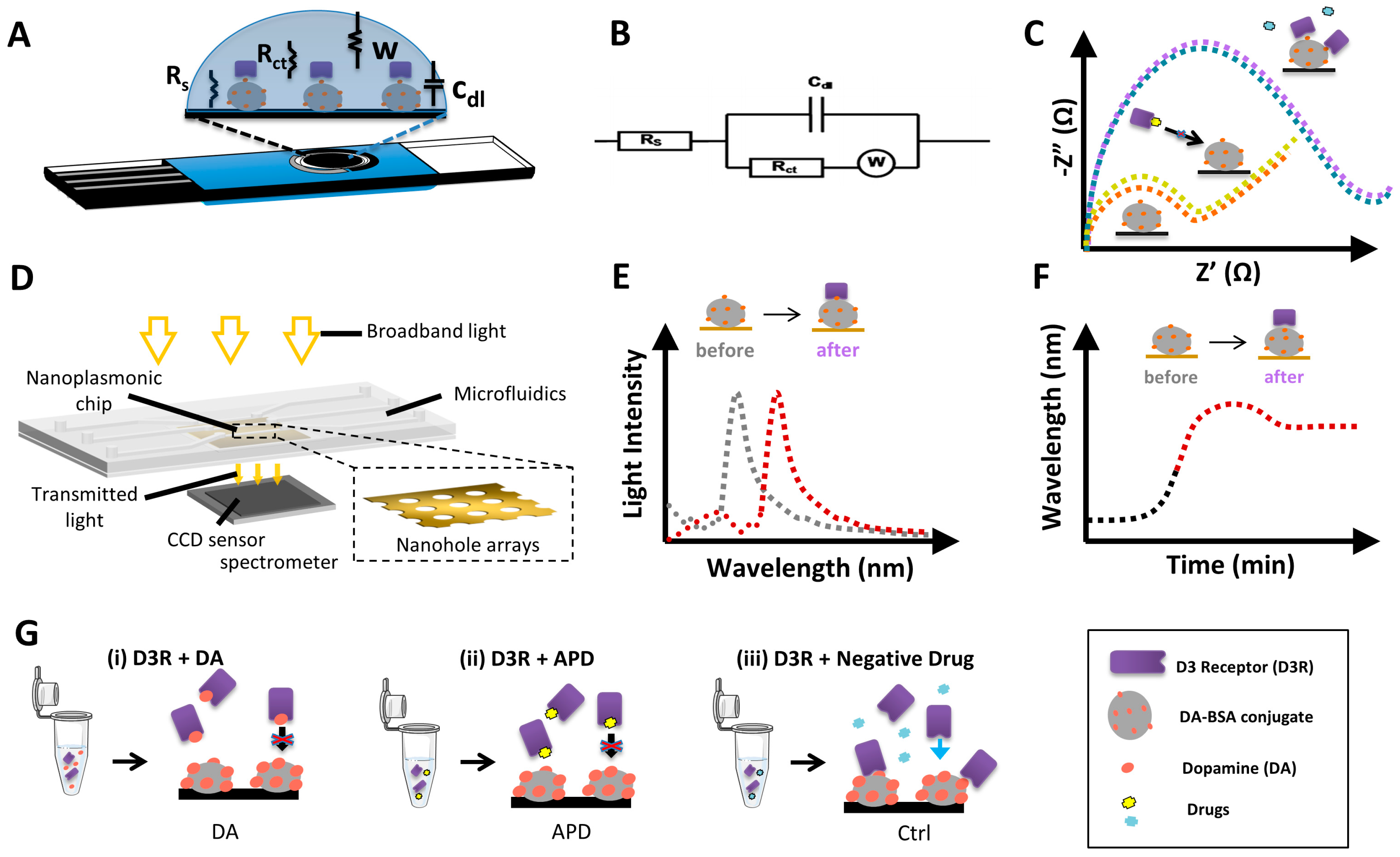
3.1. Design of Biosensor Strategies for D3R Competitive Inhibition Assay for APD Screening
3.2. Electrochemical Biosensor for APD Drug Screening
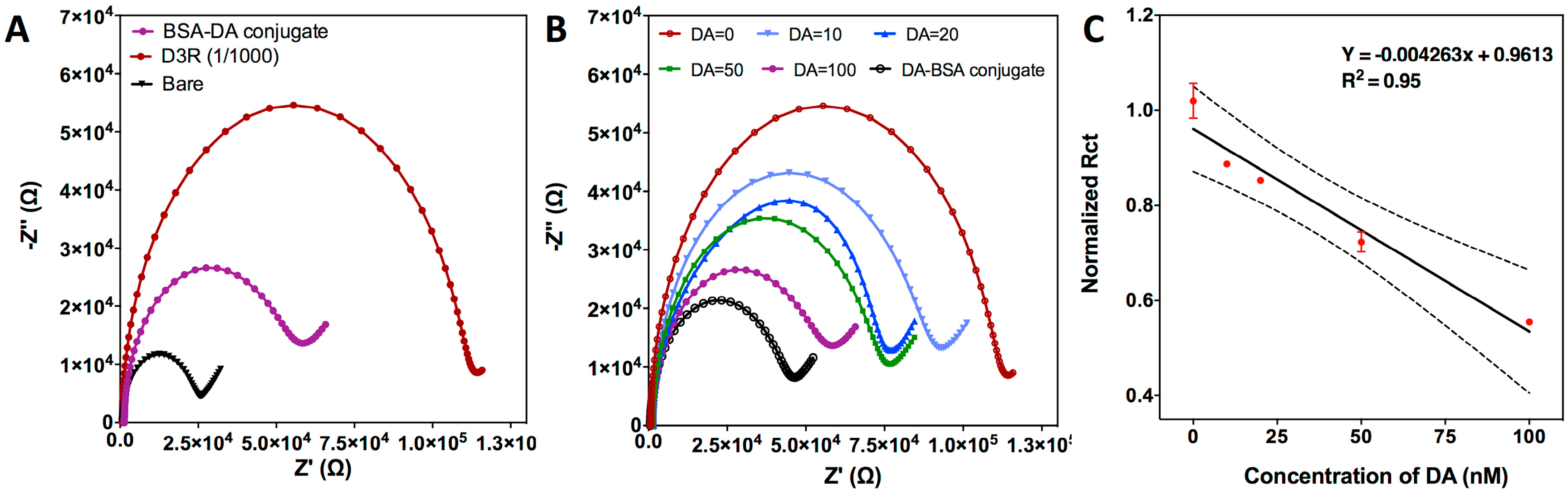
