
Nanopore-CMOS Interfaces for DNA Sequencing


Abstract
:
1. Introduction
2. Nanopore Sequencing in Context
2.1. Sanger and Next Generation Sequencing
2.2. Next Generation Sequencing CMOS Interfaces
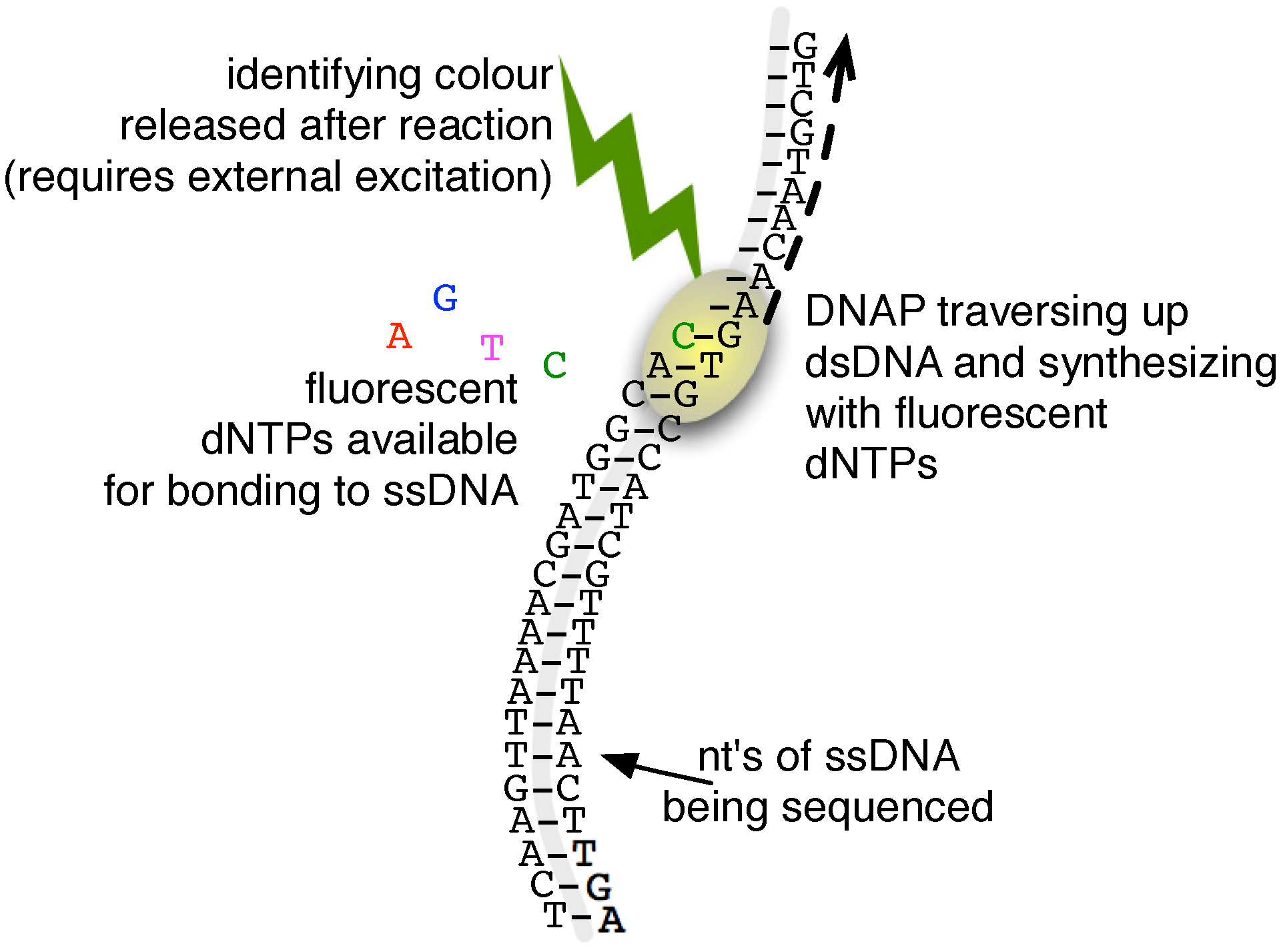
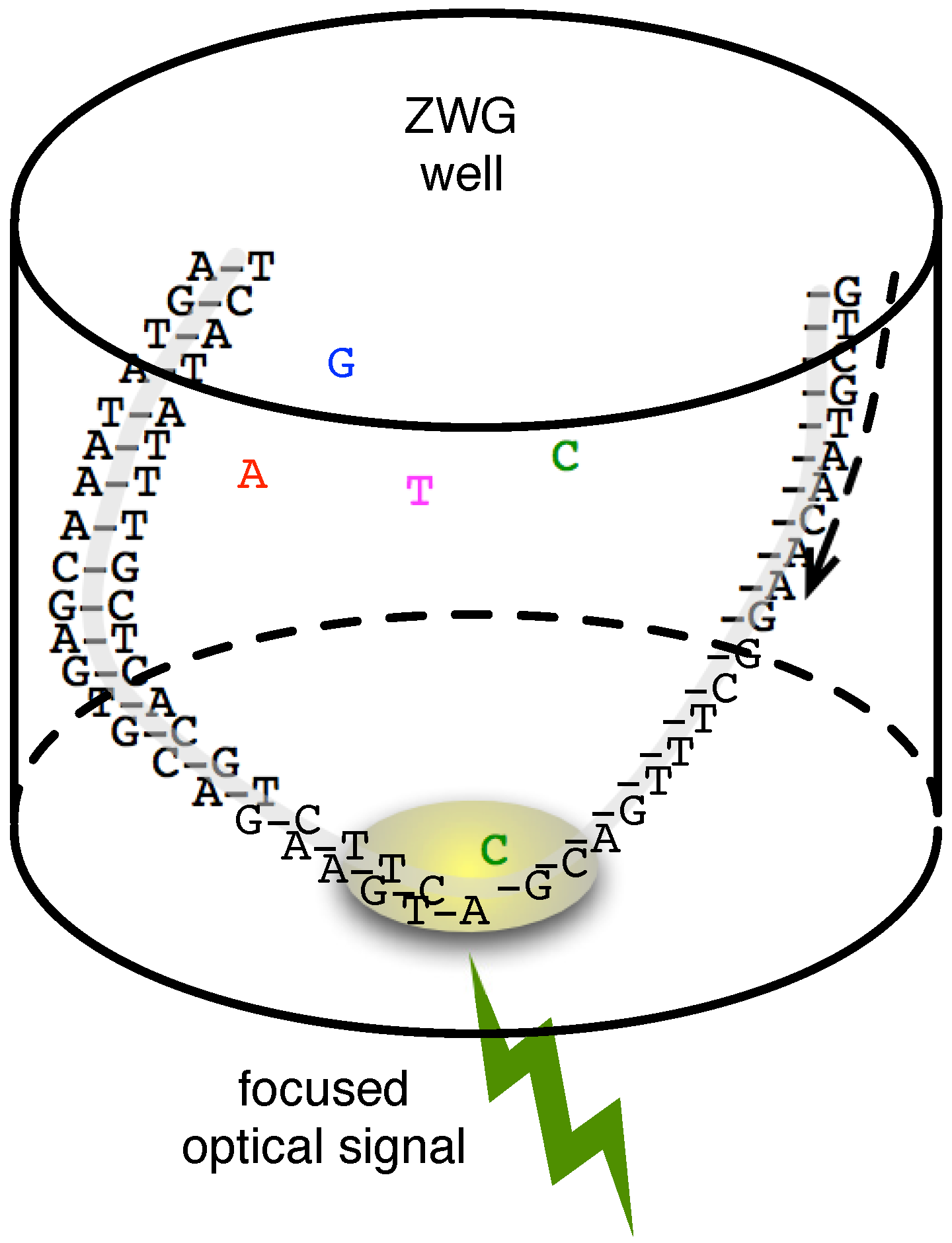
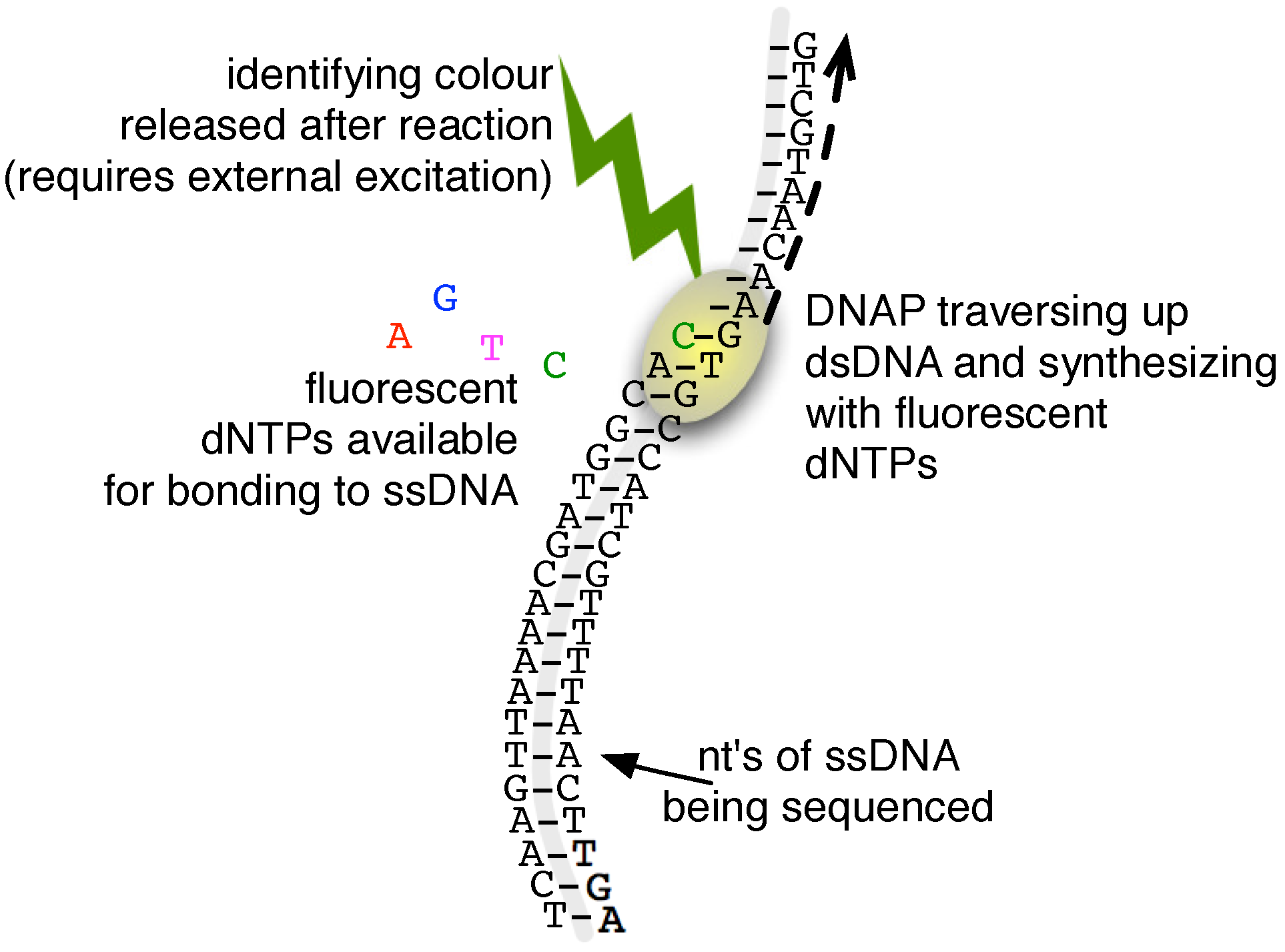
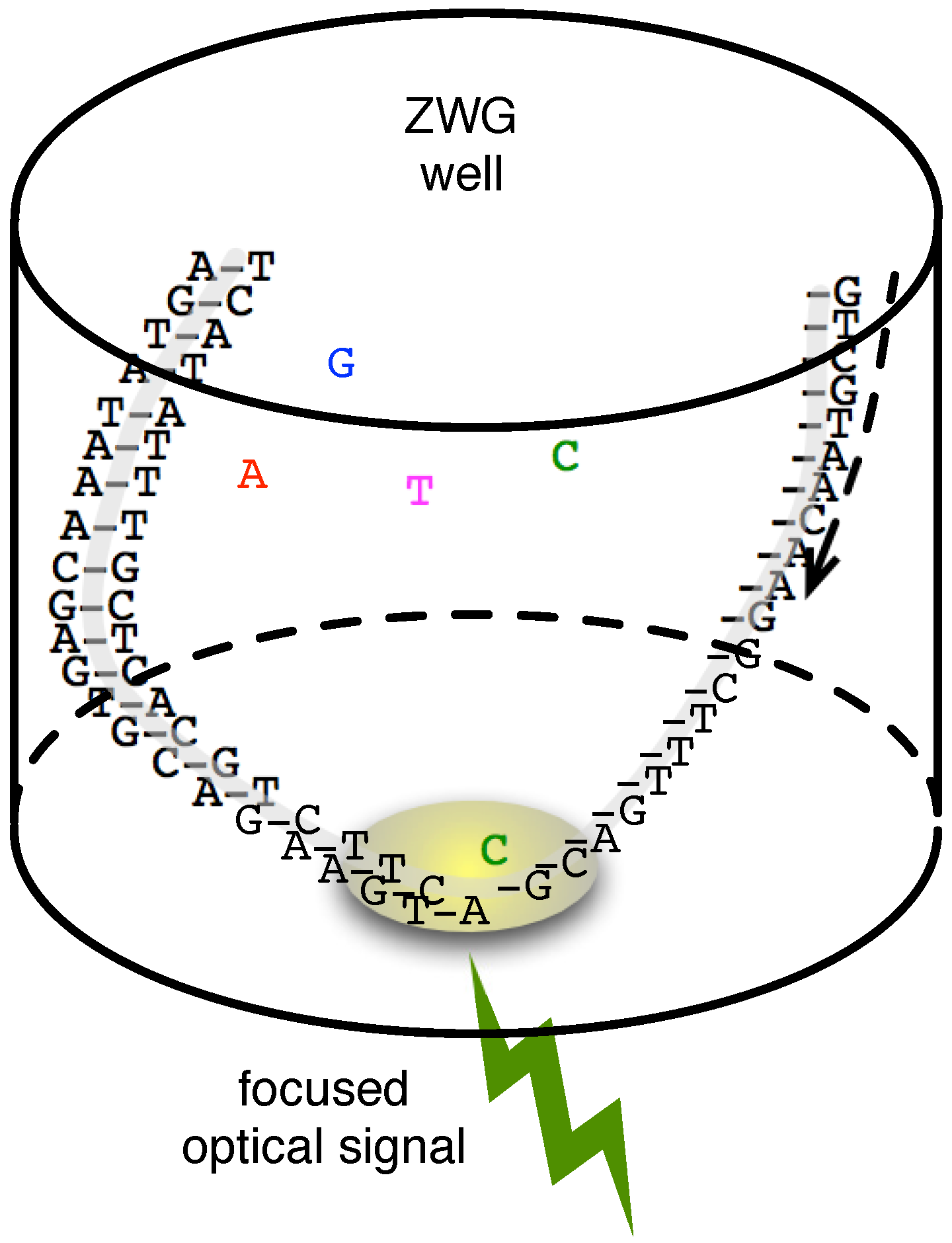
2.3. Single-Molecule Sequencing
3. Nanopores
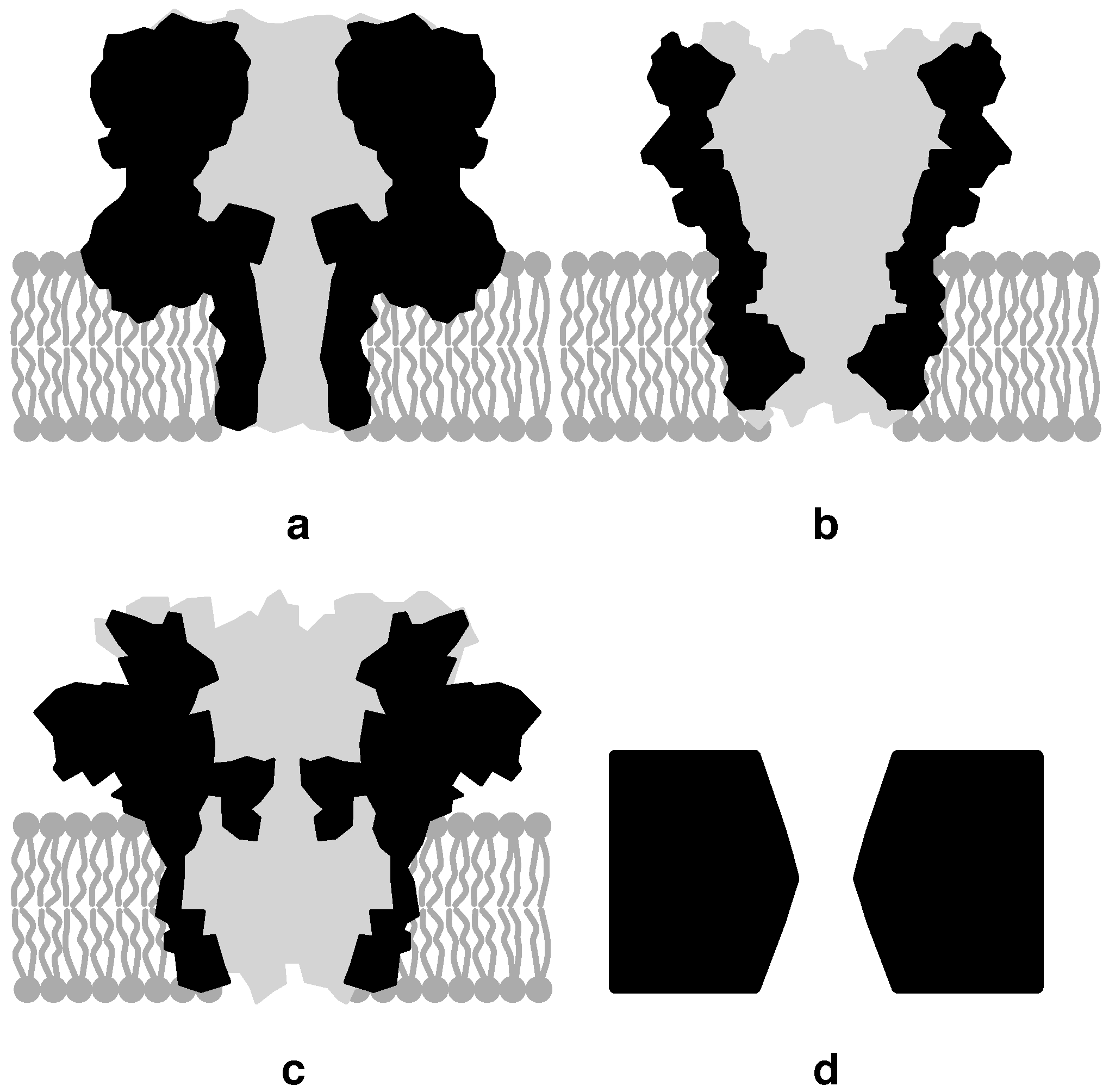
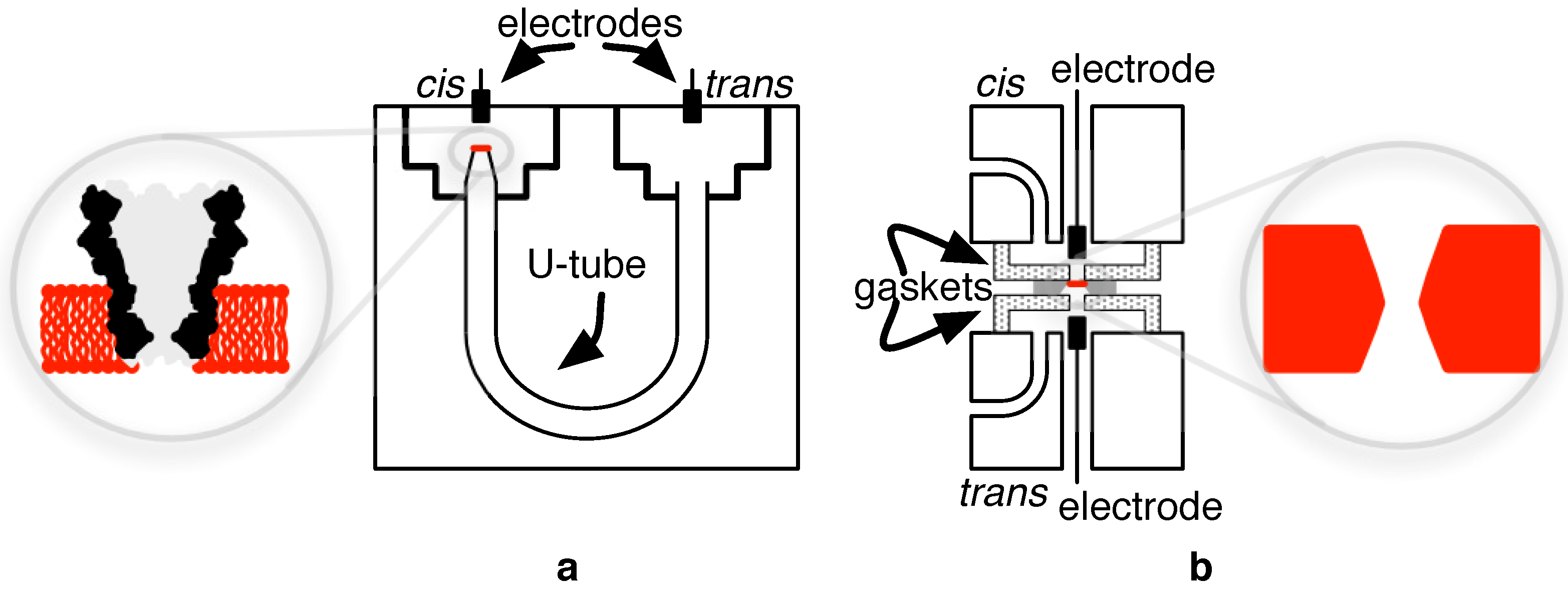
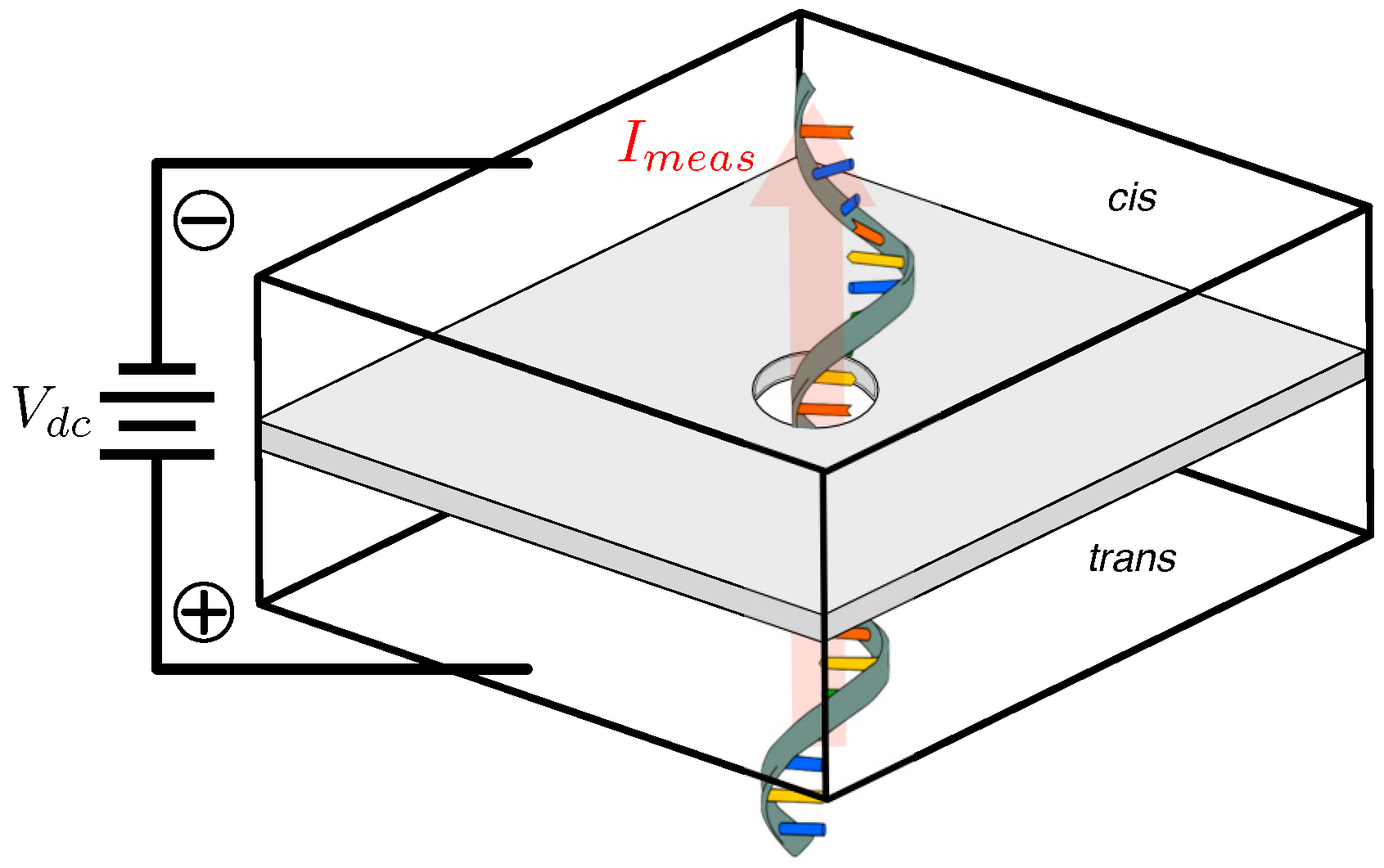
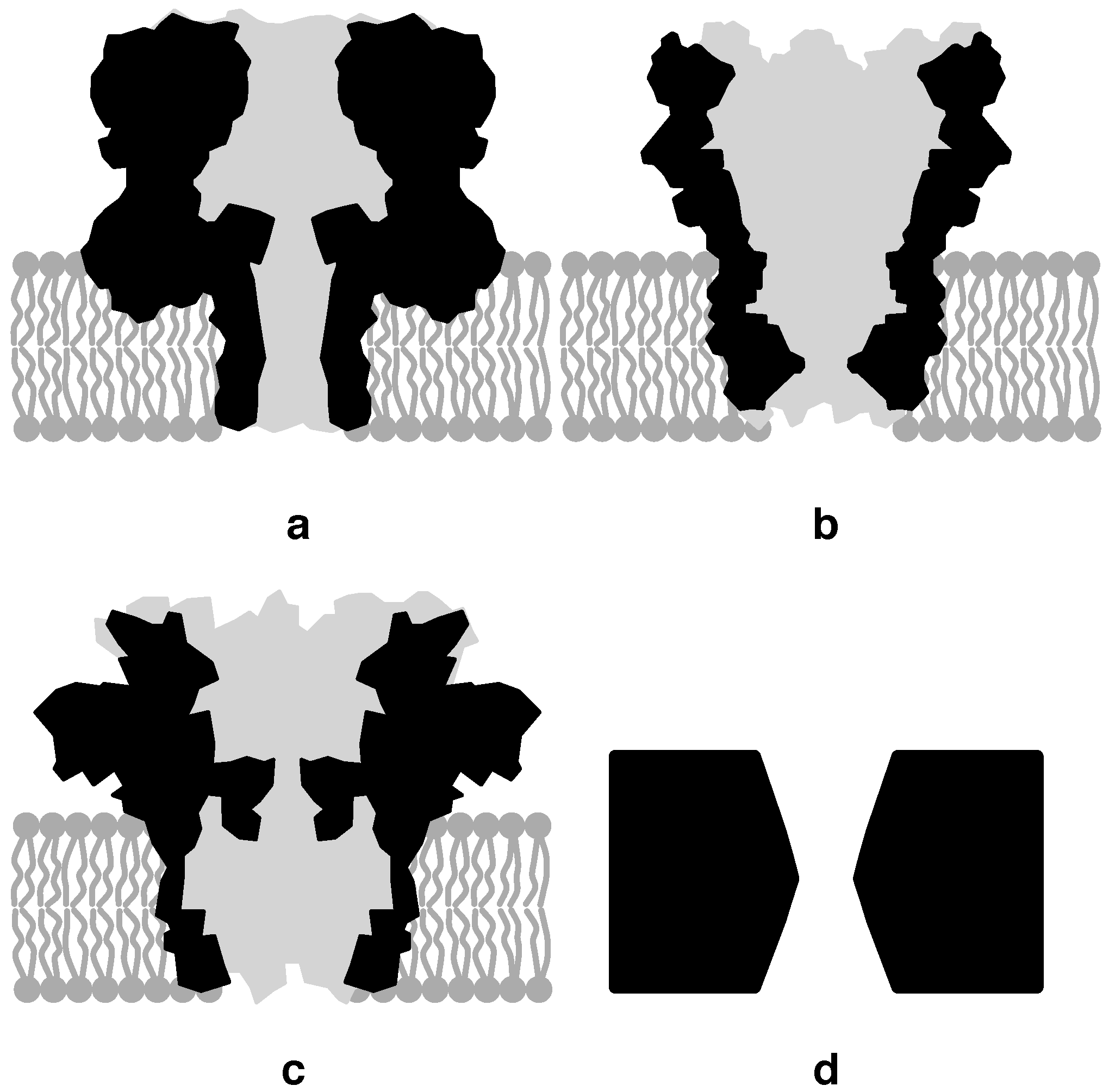
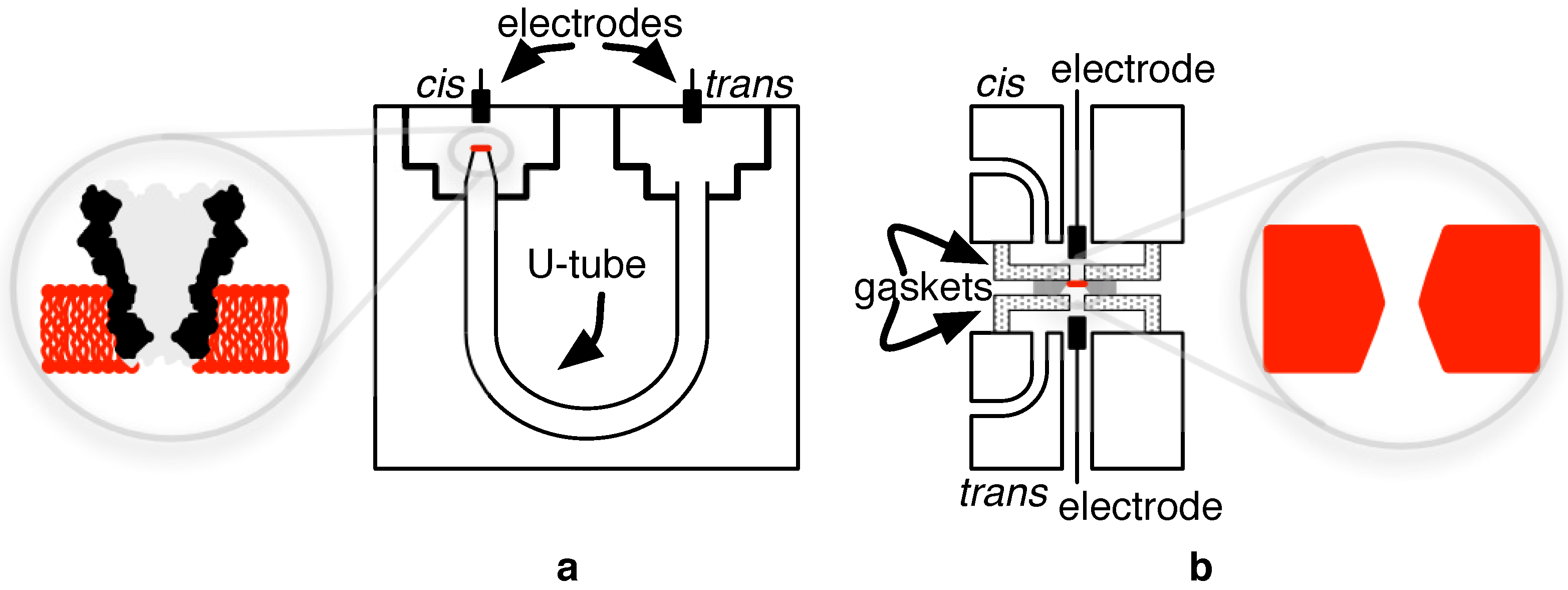
3.1. Biological Nanopores
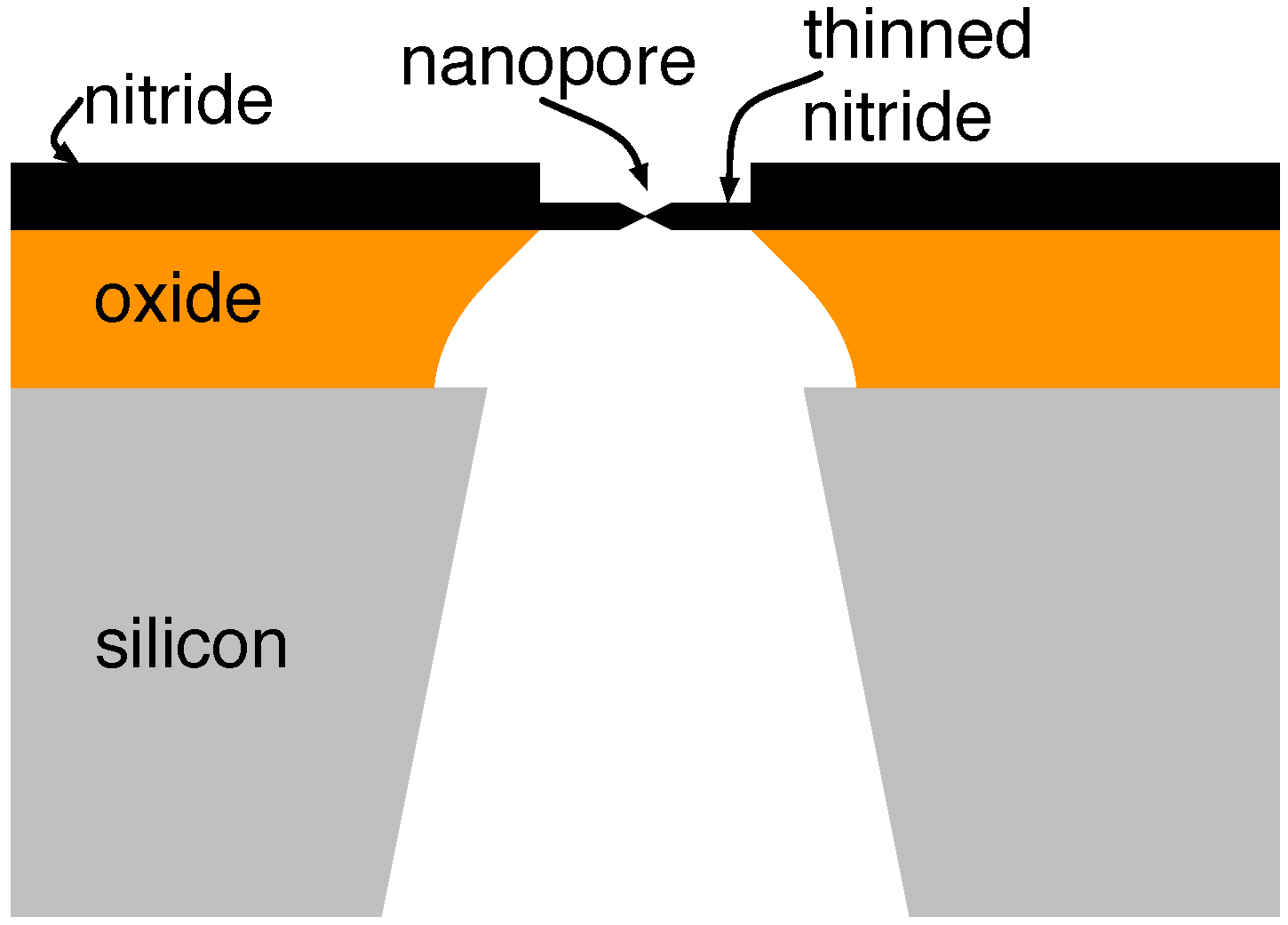
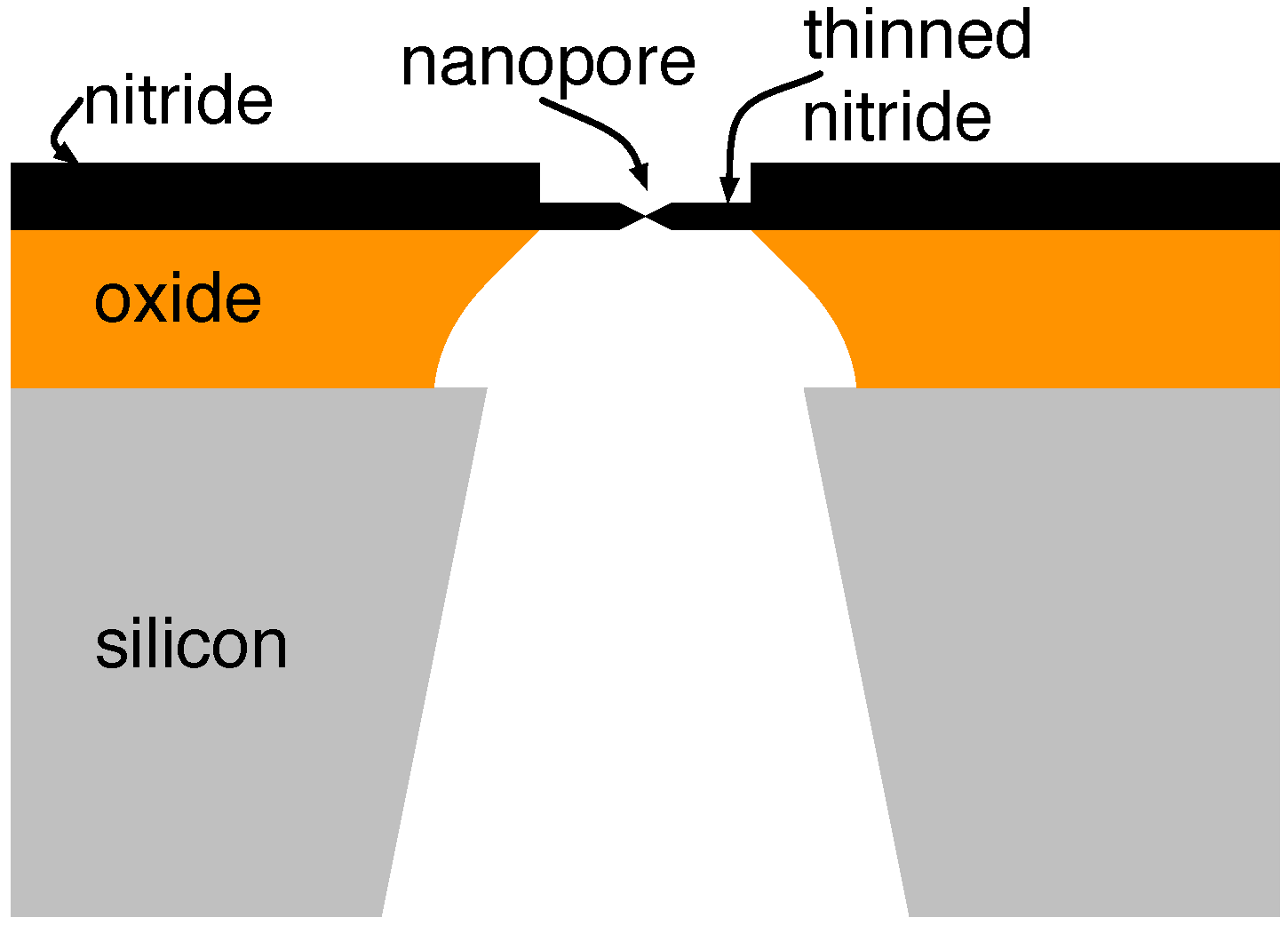
3.2. Solid-State Nanopores
3.3. Nanopore Characteristics and Models
3.3.1. Nanopore Conductance
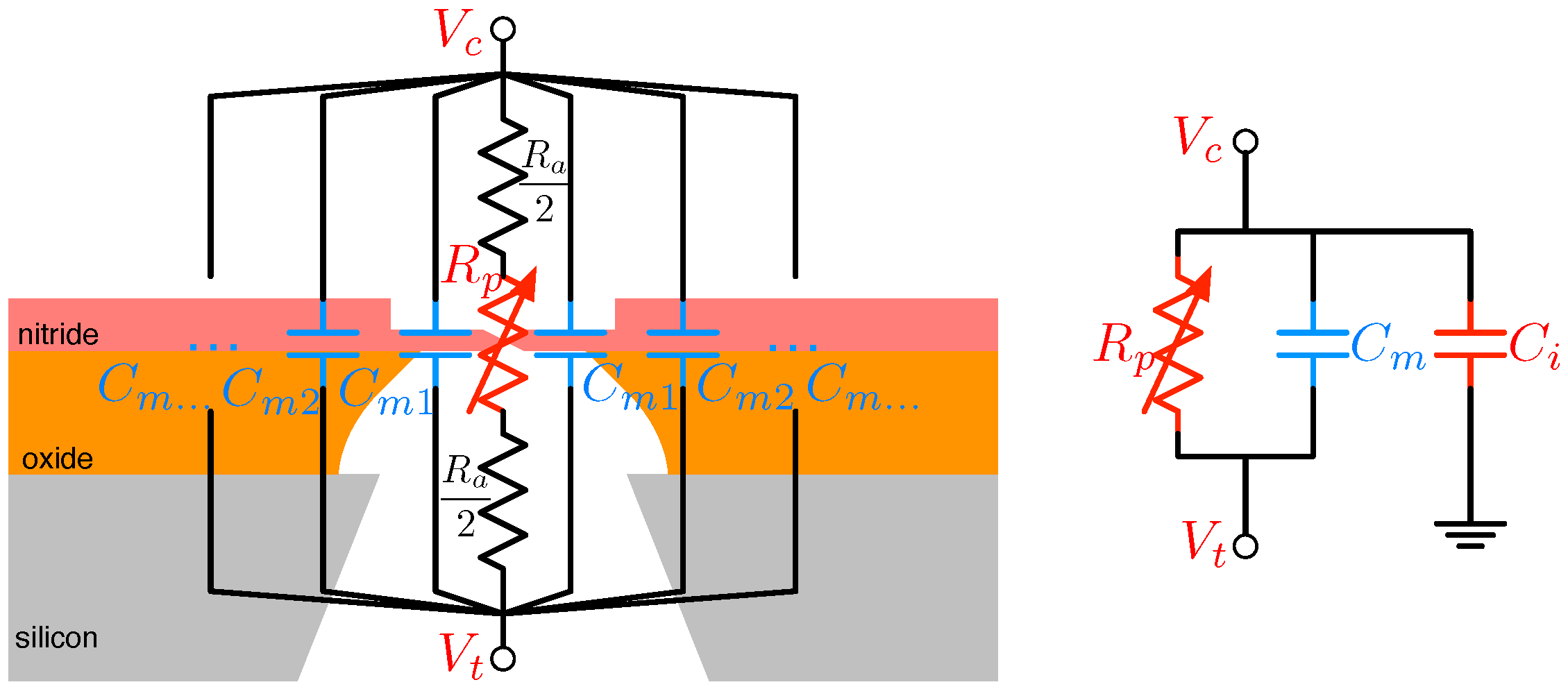
3.3.2. Nanopore Capacitance
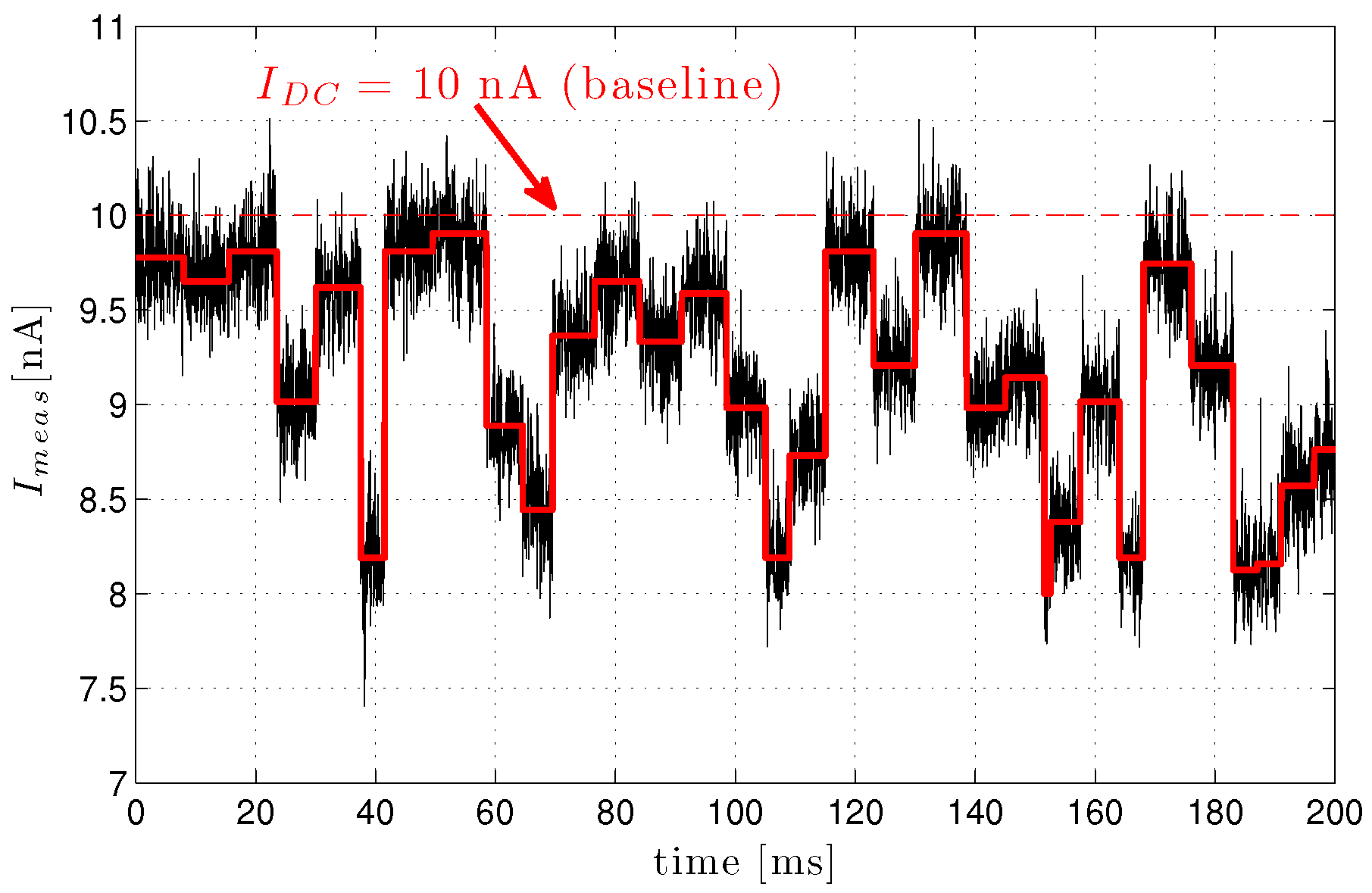
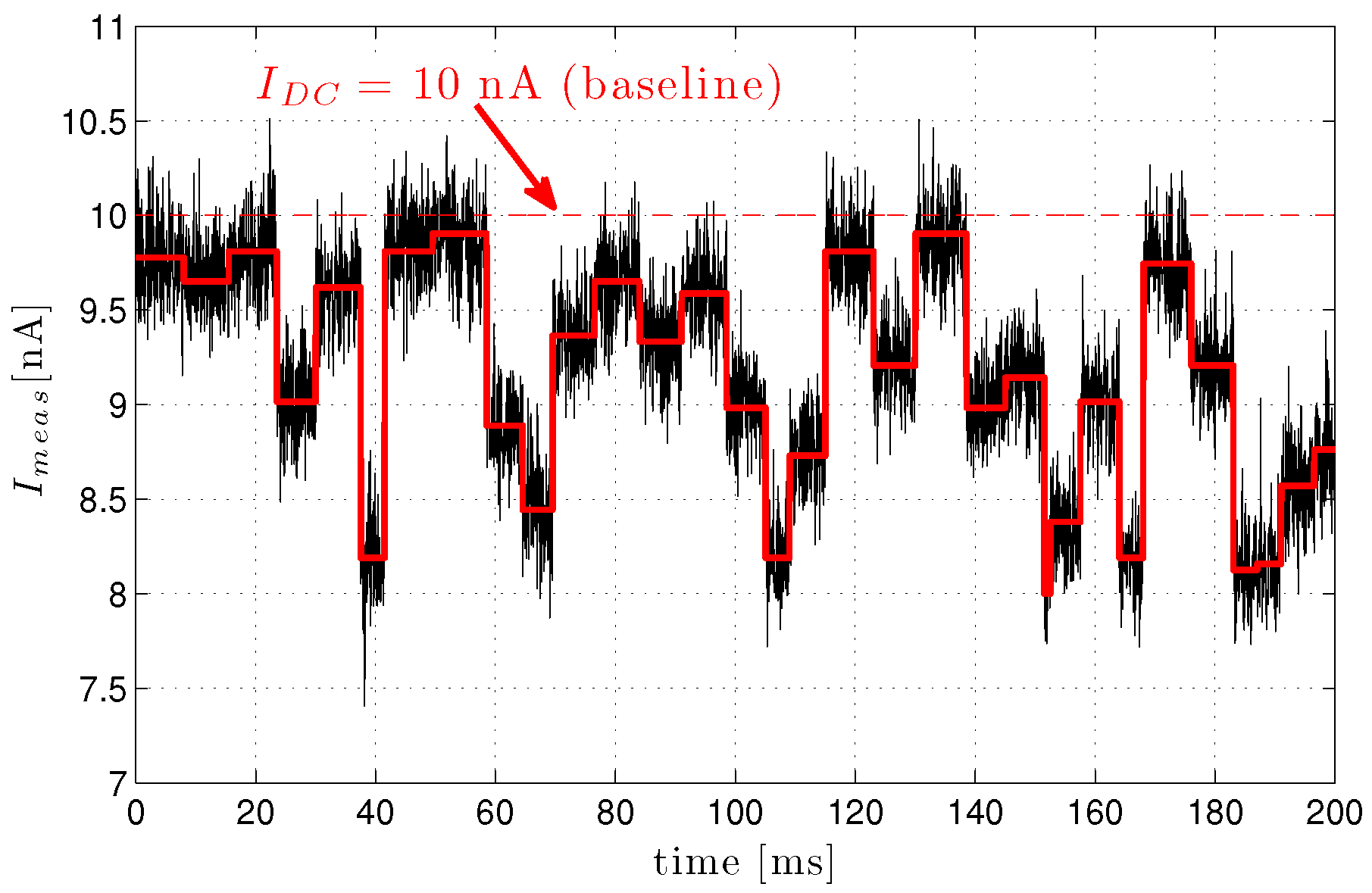
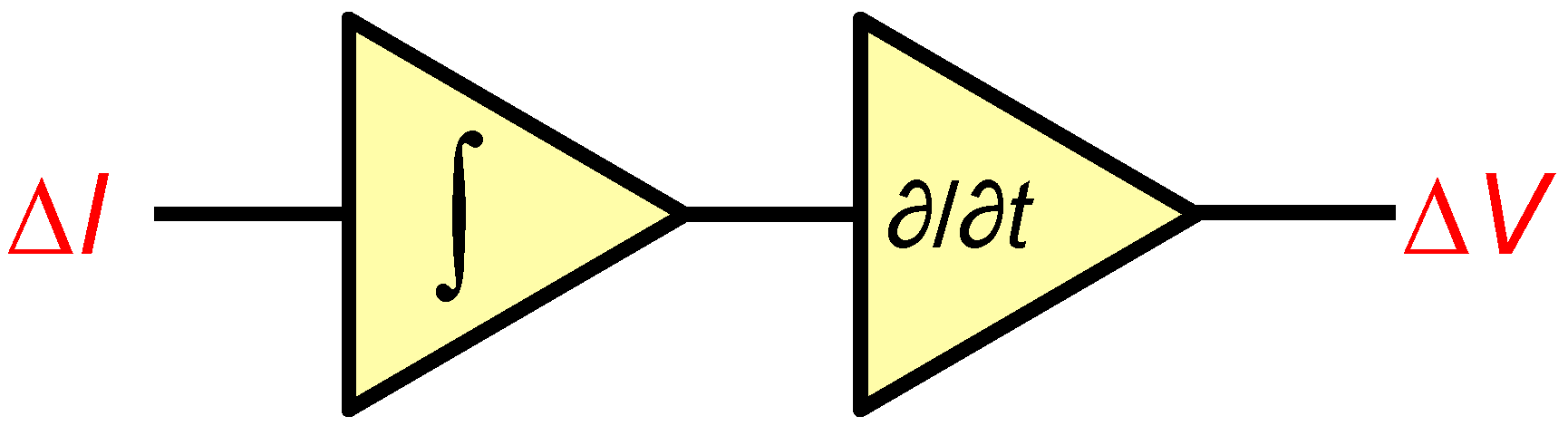
3.3.3. Nanopore Signal Dynamics
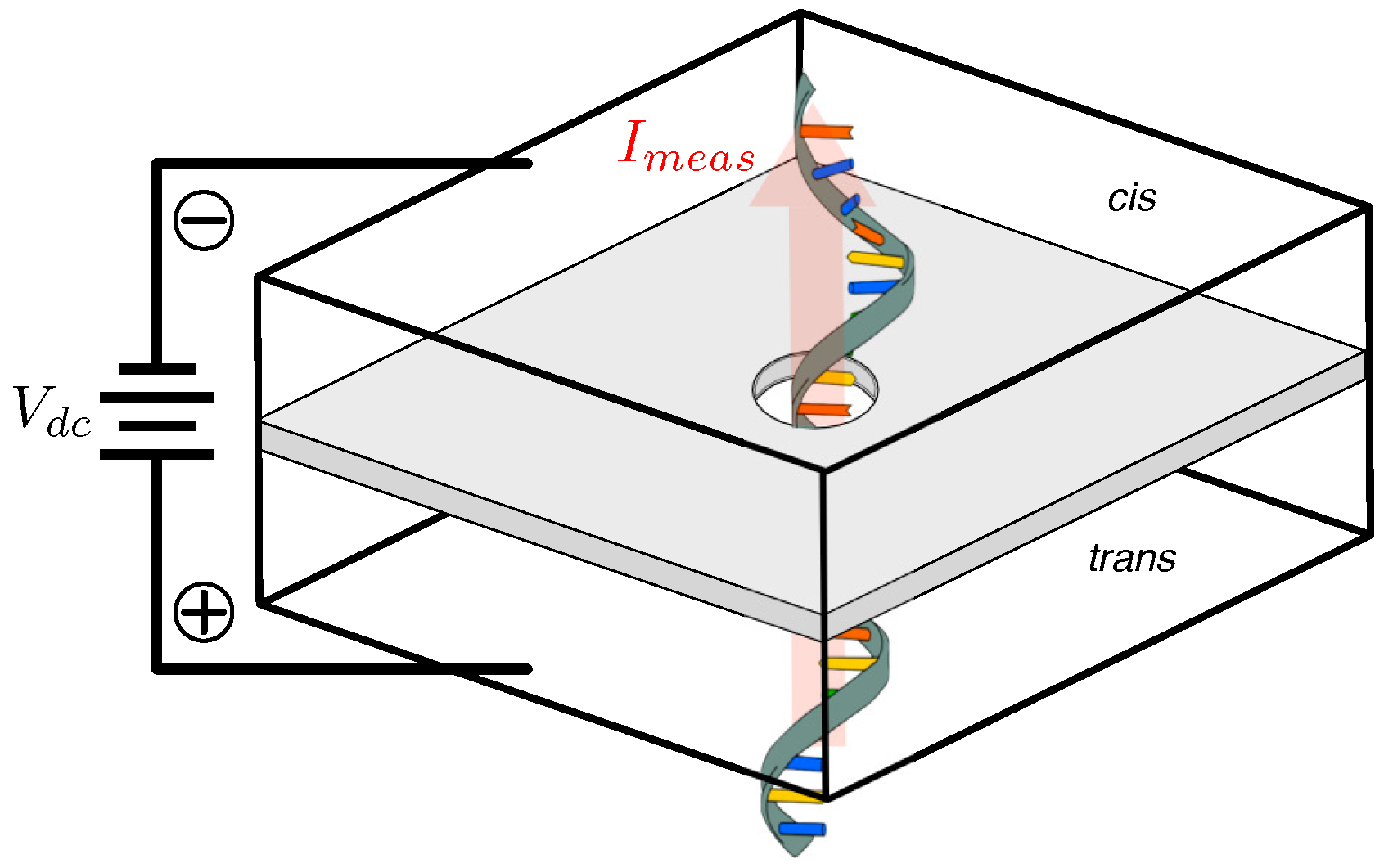
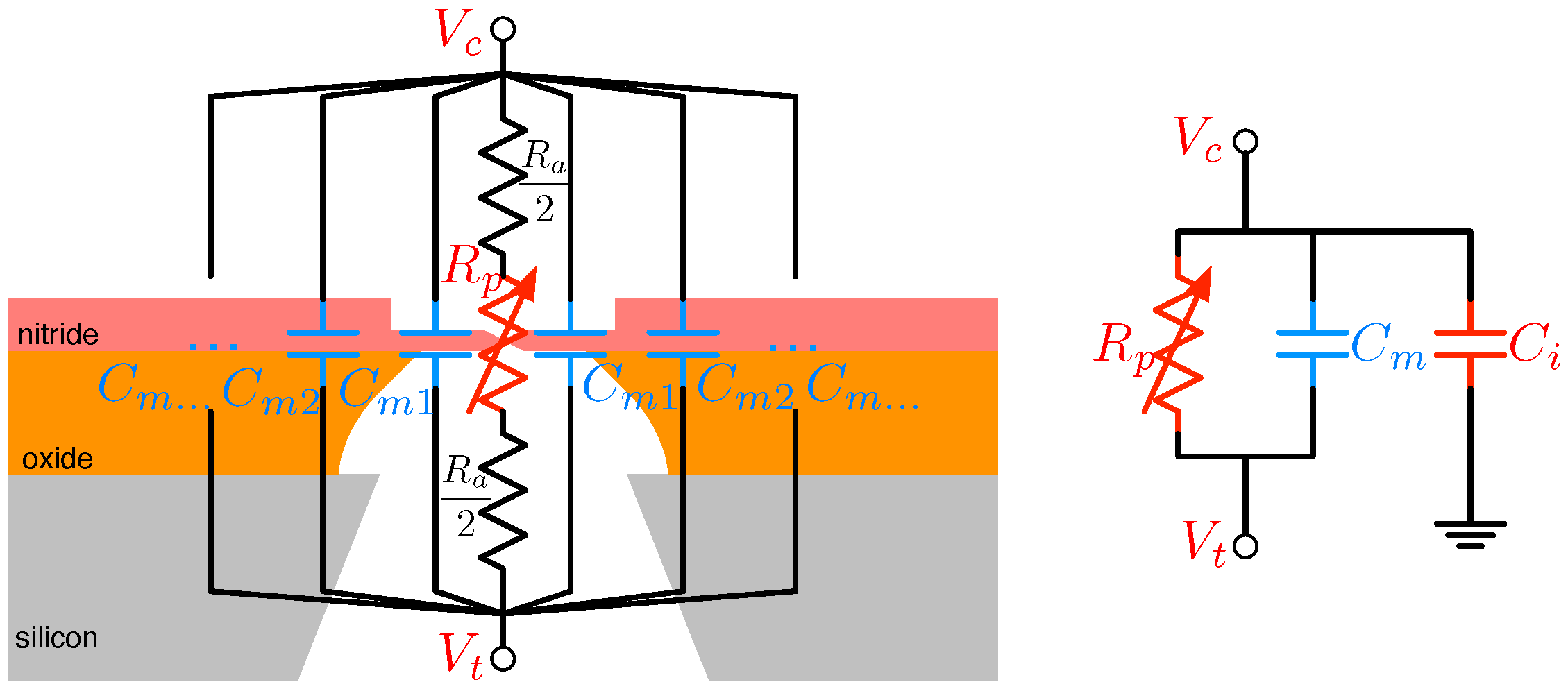
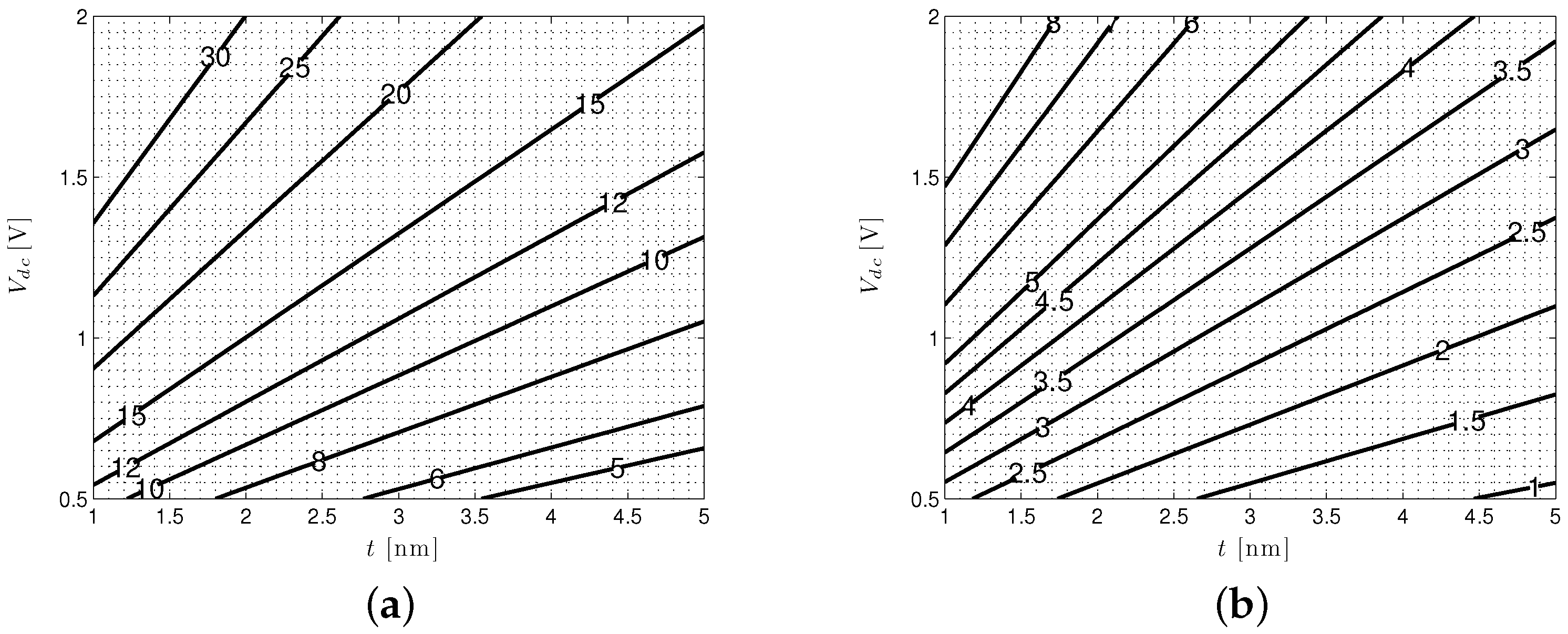
4. Nanopore-CMOS Interface
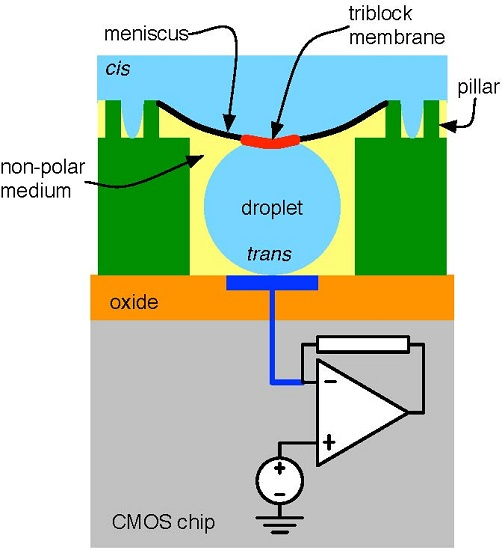
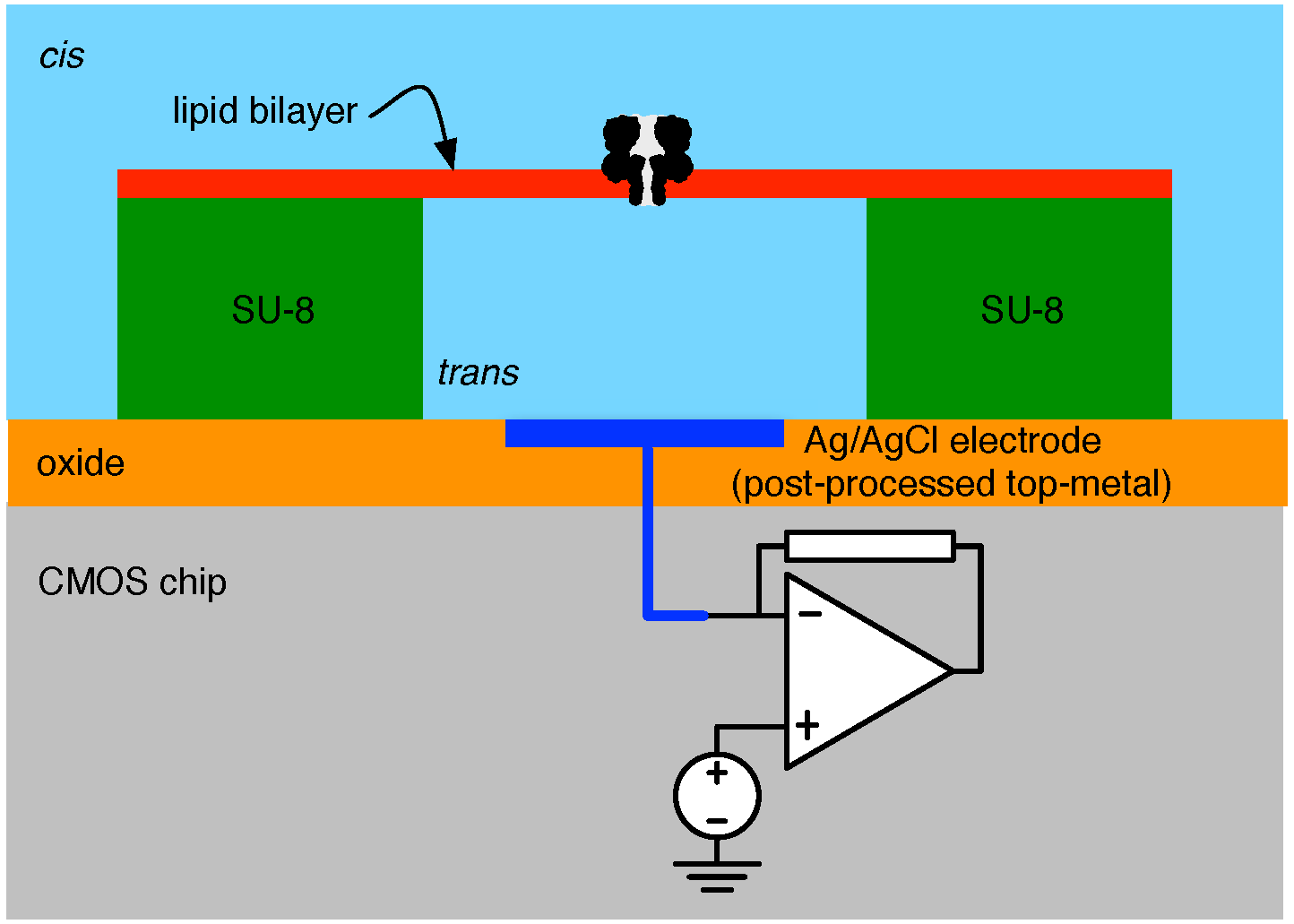
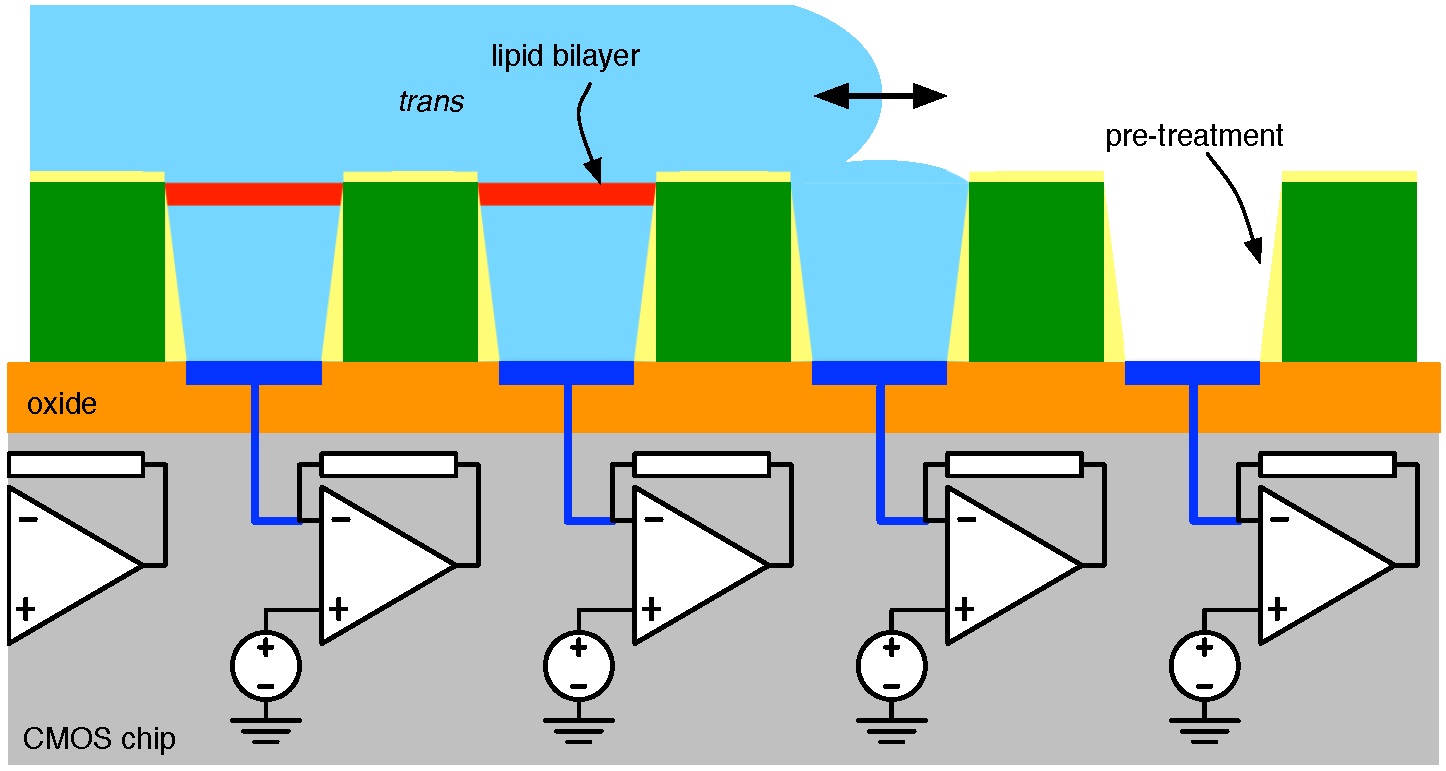
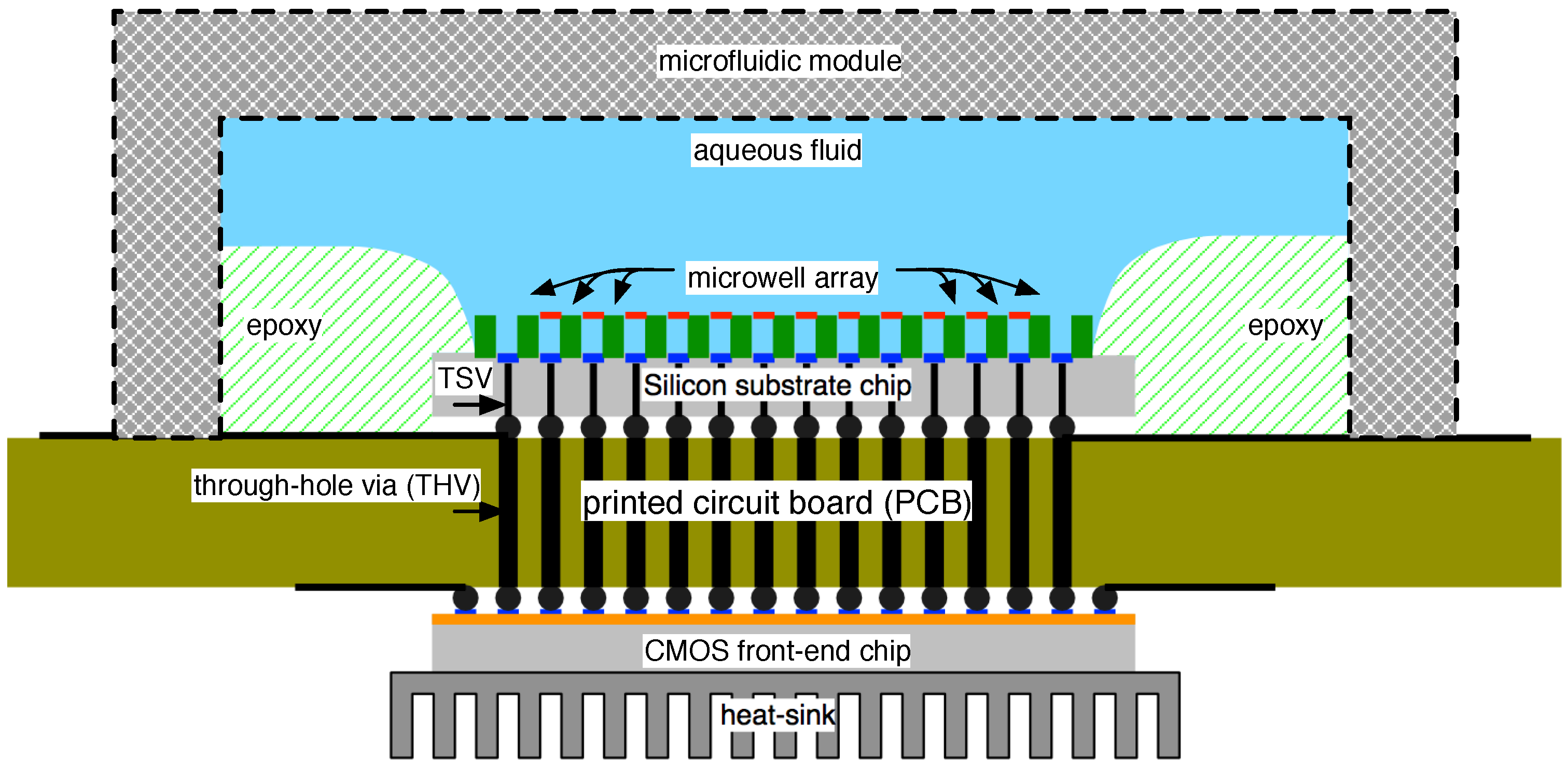
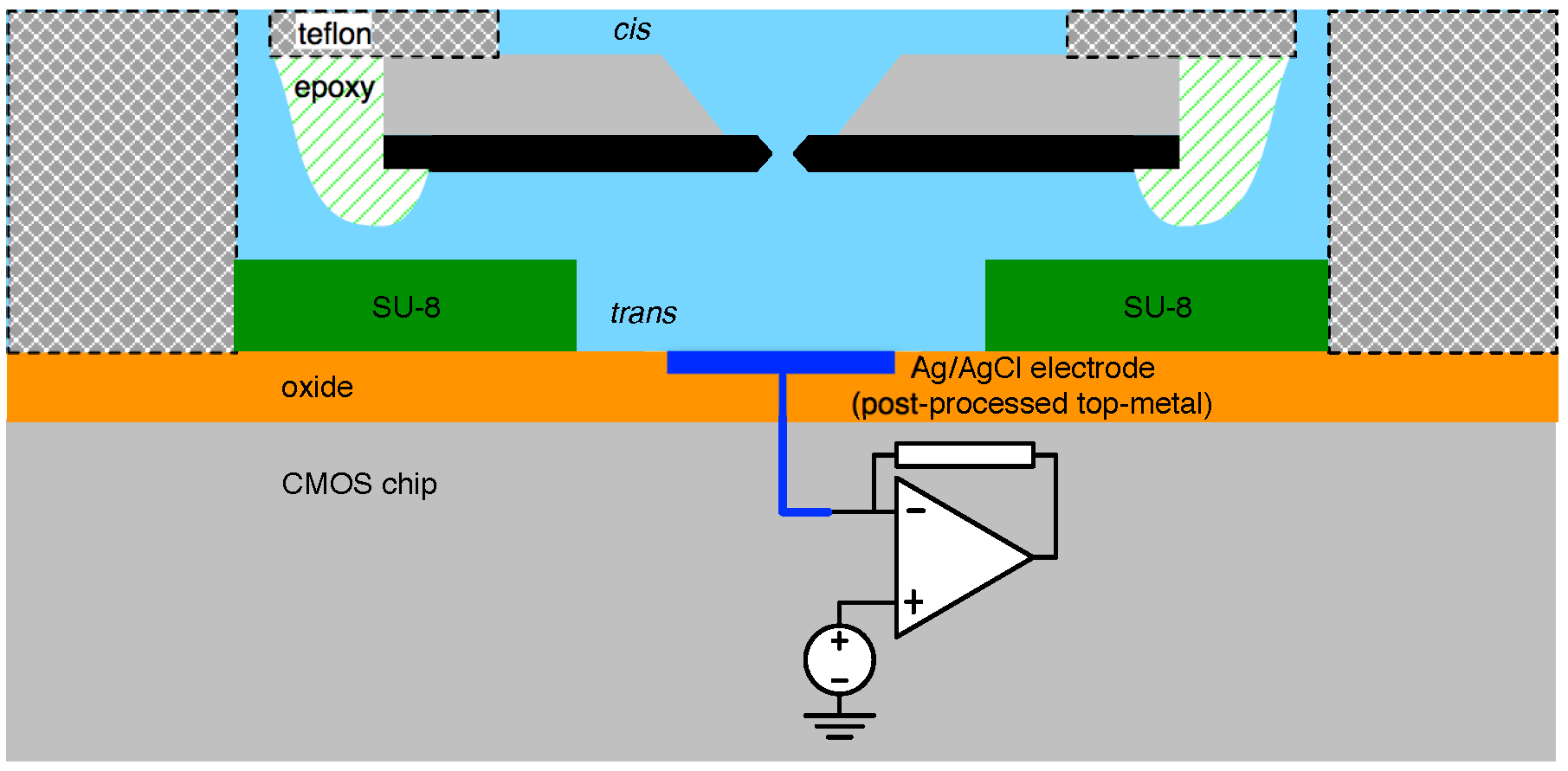
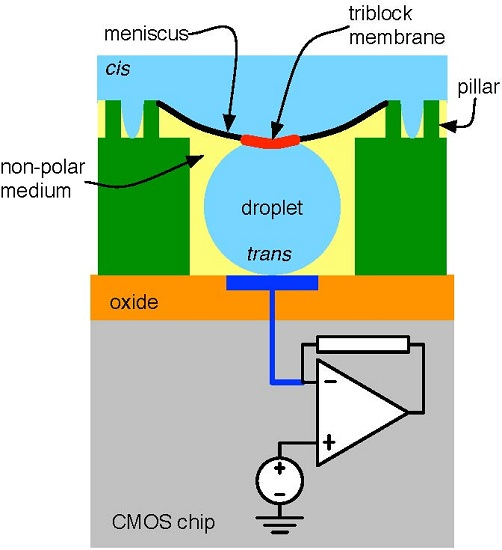
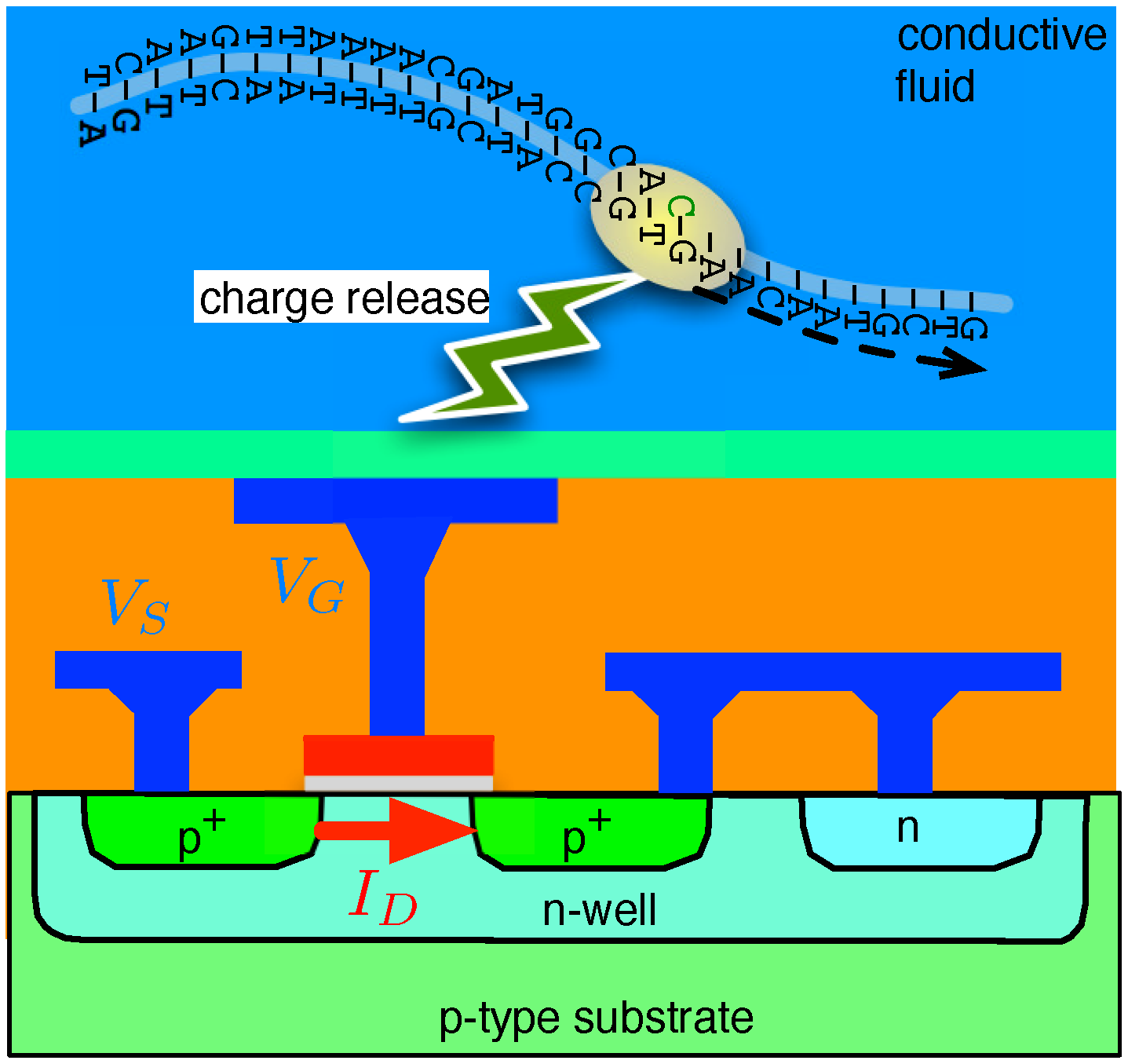
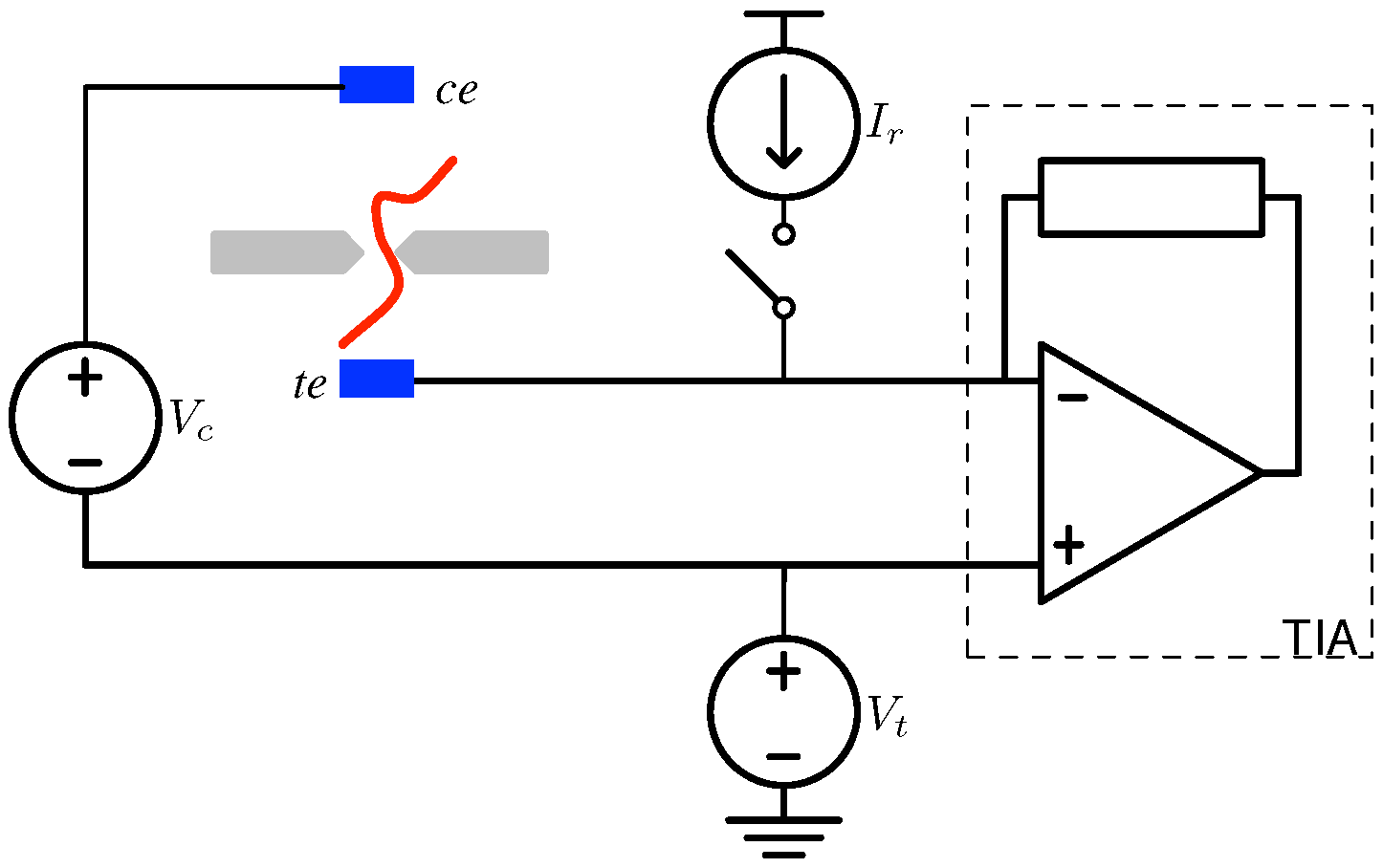
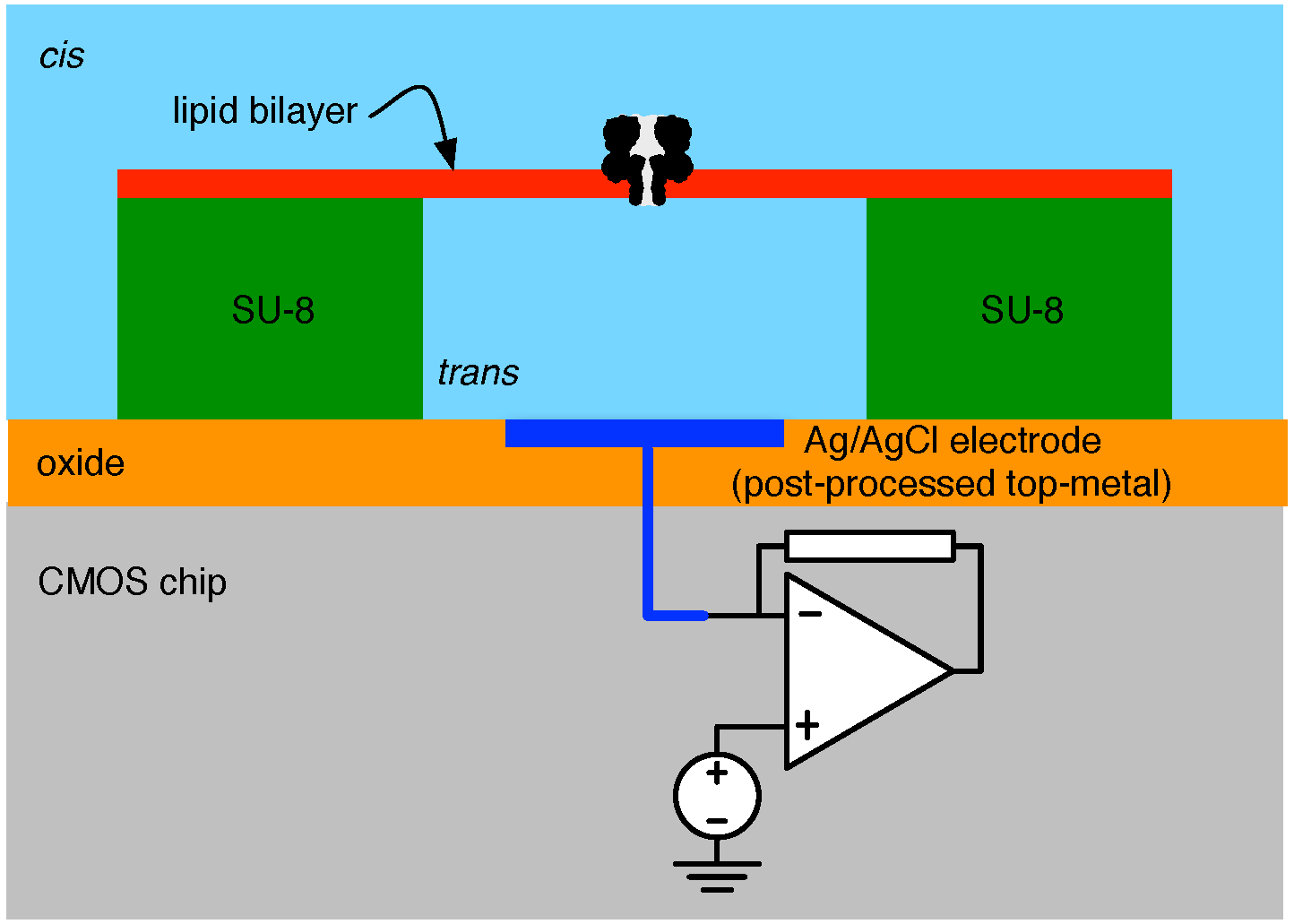
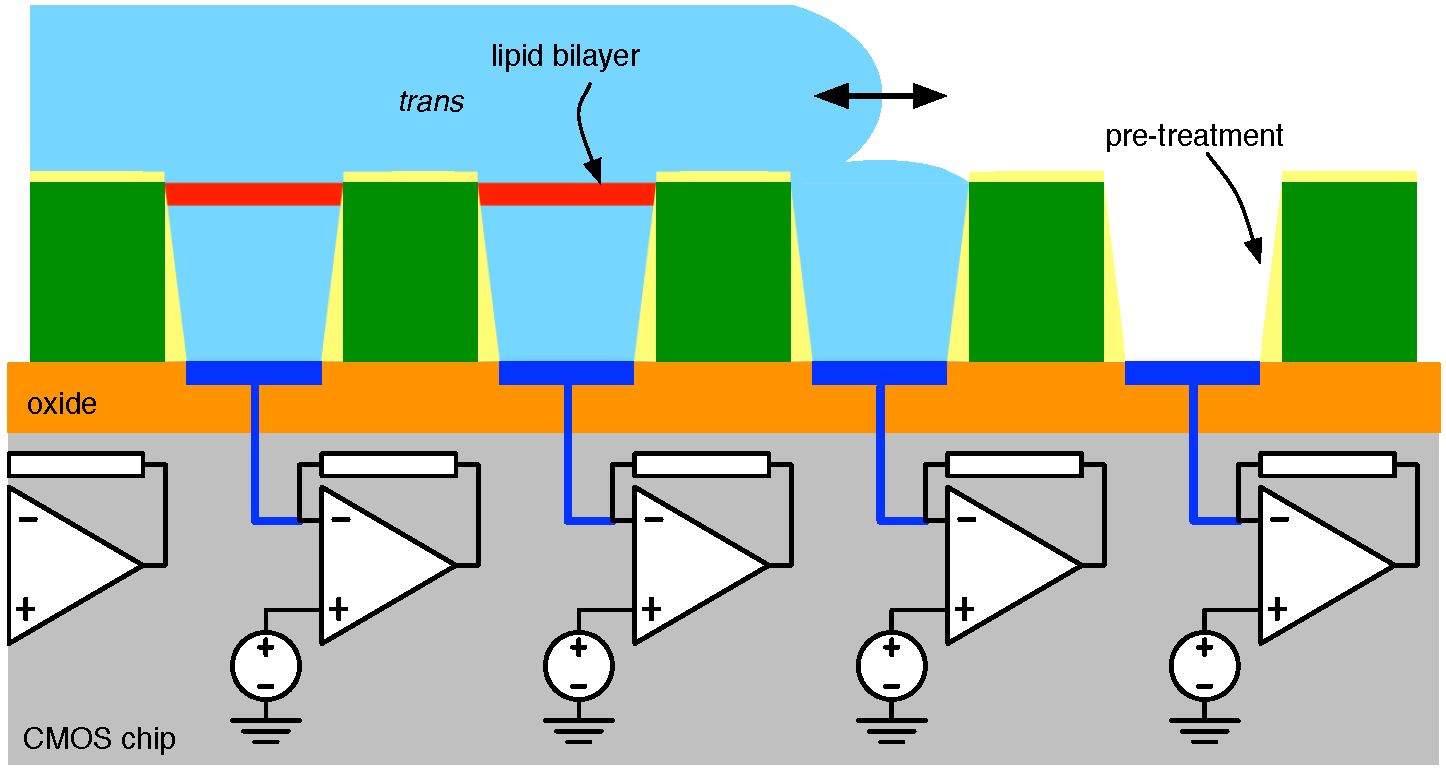
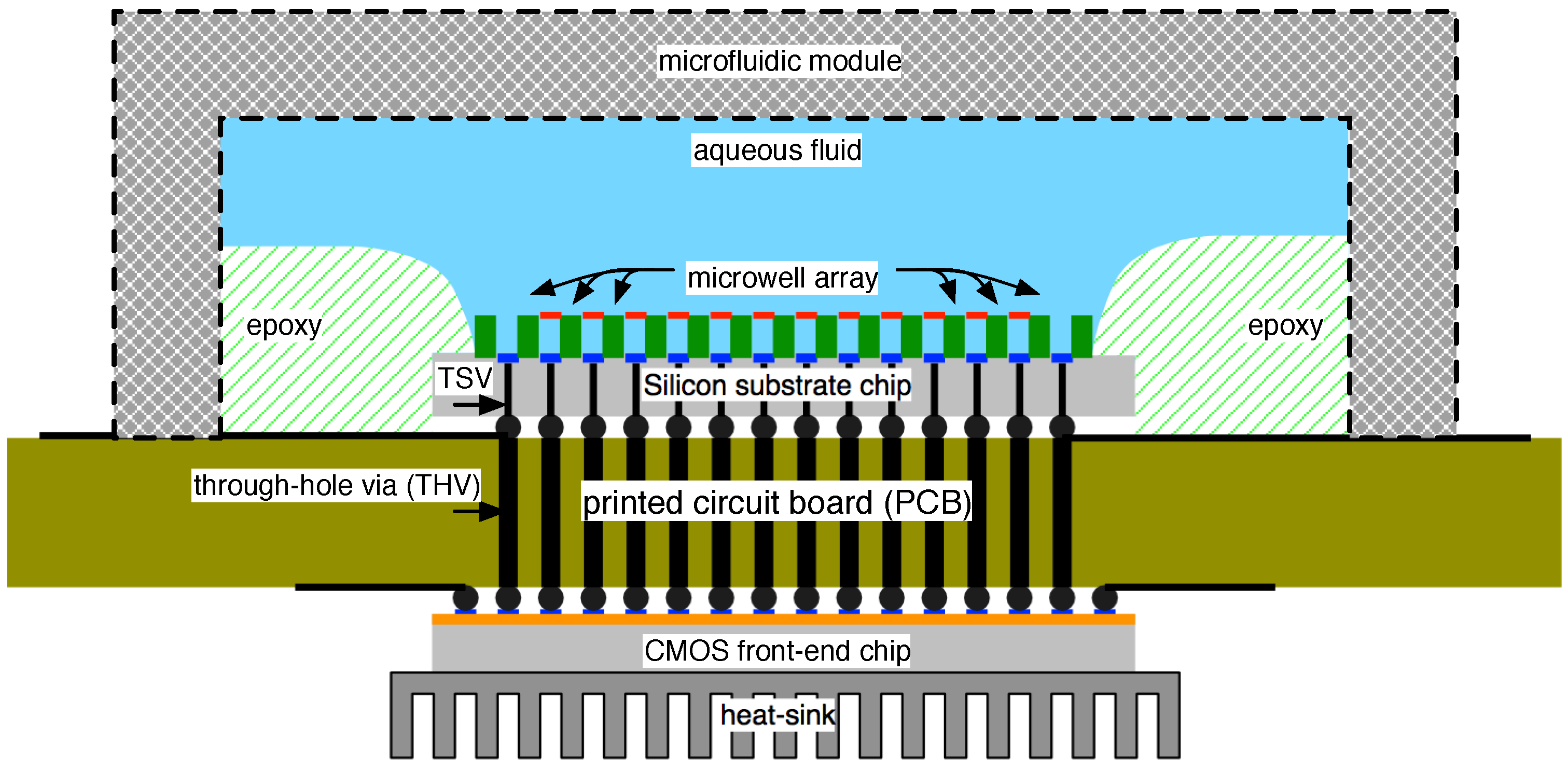
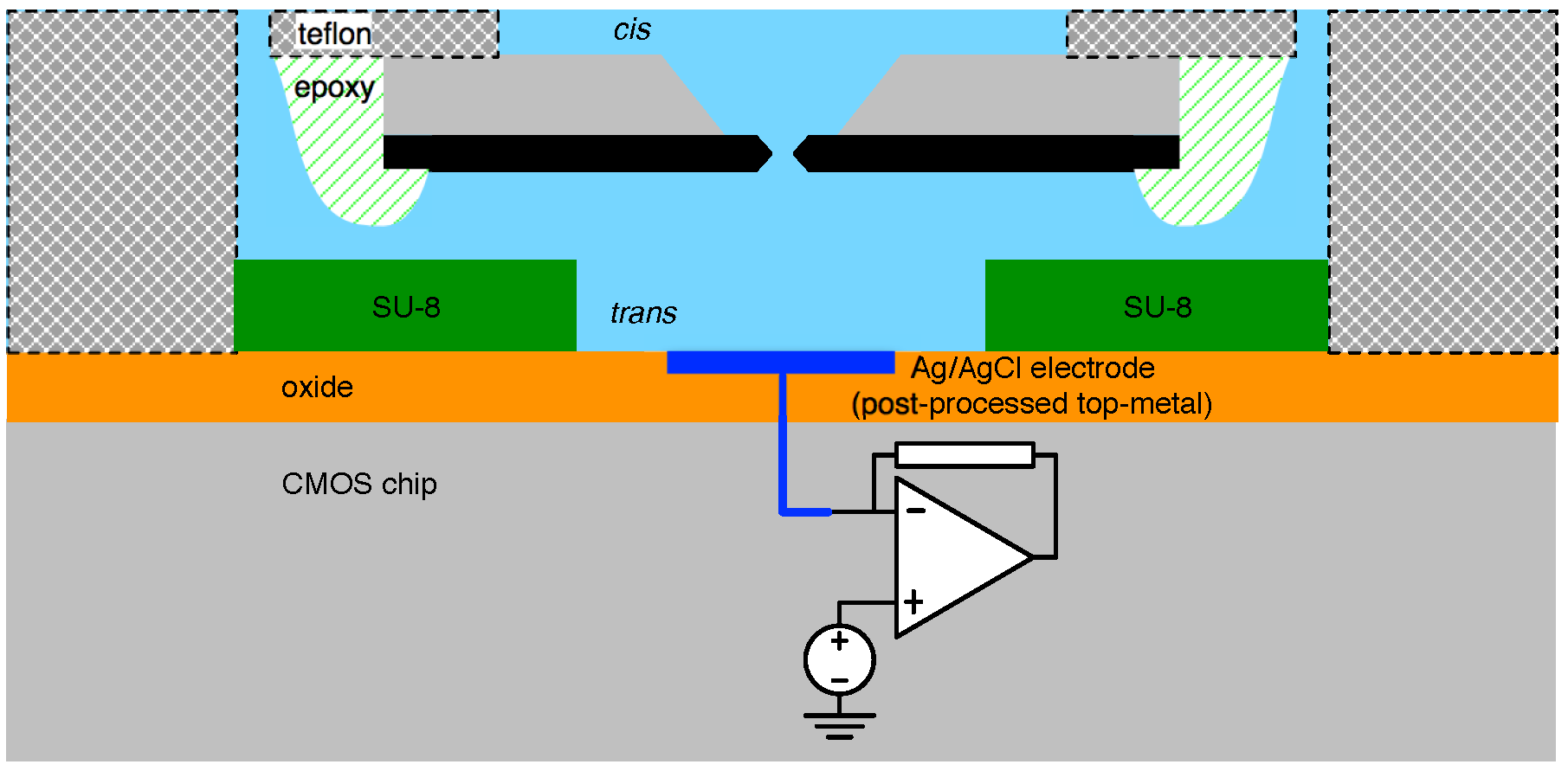
4.1. Biological Nanopore-CMOS Interfaces
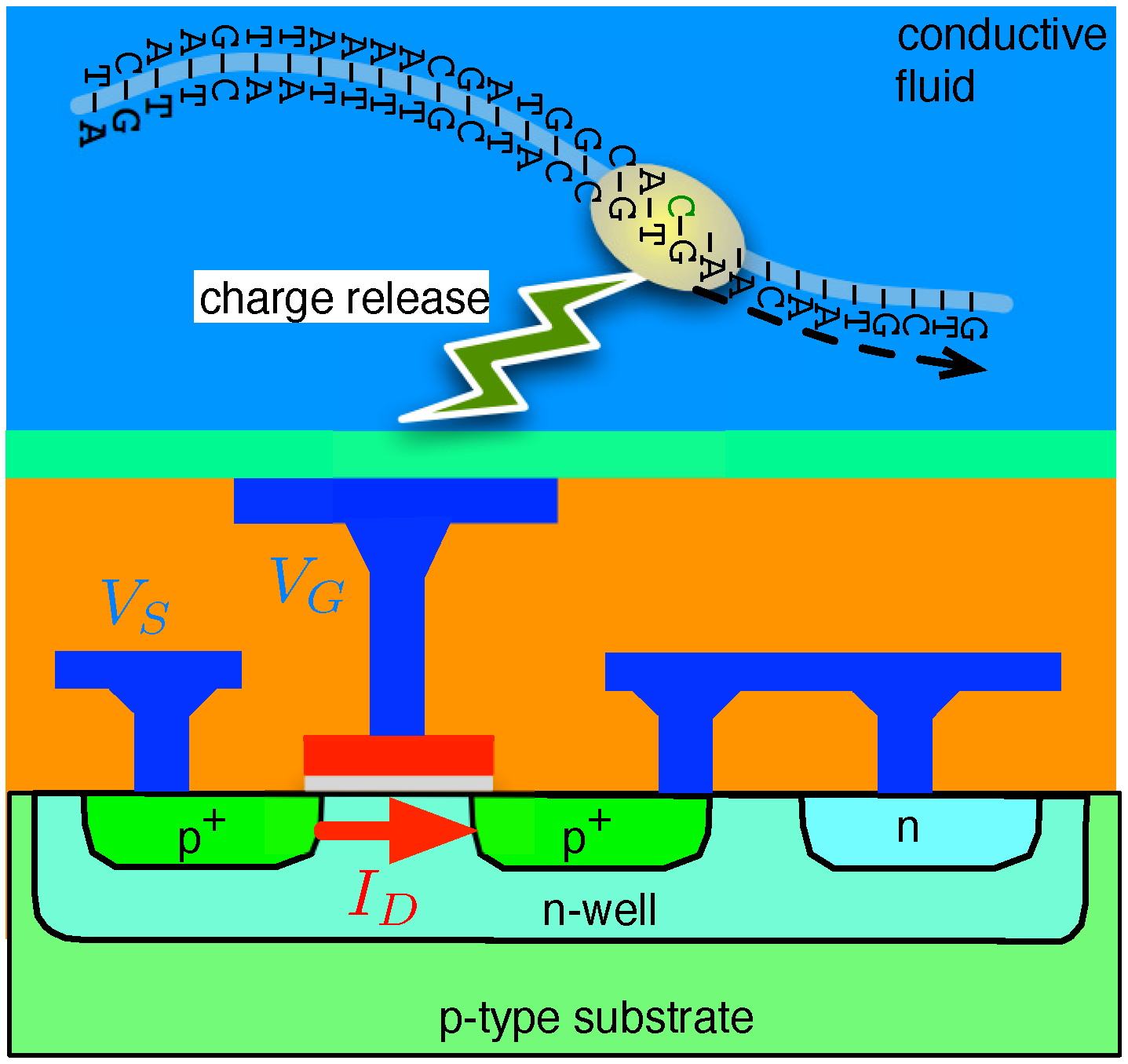
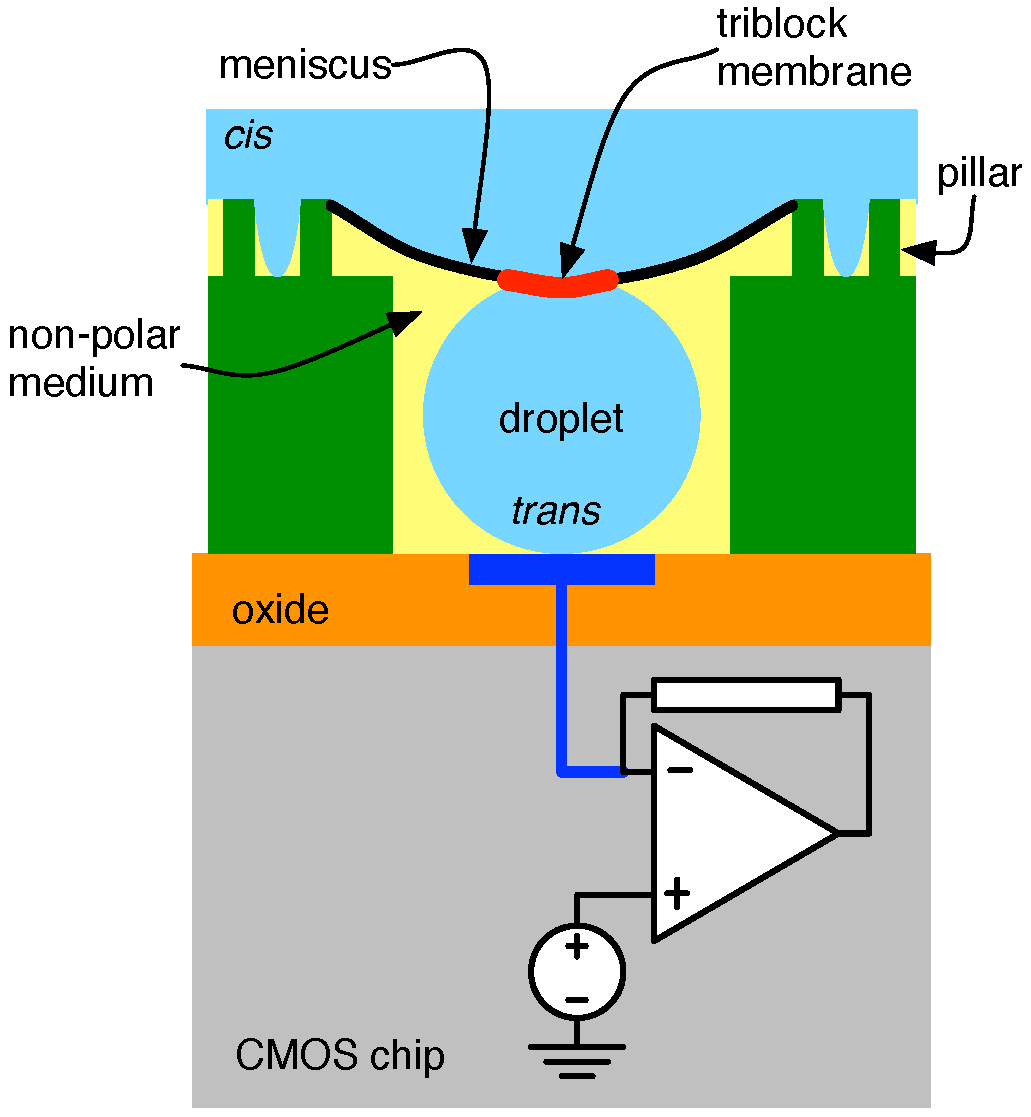
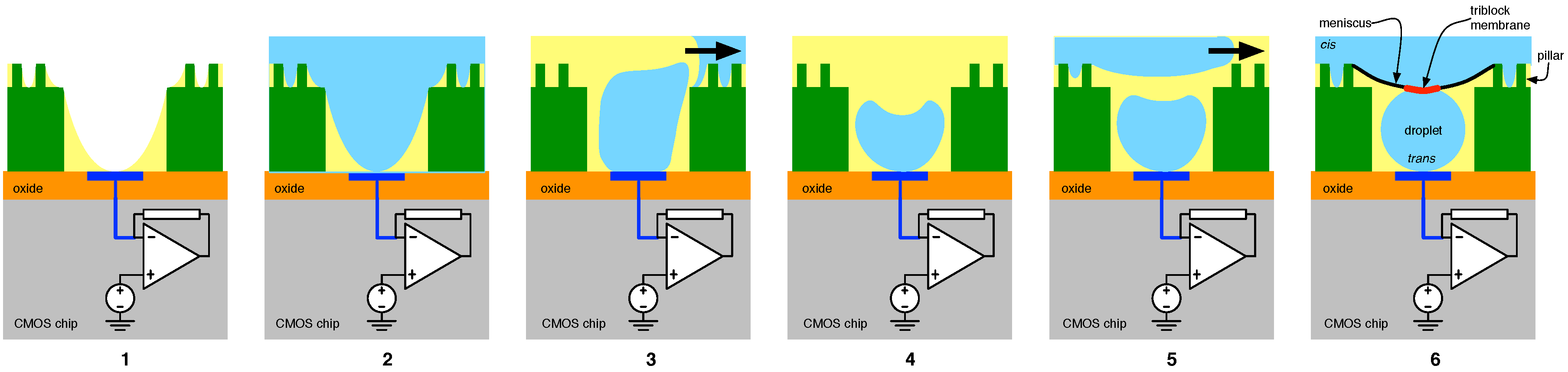
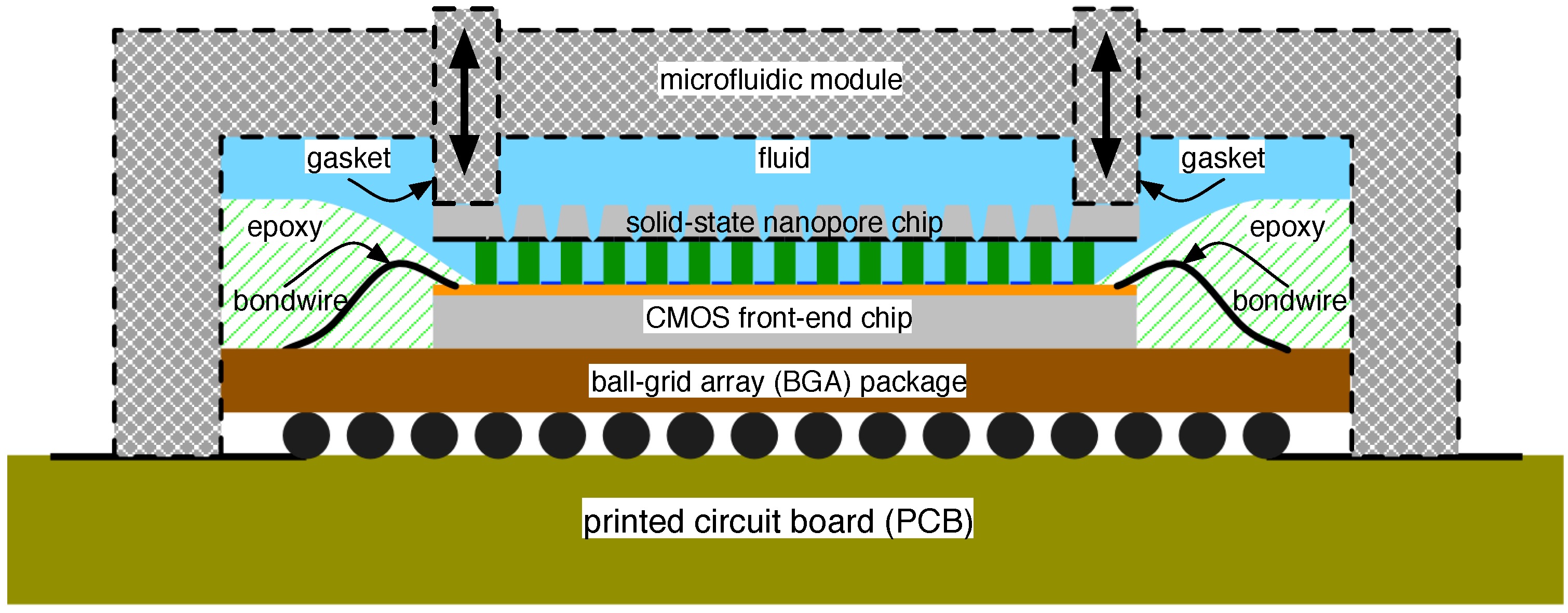
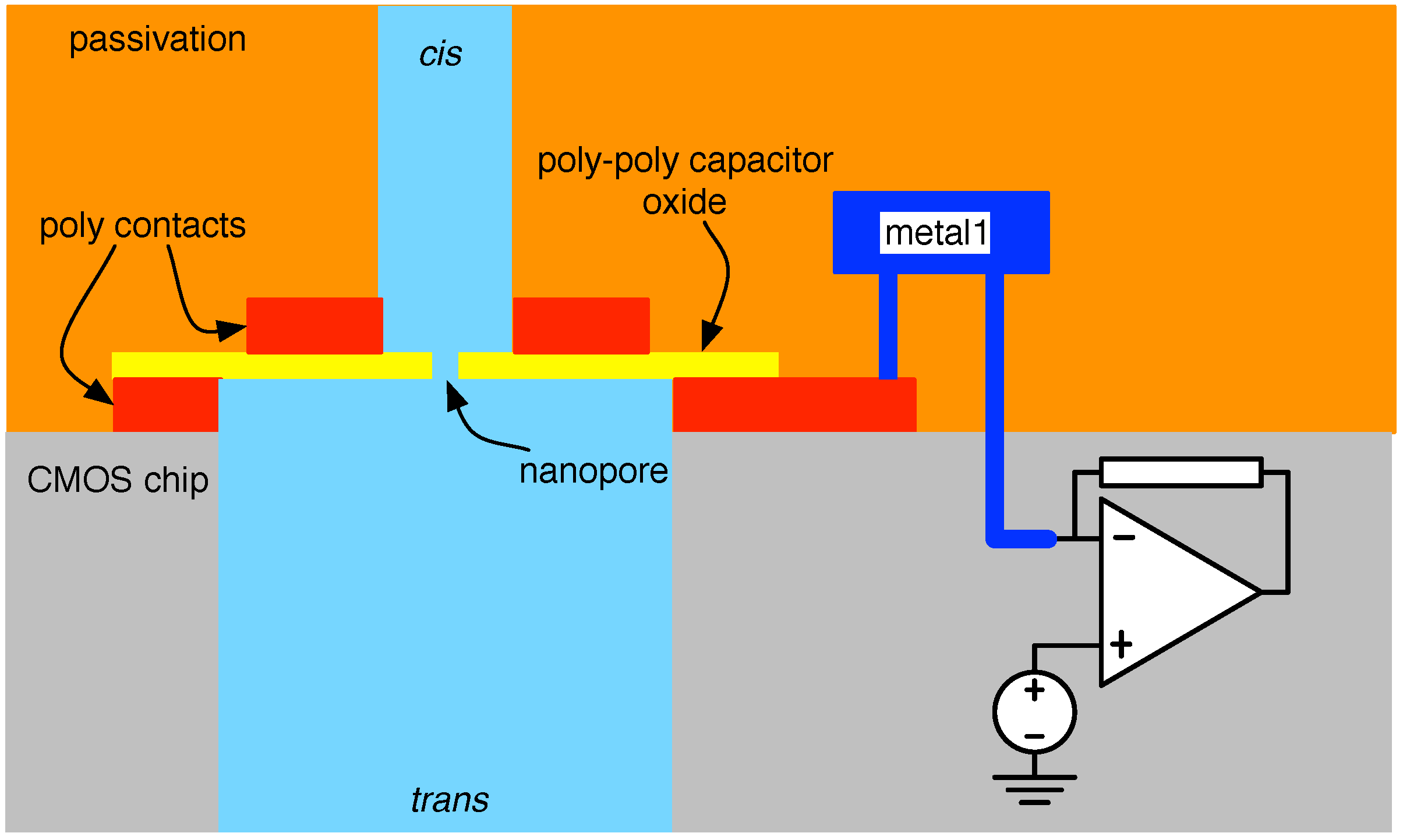
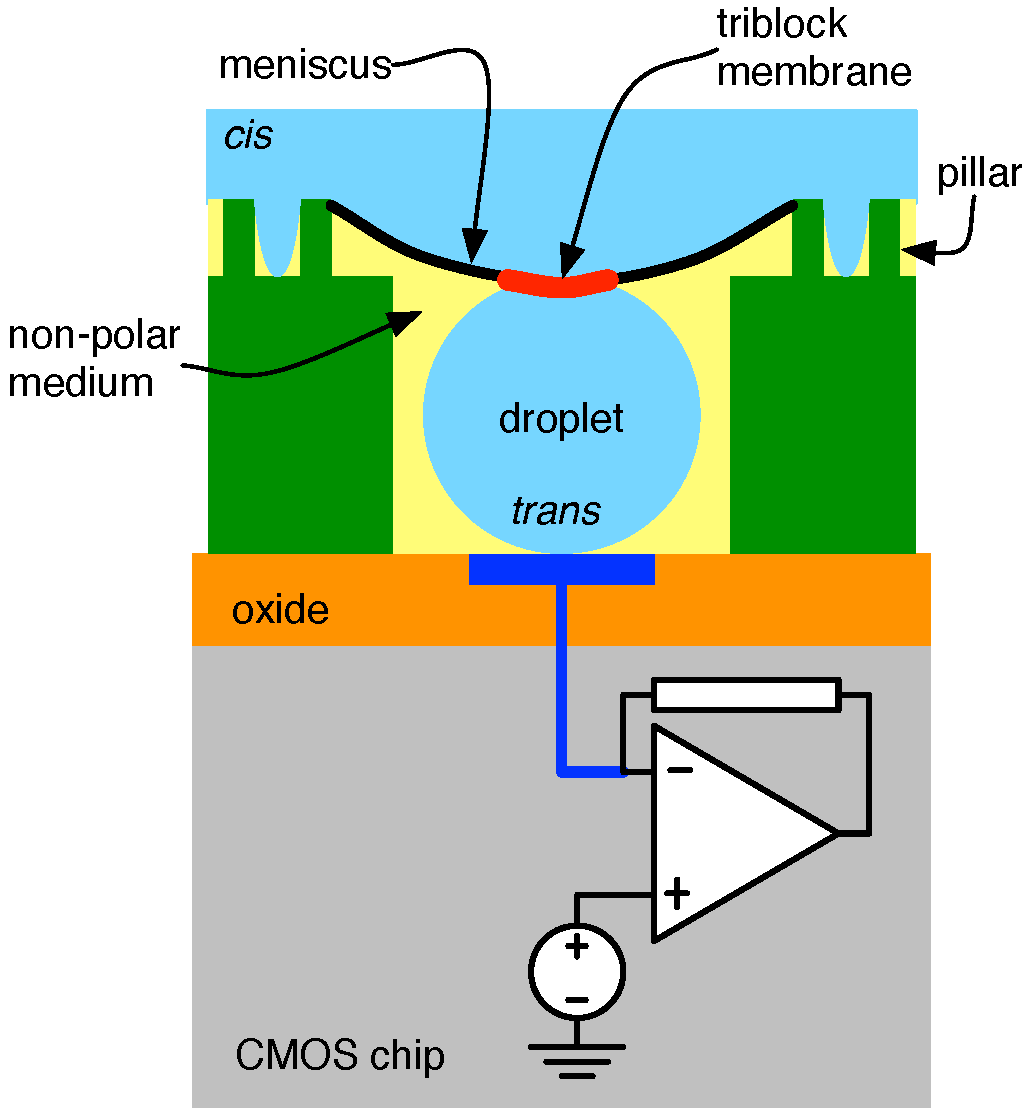