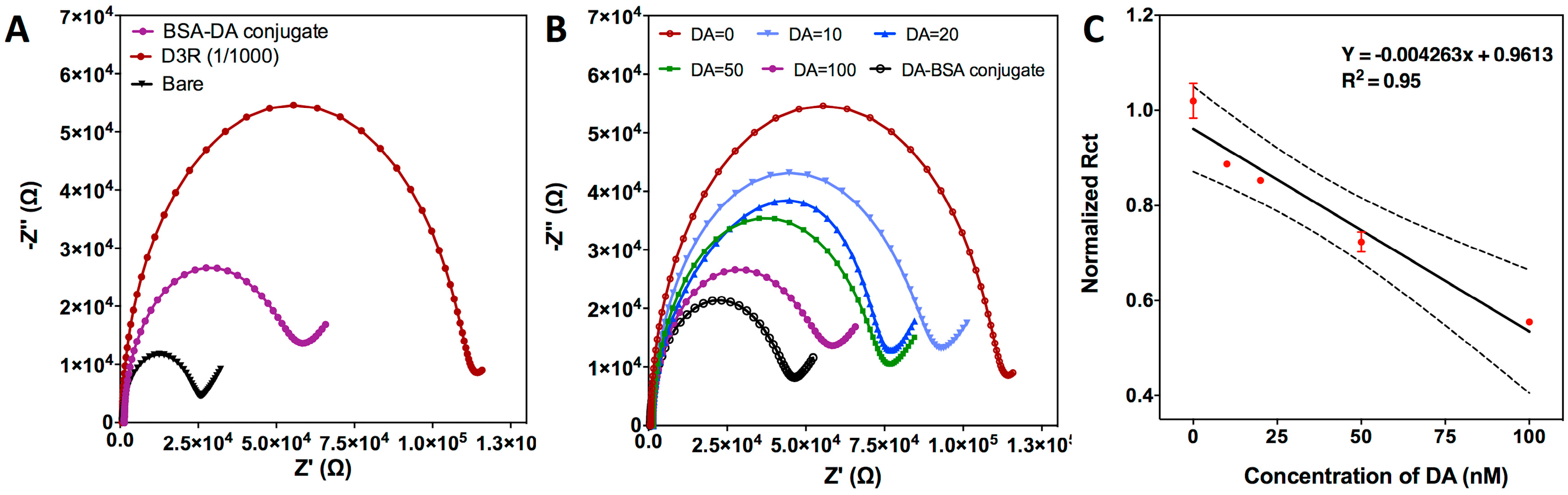
3.2.1. Characterization of the Electrochemical Biosensor
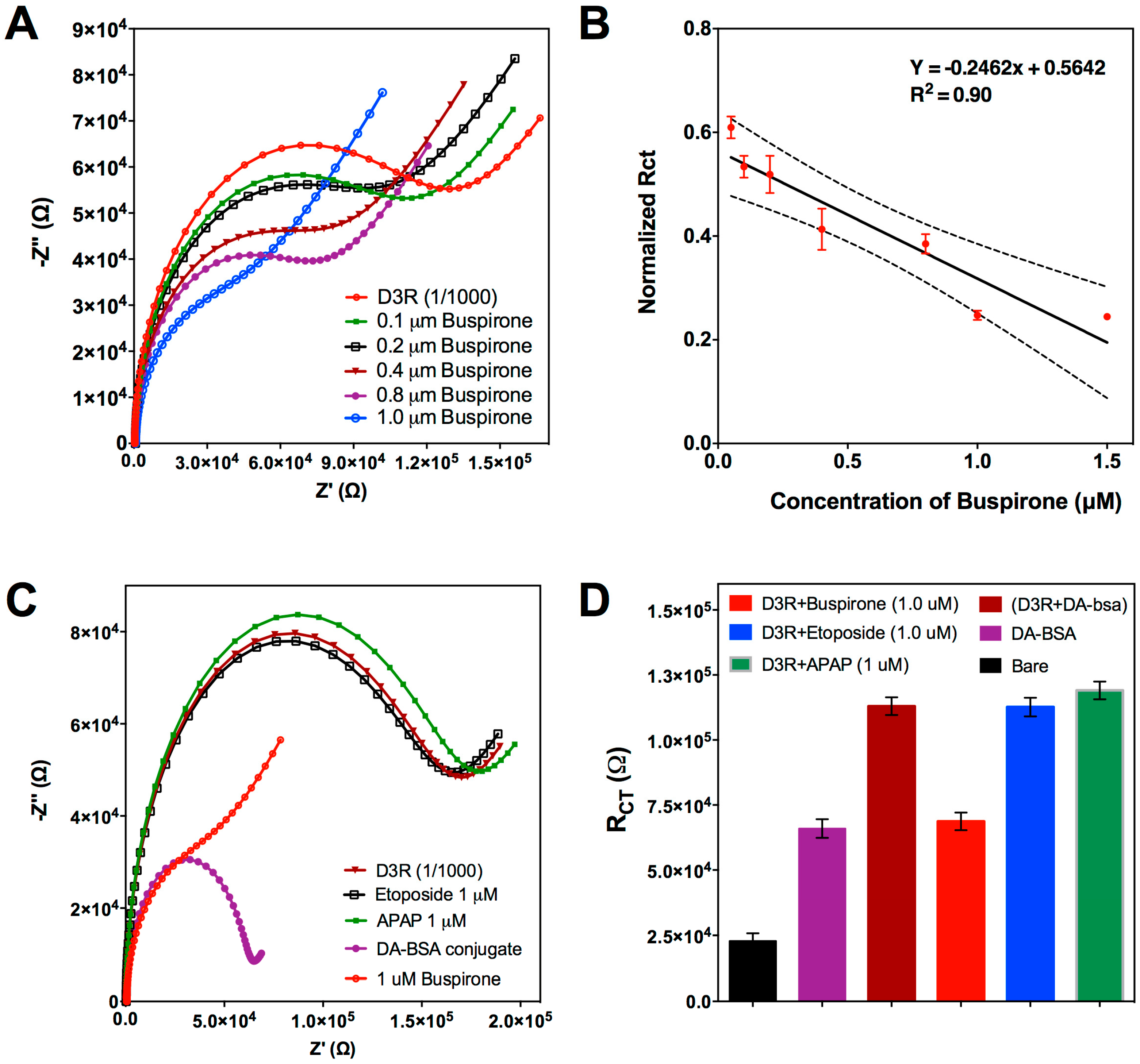
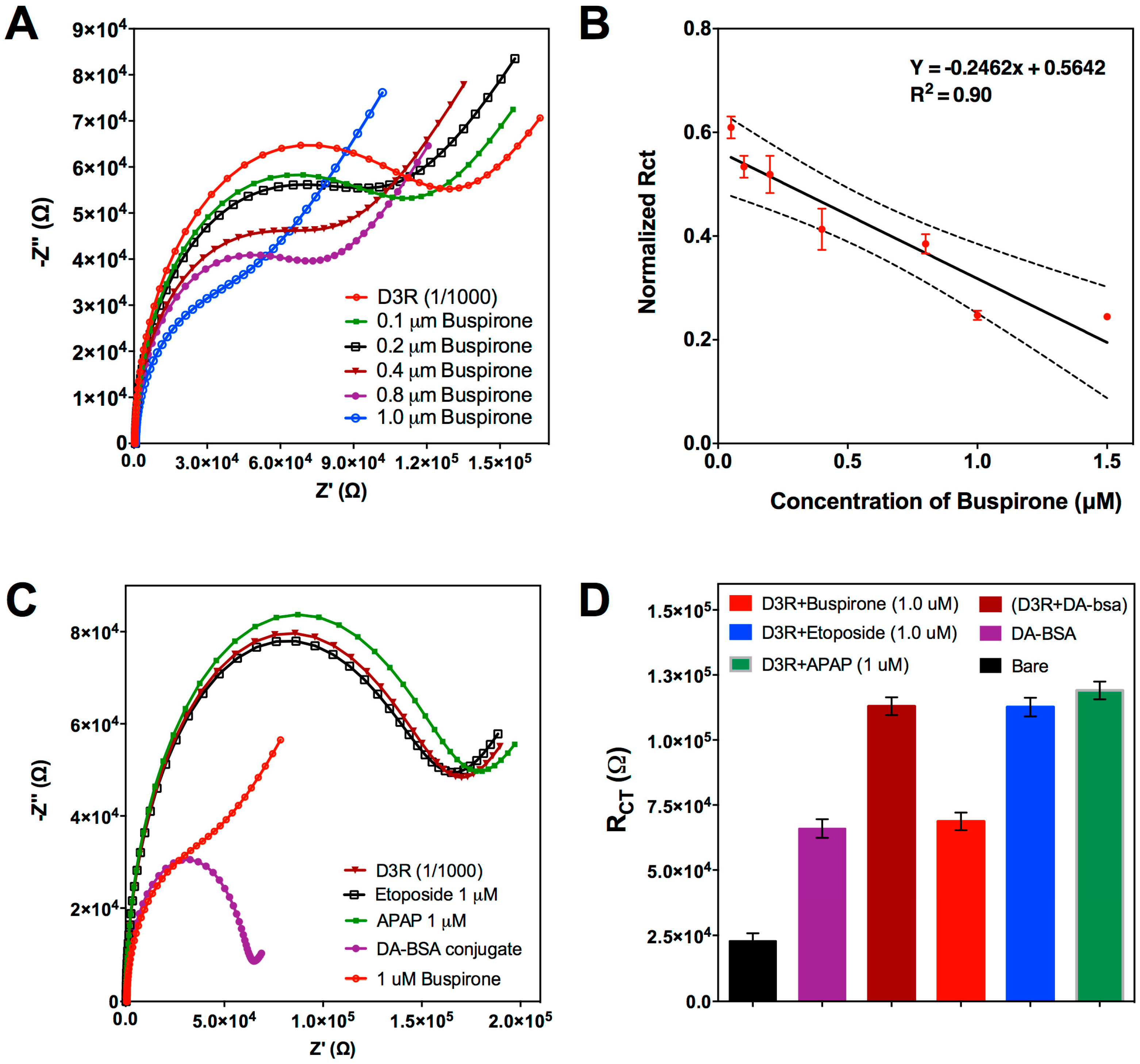