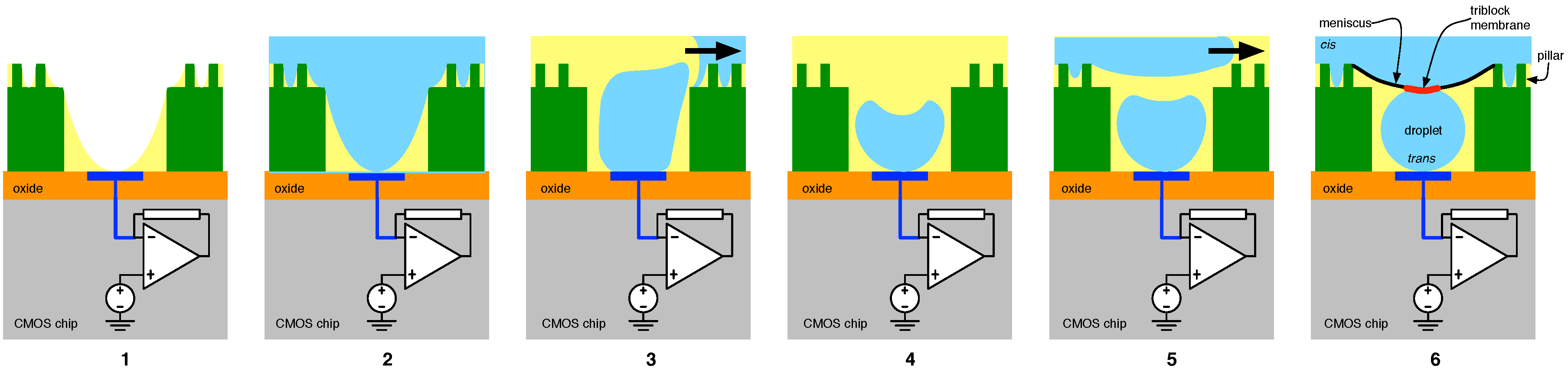
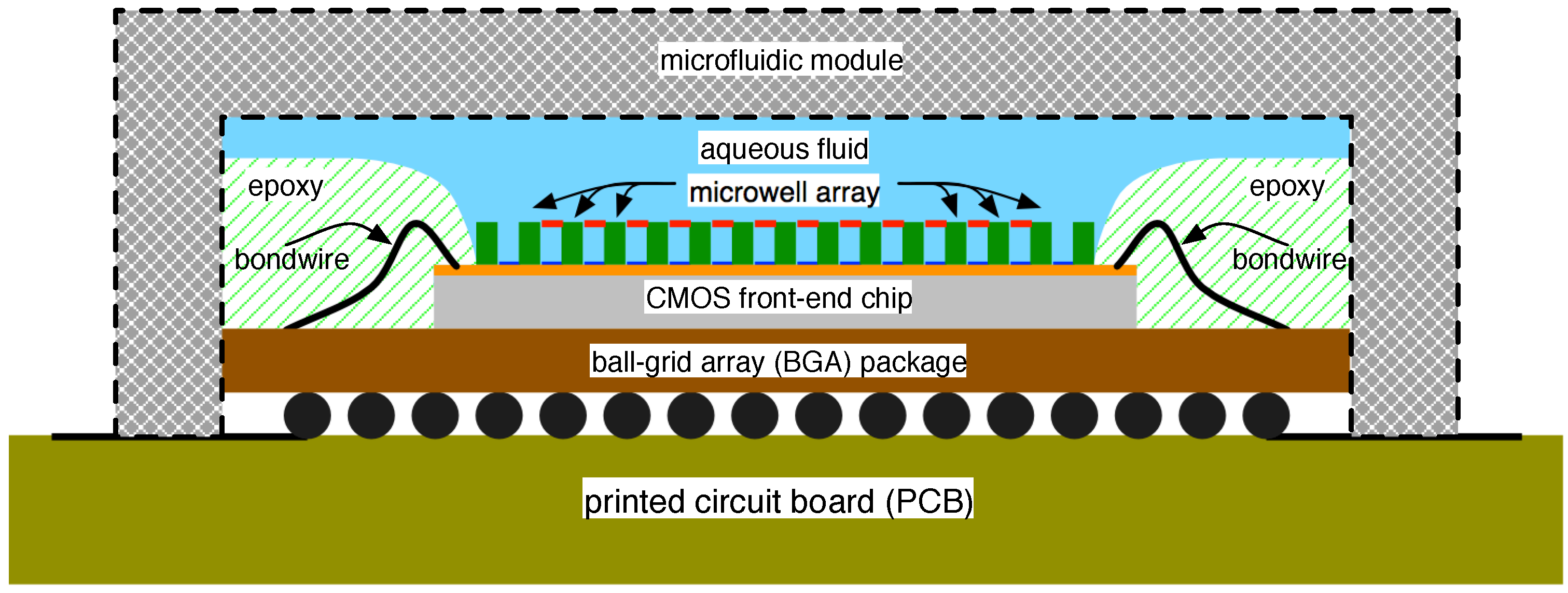
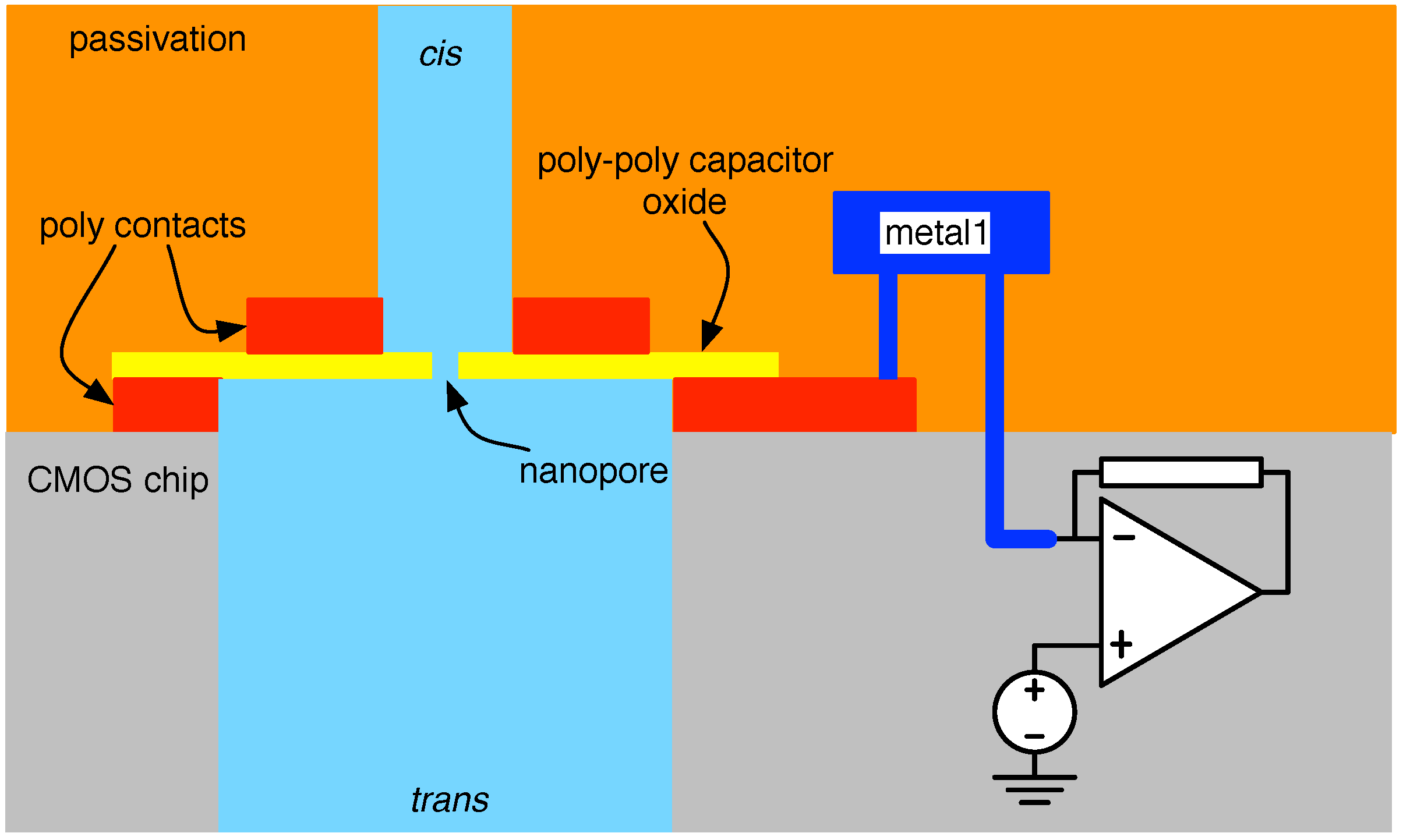
4.2. Solid-State Nanopore-CMOS Interfaces
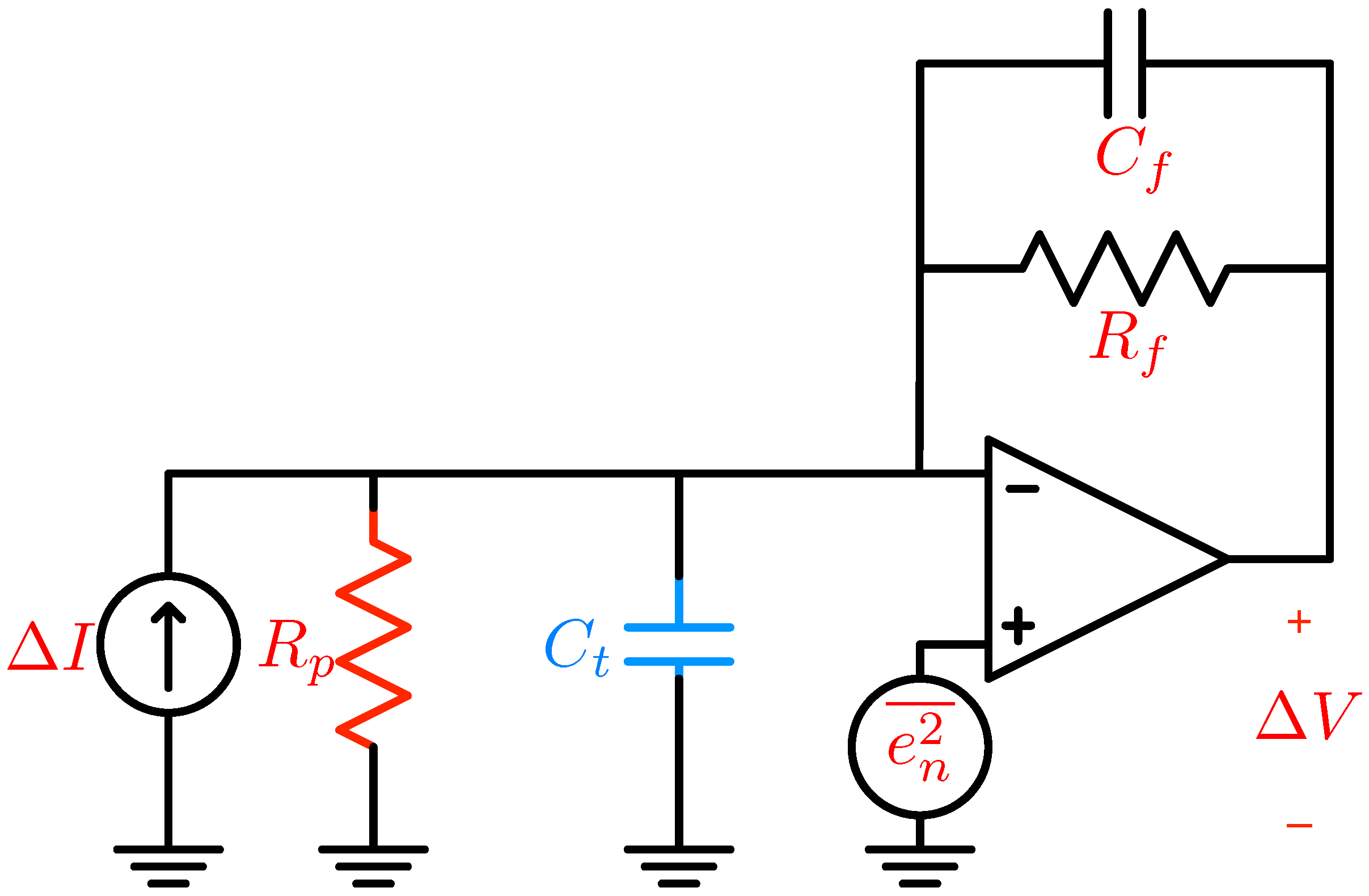
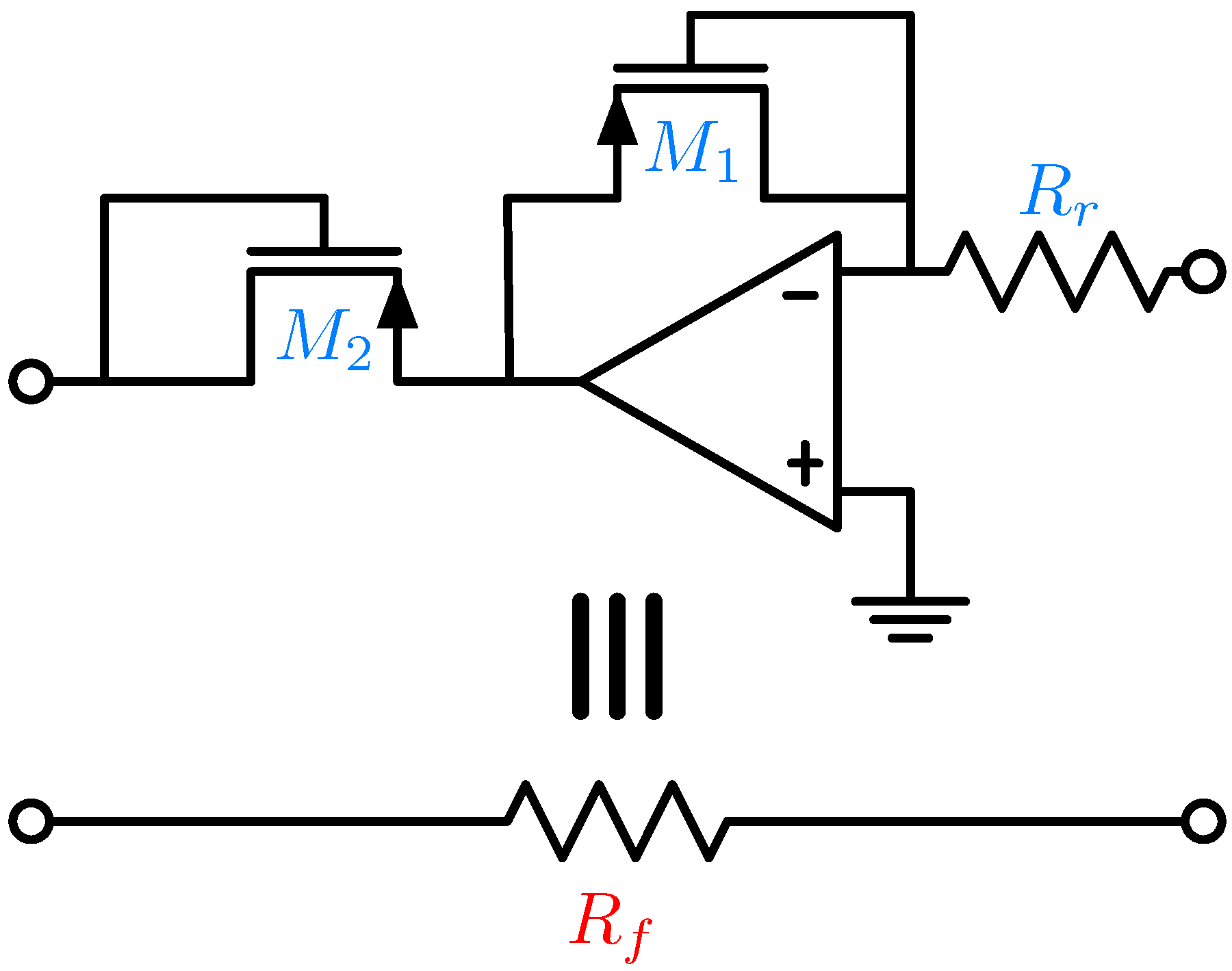
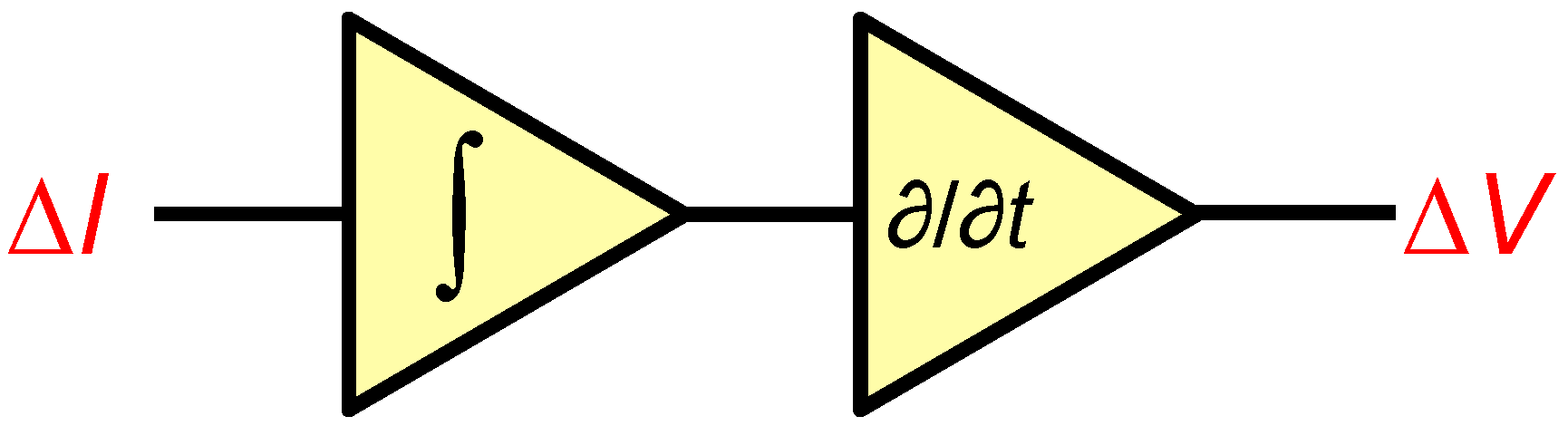
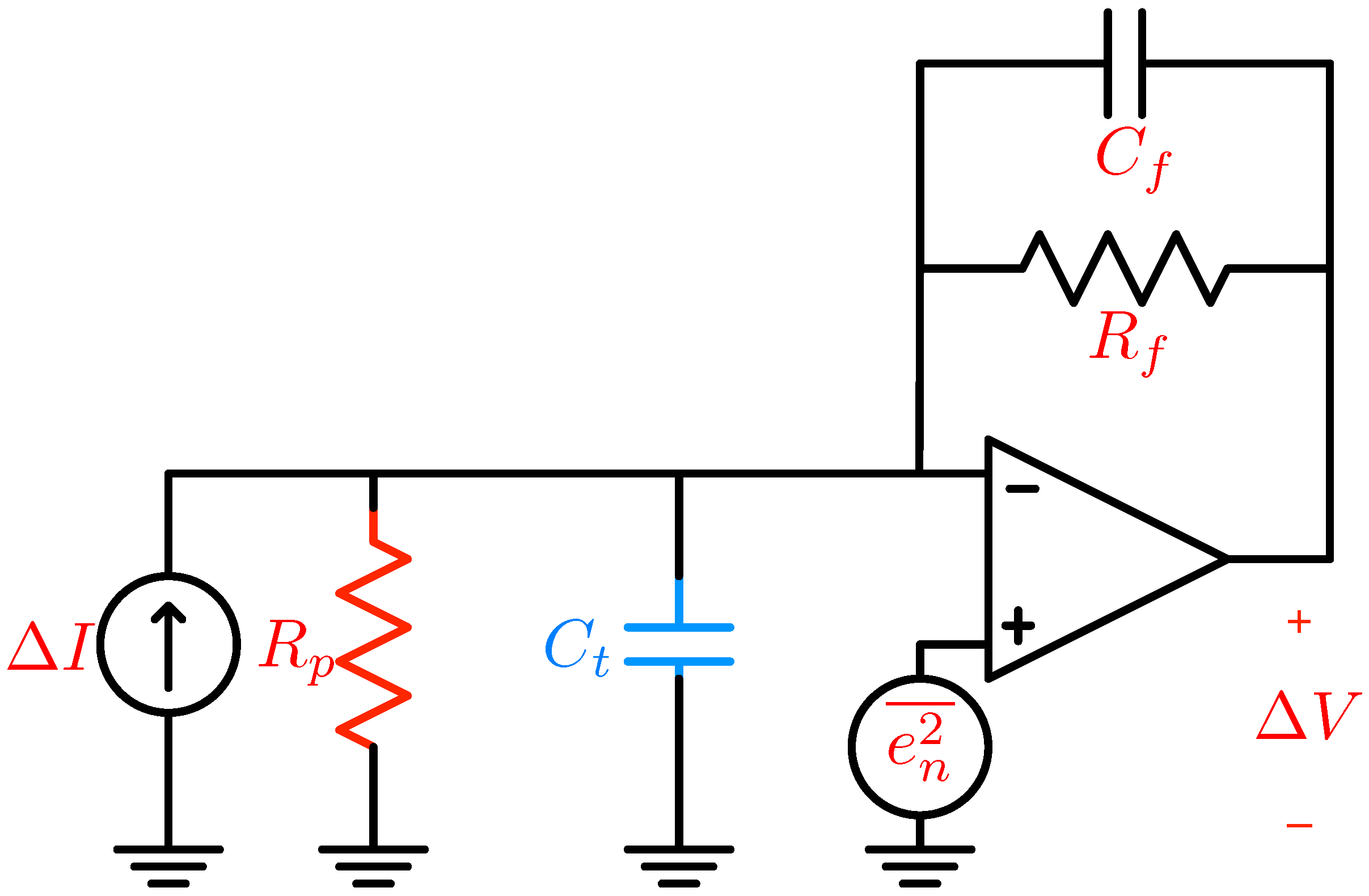
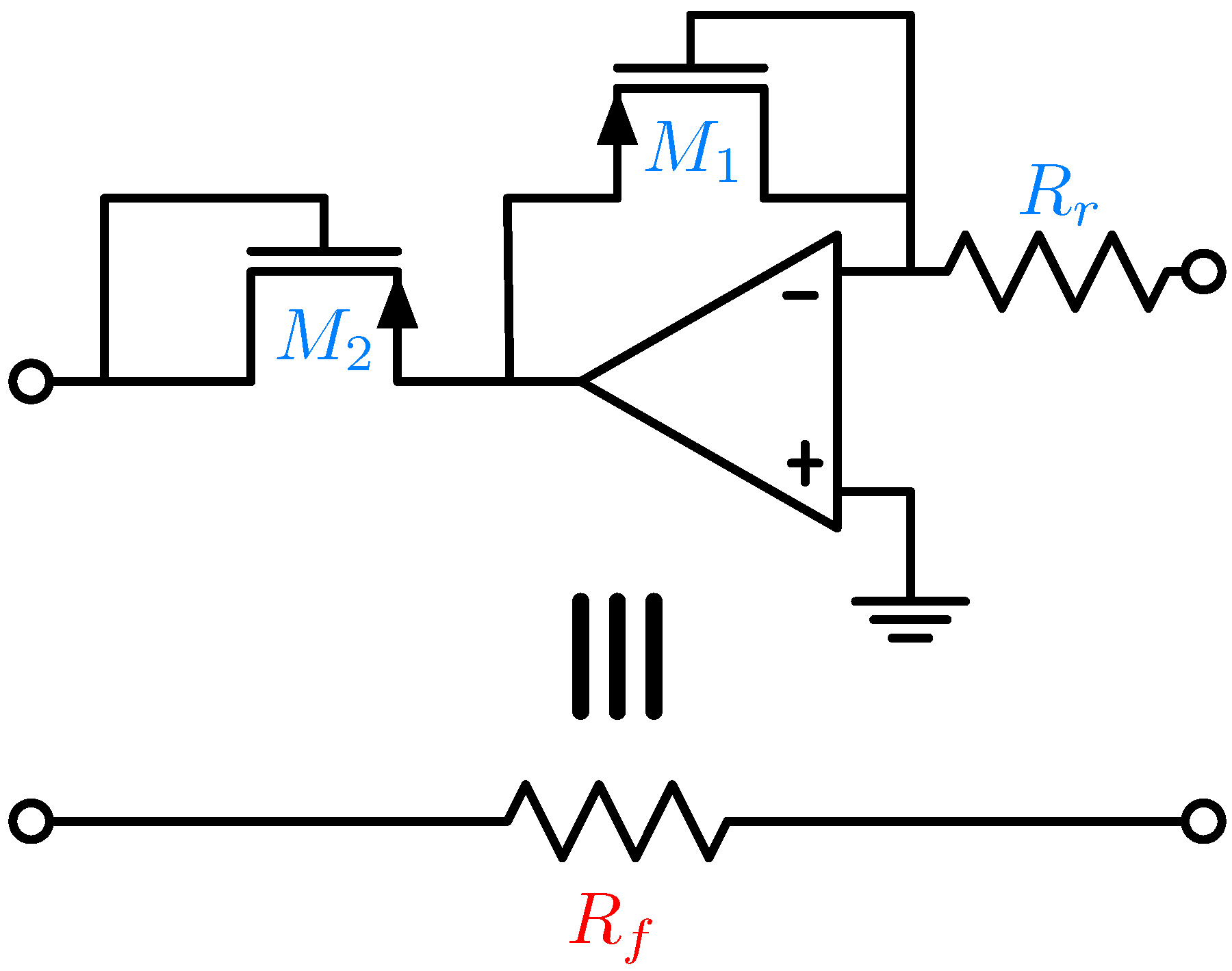
5. Electronic Amplification
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sanger, F.; Nicklen, S.; Coulson, A. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Hood, L.; Goldberg, M.L.; Reynolds, A.E.; Silver, L.M. Genetics: From Genes to Genomes, 4th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, S.B.H.; Hood, L.E. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Tu, O.; Dolnik, V.; Roach, D.; Solomon, N.; Bechtol, K.; Smietana, P.; Wang, L.; Li, X.; Cartwright, P.; et al. High-throughput DNA sequencing on a capillary array electrophoresis system. J. Capillary Electrophor. 1997, 4, 83–89. [Google Scholar] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotech. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S. Polynucleotide Sequencing. US 2003/0013101 A1, 29 September 1999. [Google Scholar]
- Nyren, P. Method of Sequencing DNA Based on the Detection of the Release of Pyrophosphate and Enzymatic Nucleotide Degradation. US 6,258,568 B1, 10 July 2001. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome Sequencing in Microfabricated High-Density Picolitre Reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An Integrated Semiconductor Device Enabling Non-Optical Genome Sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Mullis, K.B. Process for Amplifying Nucleic Acid Sequences. US 4,683,202, 28 July 1987. [Google Scholar]
- Wanunu, M.; Cohen-Karni, D.; Johnson, R.R.; Fields, L.; Benner, J.; Peterman, N.; Zheng, Y.; Klein, M.L.; Drndic, M. Discrimination of Methylcytosine from Hydroxymethylcytosine in DNA Molecules. J. Am. Chem. Soc. 2011, 133, 286–492. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Humphreys, G.I.; Venkatesan, B.M.; Munz, J.M.; Zou, X.; Sathe, C.; Schluten, K.; Kosari, F.; Nardulli, A.M.; Vasmatzis, G.; et al. Detection and Quantification of Methylation in DNA using Solid-State Nanopores. Sci. Rep. 2013, 3, 1389. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Kim, Y.; Humphreys, G.I.; Nardulli, A.M.; Kosari, F.; Vasmatzis, G.; Taylor, W.R.; Ahlquist, D.A.; Myong, S.; Bashir, R. Nanopore-Based Assay for Detection of Methylation in Double-Stranded DNA Fragments. ACS Nano 2015, 9, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-Generation Sequencing Platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Merriman, B.; Rothberg, J.M. Progress in Ion Torrent Semiconductor Chip Based Sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Marra, M.A. Applications of Next-Generation Sequencing Technologies in Functional Genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Johnson, K.L.; Bustillo, J. Methods and Apparatus for Measuring Analytes using Large Scale FET Arrays. US 7,948,015 B2, 15 October 2013. [Google Scholar]
- Bergveld, P. Development of an Ion-Sensitive Solid-State Device for Neurophysiological Measurements. IEEE Trans. Biomed. Eng. 1970, 17, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Bergveld, P. Thirty Years of ISFETOLOGY. Sens. Actuator B Chem. 2003, 88, 1–20. [Google Scholar] [CrossRef]
- Bausells, J.; Carrabina, J.; Errachid, A.; Merlos, A. Ion-Sensitive Field-Effect Transistors Fabricated in a Commercial CMOS Technology. Sens. Actuator B Chem. 1999, 57, 56–62. [Google Scholar] [CrossRef]
- Milgrew, M.J.; Cumming, D.R.S.; Hammond, P.A. The Fabrication of Scalable Multi-Sensor Arrays using Standard CMOS Technology. In Proceedings of the IEEE Custom Integrated Circuits Conference, San Jose, CA, USA, 21–24 September 2003; pp. 513–516.