3.2.2. Competitive Inhibition Assay for D3R-Antagonist Antipsychotic Drug: Detection of Buspirone and Specificity Controls
3.3. Nanoplasmonic Biosensor for APD Drug Screening
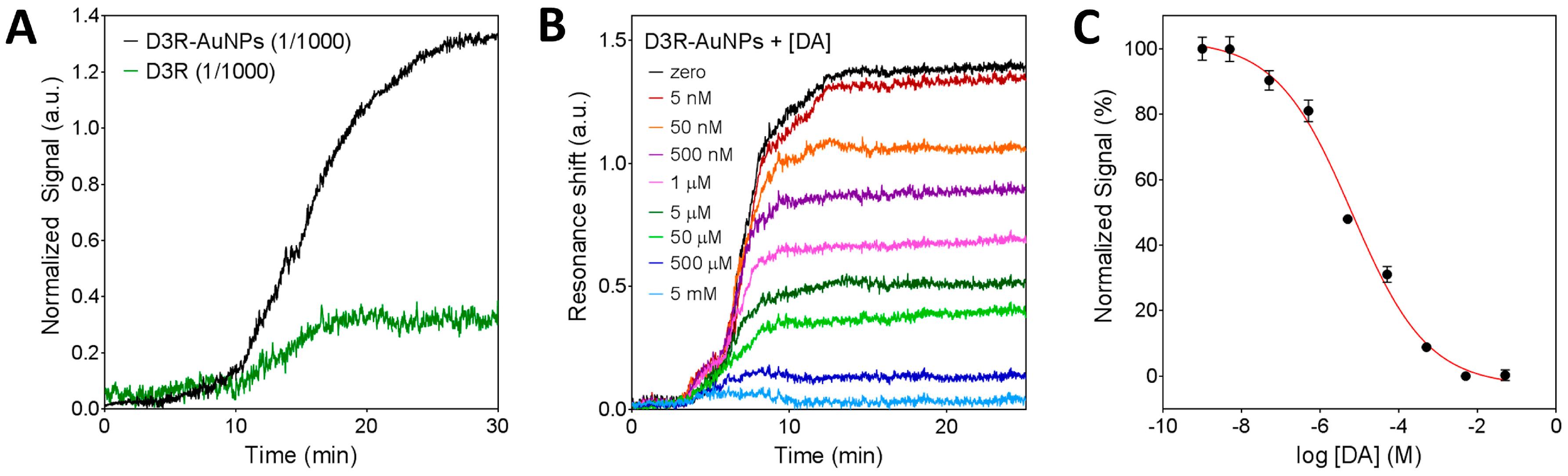