- Milgrew, M.J.; Hammond, P.A.; Cumming, D.R.S. The Development of Scalable Sensor Arrays Using Standard CMOS Technology. Sens. Actuator B Chem. 2004, 103, 37–42. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettmann, B.; et al. Real-Time DNA Sequencing form Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, P.M.; Zhong, C.F.; Zhao, P.; Tomaney, A.B.; Peluso, P.S.; Dixon, J.; Bettman, B.; Lacroix, Y.; Kwo, D.P.; McCullough, E.; et al. Parallel Confocal Detection of Single Molecules in Real-Time. Opt. Lett. 2008, 33, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.; Loose, M.; Tyson, J.; de Cesare, M.; Brown, B.; Jain, M.; Leggett, R.; Eccles, D.; Zalunin, V.; Urban, J.; et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Research 2015, 4, 1075. [Google Scholar] [CrossRef] [PubMed]
- Coulter, W.H. Means of Counting Particles Suspended in a Fluid. US 2,656,508, 20 October 1953. [Google Scholar]
- Church, G.; Deamer, D.W.; Branton, D.; Baldarelli, R.; Kasianowicz, J. Characterization of Individual Polymer Molecules Based on Monomer-Interface Interactions. US 5,795,782, 18 August 1998. [Google Scholar]
- Baldarelli, R.; Branton, D.; Church, G.; Deamer, D.W.; Akeson, M.; Kasianowicz, J. Characterization of Individual Polymer Molecules Based on Monomer-Interface Interactions. US 6,015,714, 18 January 2000. [Google Scholar]
- Denison, T.J.; Sauer, A.; Golovchenko, J.; Meller, A.; Brandin, E.; Branton, D. Characterization of Individual Polymer Molecules Based on Monomer-Interface Interactions. US 6,362,002 B1, 2 June 2015. [Google Scholar]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The Potential and Challenges of Nanopore Sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, B.M.; Bashir, R. Nanopore Sensors for Nucleic Acid Analysis. Nat. Nanotechnol. 2011, 6, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.; Akeson, M.; Branton, D. Three Decades of Nanopore Sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Song, L.Z.; Hobaugh, M.R.; Shustack, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of Staphylococcal Alpha-Hemolysin, a Heptameric Transmembrane Pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Jayasinghe, L.; Reid, T.; Bayley, H. Base-Detecting Pore. US 2011/0177498 A1, 21 July 2011. [Google Scholar]
- Derrington, I.M.; Butler, T.Z.; Collins, M.D.; Manrao, E.; Pavlenok, M.; Niederweis, M.; Gundlach, J.H. Nanopore DNA Sequencing with MspA. Proc. Natl. Acad. Sci. 2010, 107, 16060–16065. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.Z.; Pavlenok, M.; Derrington, I.M.; Niederweis, M.; Gundlach, J.H. Single-Molecule DNA Detection with an Engineered MspA Protein Nanopore. Proc. Natl. Acad. Sci. 2008, 105, 20647–20652. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Krasteva, P.V.; Gerven, N.V.; Gubellini, F.; Van den Broeck, I.; Troupiotis-Tsailaki, A.; Jonckheere, W.; Pehau-Arnaudet, G.; Pinkner, J.S.; Chapman, M.R.; et al. Structural and Mechanistic Insights into the Bacterial Amyloid Secretion Channel CsgG. Nature 2014, 516, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Manrao, E.A.; Derrington, I.M.; Laszlo, A.H.; Langford, K.W.; Hopper, M.K.; Gillgren, N.; Pavlenok, M.; Niederweis, M.; Gundlach, J.H. Reading DNA at a Single-Nucleotide Resolution with a Mutant MspA nanopore and phi29 DNA Polymerase. Nat. Biotech. 2012, 30, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, K.R.; Cherf, G.M.; Moody, M.J.; Olasagasti, F.; Kolodji, Y.; Akeson, M. Progressive Replication of Single DNA Molecules in a Nanopore Catalyzed by phi29 DNA Polymerase. J. Am. Chem. Soc. 2010, 132, 17961–17972. [Google Scholar] [CrossRef] [PubMed]
- Cherf, G.M.; Lieberman, K.R.; Rashid, H.; Lam, C.E.; Karplus, K.; Akeson, M. Automated Forward and Reverse Ratcheting of DNA in a Nanopore at 5-ÅPrecision. Nat. Biotechnol. 2012, 30, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Ventra, M.D.; Taniguchi, M. Decoding DNA, RNAand peptides with quantum tunnelling. Nat. Nanotechnol. 2016, 11, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Zwolak, M.; Ventra, M.D. Physical Approaches to DNA Sequencing and Detection. Rev. Mod. Phys. 2008, 80, 141–165. [Google Scholar] [CrossRef]