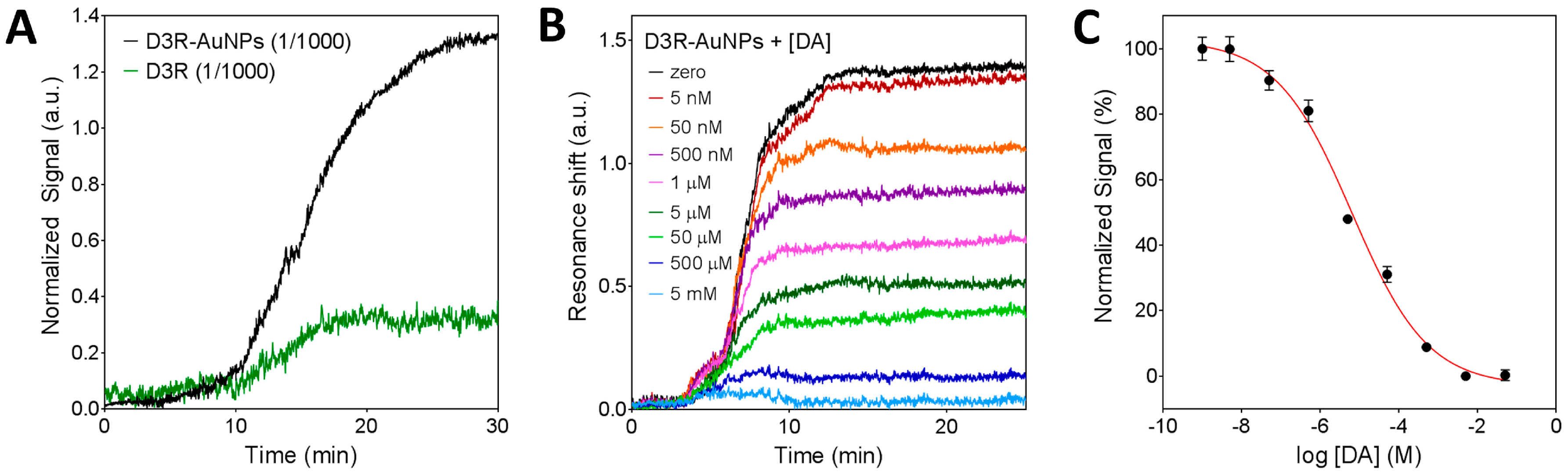
3.3.1. Characterization of the Nanoplasmonic Biosensor
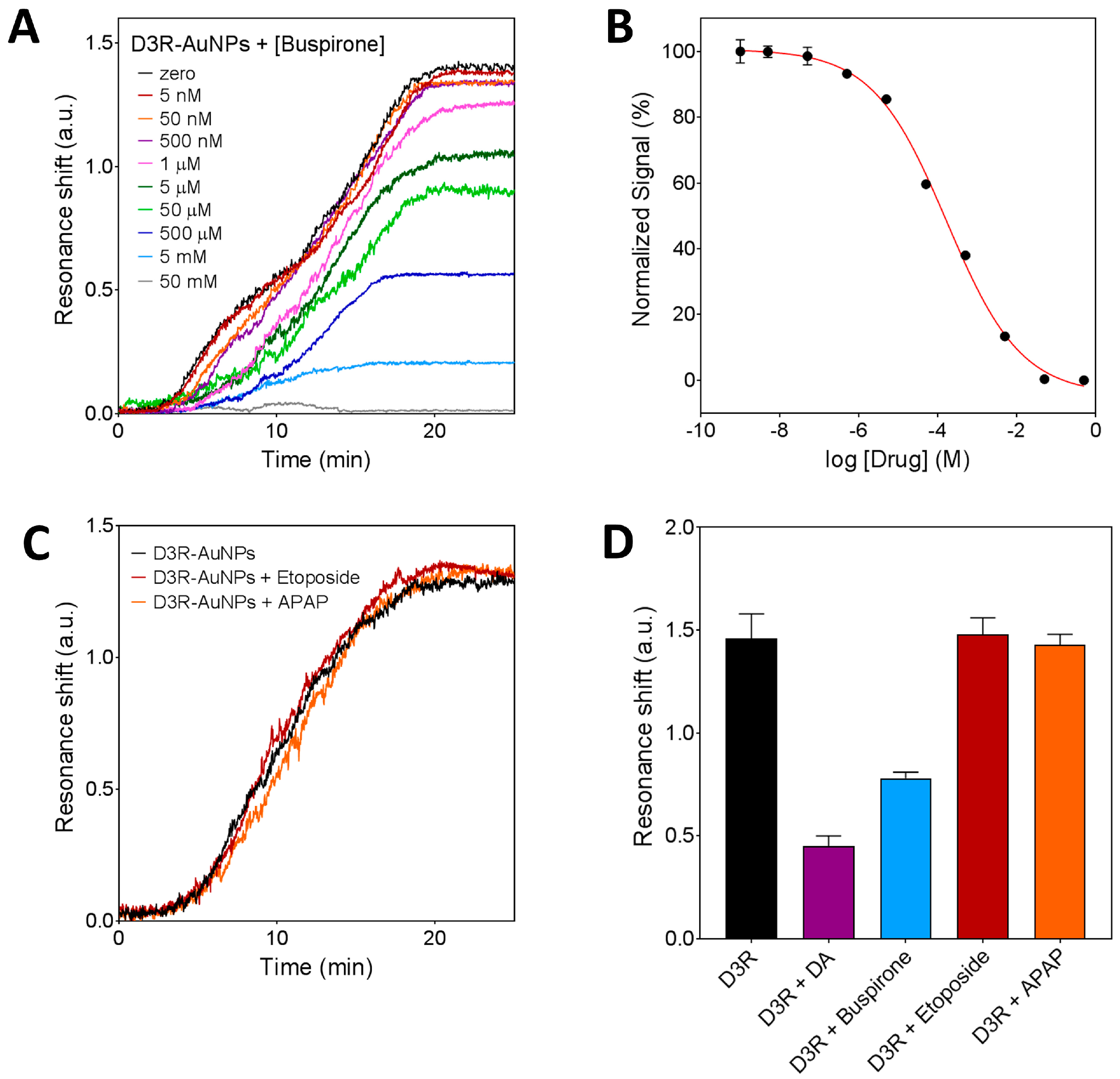
3.3.2. Inhibition Assay for D3R-Antagonist Antipsychotic Drug: Detection of Buspirone and Specificity Controls
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bunnage, M.E. Getting pharmaceutical R&D back on target. Nat. Chem. Biol. 2011, 7, 335–339. [Google Scholar] [PubMed]
- Biotechnology Innovation Organization. Clinical Development Success Rates 2006–2015. Available online: https://www.bio.org/sites/default/files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf (accessed on 8 January 2018).