- Zwolak, M.; Ventra, M.D. DNA Sequencing via Electron Tunnelling. In Proceedings of the 2012 IEEE International Symposium on Circuits and Systems (ISCAS), Seoul, Korea, 20–23 May 2012; pp. 2295–2298.
- Tsutsui, M.; Taniguchi, M.; Yokota, K.; Kawai, T. Identifying Single Nucleotides by Tunnelling Current. Nat. Nanotechnol. 2010, 5, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; He, J.; Chang, S.; Zhang, P.; Liang, F; Li, S.; Tuchband, M.; Fuhrmann, A.; Ros, R.; Lindsay, S. Identifying Single Bases in a DNA Oligomer with Electron Tunnelling. Nat. Nanotechnol. 2010, 5, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.P.; Freedman, K.J.; Kim, M.J.; Albrecht, T.; Edel, J.B. High Precision Fabrication and Positioning of Nanoelectrodes in a Nanopore. ACS Nano 2014, 8, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.B.; Aksimentiev, A.; Ho, C.; Dimitrov, V.; Sorsch, T.W.; Miner, J.F.; Mansfield, W.M.; Schulten, K.; Timp, G. Beyond the Gene Chip. Bell Labs Tech. J. 2005, 10, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, G.; Comer, J.; Timp, G.; Aksimentiev, A. Detection of DNA Sequences Using and Alternating Electric Field in a Nanopore Capacitor. Nano Lett. 2008, 8, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Gracheva, M.E.; Xiong, A.; Aksimentiev, A.; Schulten, K.; Timp, G.; Leburton, J.P. Simulation of the Electric Response of DNA Translocation Through a Semiconductor Nanopore-Capacitor. Nanotechnology 2006, 17, 622–633. [Google Scholar] [CrossRef]
- Leroux, A.; Destine, J.; Vanderheyden, B.; Gracheva, M.E.; Leburton, J.P. SPICE Circuit Simulation of the Electrical Response of a Semiconductor Membrane to a Single-Stranded DNA Translocating Through a Nanopore. IEEE Trans. Nanotech. 2010, 9, 322–329. [Google Scholar] [CrossRef]
- Li, J.; Stein, D.; McMullan, C.; Branton, D.; Aziz, M.J.; Golovchenko, J.A. Ion-Beam Sculpting at Nanometre Length Scales. Nature 2001, 412, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Heerema, S.J.; Dekker, C. Graphene Nanodevices for DNA Sequencing. Nat. Nanotechnol. 2016, 11, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Venta, K.; Shemer, G.; Puster, M.; Rodriguez-Manzo, J.A.; Balan, A.; Rosenstein, J.K.; Shepard, K.; Drndic, M. Differentiation of Short, Single-Stranded DNA Homopolymers in Solid-State Nanopores. ACS Nano 2013, 7, 4629–4636. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manzo, J.A.; Puster, M.; Nicolai, A.; Meunier, V.; Drndic, M. DNA Translocation in Nanometer Thick Silicon Nanopores. ACS Nano 2015, 9, 6555–6564. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, V.; Mirsaidov, U.; Wang, D.; Sorsch, T.; Mansfield, W.; Miner, J.; Klemens, F.; Cirelli, R.; Yemenicioglu, S.; Timp, G. Nanopores in Solid-State Membranes Engineered for Single-Molecule Detection. Nanotechnology 2010, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, J.; Ni, Z.; Zhang, L.; Hu, G. Effects of Access Resistance on the Resistive-pulse Caused by Translocating of a Nanoparticle through a Nanopore. RCS Adv. 2014, 4, 7601–7610. [Google Scholar] [CrossRef]
- Carlsen, A.T.; Zahid, O.K.; Ruzicka, J.; Taylor, E.W.; Hall, A.R. Interpreting the Conductance Blockades of DNA Translocations through Solid-State Nanopores. ACS Nano 2014, 8, 4754–4760. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, S.W.; Grosberg, A.Y.; Rabin, Y.; Dekker, C. Modeling the Conductance and DNA Blockade of Solid-State Nanopores. Nanotechnology 2011, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Willmott, G.R.; Smith, B.G. Comment on ‘Modeling the Conductance and DNA Blockade of Solid-State Nanopores’. Nanotechnology 2012, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, S.W.; Grosberg, A.Y.; Rabin, Y.; Dekker, C. Reply to Comment on ‘Modeling the Conductance and DNA Blockade of Solid-State Nanopores’. Nanotechnology 2012, 23, 1. [Google Scholar] [CrossRef]