- Arrowsmith, J. A decade in drug discovery. Nat. Rev. Drug Discov. 2012, 11, 3. [Google Scholar]
- Middelkamp, H.H.T.; van der Meer, A.D.; Marjan, H.J.; Stamatialis, D.F.; Mummery, C.L.; Passier, R.; IJzerman, M.J. Organs-on-Chips in Drug Development: The Importance of Involving Stakeholders in Early Health Technology Assessment. Appl. In Vitro Toxicol. 2016, 2, 74–81. [Google Scholar] [CrossRef]
- Low, L.A.; Tagle, D.A. Microphysiological Systems (“Organs-on-Chips”) for Drug Efficacy and Toxicity Testing. Clin. Transl. Sci. 2017, 10, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Mousavi Shaegh, S.A.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.; Hargreaves, R. Clinical biomarkers in drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B. Functional genomics to uncover drug mechanism of action. Nat. Chem. Biol. 2015, 11, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Kato, G. Use of the Drug-Receptor Binding Assay for Drug Screening. Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 569–572. [Google Scholar] [CrossRef]
- Bateman, D.N. Antipsychotic drugs. Medicine 2012, 40, 105–106. [Google Scholar] [CrossRef]
- Carlsson, A. Antipsychotic drugs, neurotransmitters, and schizophrenia. Am. J Psychiatry 1978, 135, 164–173. [Google Scholar] [CrossRef]
- Kinon, B.J.; Lieberman, J.A. Mechanisms of action of atypical antipsychotic drugs: A Critical analysis. Psychopharmacology 1996, 124, 2–34. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Cortés, R.; Gueye, B.; Probst, A.; Palacios, J.M. Dopamine receptors in human brain: Autoradiographic distribution of D2 sites. Neuroscience 1989, 28, 275–290. [Google Scholar] [CrossRef]
- Deutch, A.Y.; Duman, R.S. The effects of antipsychotic drugs on Fos protein expression in the prefrontal cortex: Cellular localization and pharmacological characterization. Neuroscience 1996, 70, 377–389. [Google Scholar] [CrossRef]
- Fang, Y. Ligand–receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 2012, 7, 969–988. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Blankert, B.; Viré, J.C.; Kauffmann, J.M. Biosensors in Drug Discovery and Drug Analysis. Anal Lett. 2005, 38, 1687–1701. [Google Scholar] [CrossRef]
- Cooper, M.A. Optical biosensors in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yoav, H.; Winkler, T.E.; Kim, E.; Chocron, S.E.; Kelly, D.L.; Payne, G.F.; Ghodssi, R. Redox cycling-based amplifying electrochemical sensor for in situ clozapine antipsychotic treatment monitoring. Electrochim. Acta 2014, 130, 497–503. [Google Scholar] [CrossRef]
- Sanghavi, B.J.; Wolfbeis, O.S.; Hirsch, T.; Swami, N.S. Nanomaterial-based electrochemical sensing of neurological drugs and neurotransmitters. Microchim. Acta 2015, 182, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Carrara, S. Electrochemical detection of a novel therapeutic compound for Schizophrenia. Sens. IEEE 2016, 8–10. [Google Scholar] [CrossRef]
- Kim, Y.R.; Bong, S.; Kang, Y.J.; Yang, Y.; Mahajan, R.K.; Kim, J.S.; Kim, H. Electrochemical detection of dopamine in the presence of ascorbic acid using graphne modified electrodes. Biosens. Bioelectron. 2010, 25, 2366–2369. [Google Scholar] [CrossRef] [PubMed]
- Rozniecka, E.; Jonsson-Niedziolka, M.; Celebanska, A.; Niedziolka-Jonsson, J.; Opallo, M. Selective electrochemical detection of dopamine in a microfluidic channel on carbon nanoparticulate electrodes. Analyst 2014, 139, 2896–2903. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Nazal, M.K.; Mansha, M.; Alsharaa, A.; Jillani, S.M.S.; Basheer, C. Chemically modified electrodes for electrochemical detection of dopamine in the presence of uric acid and ascorbic acid: A review. Trends Anal. Chem. 2016, 76, 15–29. [Google Scholar] [CrossRef]
- Yusoff, N.; Pandikumar, A.; Ramaraj, R.; Lim, H.N.; Huang, N.M. Gold nanoparticle based optical and electrochemical sensing of dopamine. Microchim. Acta 2015, 182, 2091–2114. [Google Scholar] [CrossRef]
- Raj, D.R.; Prasanth, S.; Vineeshkumar, T.V.; Sudarsanakumar, C. Surface plasmon resonance based fiber optic dopamine sensor using green synthesized silver nanoparticles. Sens. Actuator B Chem. 2016, 224, 600–606. [Google Scholar]
- Kilic, T.; Brunner, V.; Audoly, L.; Carrara, S. A novel psychoanalytical approach: An electrochemical ligand-binding assay to screen antipsychotics. Biosens. Bioelectron. 2017, 100, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Belushkin, A.; Cavallini, A.; Kebbi-Beghdadi, C.; Greub, G.; Altug, H. Multiplexed nanoplasmonic biosensor for one-step simultaneous detection of Chlamydia trachomatis and Neisseria gonorrhoeae in urine. Biosens. Bioelectron. 2017, 94, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Mattson, G.; Conklin, E.; Desai, S.; Nielander, G.; Savage, M.D.; Morgensen, S. A practical approach to crosslinking. Mol. Biol. Rep. 1993, 17, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Kumbhat, S.; Shankaran, D.R.; Kim, S.J.; Gobi, K.V.; Joshi, V.; Miura, N. Surface plasmon resonance biosensor for dopamine using D3 dopamine receptor as a biorecognition molecule. Biosens. Bioelectron. 2007, 23, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.R.; Zhang, Y.S.; Kim, D.J.; Manbohi, A.; Avci, H.; Silvestri, A.; Aleman, J.; Hu, N.; Kilic, T.; Keung, W.; et al. Aptamer-Based Microfluidic Electrochemical Biosensor for Monitoring Cell-Secreted Trace Cardiac Biomarkers. Anal. Chem. 2016, 88, 10019–10027. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.R.; Kilic, T.; Zhang, Y.S.; Avci, H.; Hu, N.; Kim, D.; Branco, C.; Aleman, J.; Massa, S.; Silvestri, A.; et al. Label-Free and Regenerative Electrochemical Microfluidic Biosensors for Continual Monitoring of Cell Secretomes. Adv. Sci. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Soler, M.; Özdemir, C.I.; Belushkin, A.; Yesilkoy, F.; Altug, H. Plasmonic nanohole array biosensor for label-free and real-time analysis of live cell secretion. Lab Chip 2017, 17, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Bath, B.D.; Michael, D.J.; Trafton, B.J.; Joseph, J.D.; Runnels, P.L.; Wightman, R.M. Subsecond adsorption and desorption of dopamine at carbon-fiber microelectrodes. Anal. Chem. 2000, 72, 5994–6002. [Google Scholar] [CrossRef] [PubMed]
- Günzler, H.; Williams, A. Handbook of Analytical Techniques. Evolution 2001, 1, 1–2. [Google Scholar]
- Springer, T.; Ermini, M. L.; Spackova, B.; Jablonku, J.; Homola, J. Enhancing sensitivity of surface plasmon resonance biosensors by functionalized gold nanoparticles: Size matters. Anal. Chem. 2014, 86, 10350–10356. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Payer, D.; Di Ciano, P.; Guranda, M.; Nakajima, S.; Tong, J.; Mansouri, E.; Wilson, A.A.; Houle, S.; Meyer, J.F.; et al. Occupancy of Dopamine D3 and D2 Receptors by Buspirone: A [11C]-(+)-PHNO PET Study in Humans. Neuropsychopharmacology 2015, 41, 529. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, Y.; Zhou, J.; Li, Y.; Chen, S.; Zhang, L.; Ma, Y.; Wang, L.; Yan, X. Three-dimensional nitrogen-doped graphene as an ultrasensitive electrochemical sensor for the detection of dopamine. Nanoscale 2015, 7, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilic, T.; Soler, M.; Fahimi-Kashani, N.; Altug, H.; Carrara, S. Mining the Potential of Label-Free Biosensors for In Vitro Antipsychotic Drug Screening. Biosensors 2018, 8, 6. https://doi.org/10.3390/bios8010006
Kilic T, Soler M, Fahimi-Kashani N, Altug H, Carrara S. Mining the Potential of Label-Free Biosensors for In Vitro Antipsychotic Drug Screening. Biosensors. 2018; 8(1):6. https://doi.org/10.3390/bios8010006
Chicago/Turabian StyleKilic, Tugba, Maria Soler, Nafiseh Fahimi-Kashani, Hatice Altug, and Sandro Carrara. 2018. "Mining the Potential of Label-Free Biosensors for In Vitro Antipsychotic Drug Screening" Biosensors 8, no. 1: 6. https://doi.org/10.3390/bios8010006
APA StyleKilic, T., Soler, M., Fahimi-Kashani, N., Altug, H., & Carrara, S. (2018). Mining the Potential of Label-Free Biosensors for In Vitro Antipsychotic Drug Screening. Biosensors, 8(1), 6. https://doi.org/10.3390/bios8010006