- Rosenstein, J.K.; Ramakrishnan, S.; Roseman, J.; Shepard, K.L. Single Ion Channel Recordings with CMOS-Anchored Lipid Membranes. Nano Lett. 2013, 13, 2682–2686. [Google Scholar] [CrossRef] [PubMed]
- Balan, A.; Machielse, B.; Niedzwiecki, D.; Lin, J.; Ong, P.; Engelke, R.; Shepard, K.L.; Drndic, M. Improving Signal-to-Noise Performance for DNA Translocation in Solid-State Nanopores at MHz Bandwidths. Nano Lett. 2014, 14, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- Smeets, R.M.M.; Keyser, U.F.; Dekker, N.H.; Dekker, C. Noise in Solid-State Nanopores. Proc. Natl. Acad. Sci. USA 2008, 105, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Wanunu, M.; Sutin, J.; McNally, B.; Chow, A.; Meller, A. DNA Translocation Governed by Interactions with Solid-State Nanopores. Biophys. J. 2008, 95, 4716–4725. [Google Scholar] [CrossRef] [PubMed]
- Montal, M.; Mueller, P. Formation of Bimolecular Membranes from Lipid Monolayers and a Study of Their Electrical Properties. Proc. Natl. Acad. Sci. USA 1972, 69, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Gutsmann, T.; Heimburg, T.; Keyser, U.; Mahendran, K.R.; Winterhalter, M. Protein Reconstitution Into Freestanding Planar Lipid Membranes for Electrophysiological Characterization. Nat. Protoc. 2015, 10, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Akeson, M.; Branton, D.; Kasianowicz, J.J.; Brandin, E.; Deamer, D.W. Microsecond Time-Scale Discrimination Among Polycytidylic Acid, Polyadenylic Acid, and Polyuridylic Acid as Homopolymers or as Segments Within Single RNA Molecules. Biophys. J. 1999, 77, 3227–3233. [Google Scholar] [CrossRef]
- Baaken, G.; Sondermann, M.; Schlemmer, C.; Ruhe, J.; Behrends, J.C. Planar microelectrode-cavity array for high-resolution and parallel electrical recording of membrane ionic currents. Lab Chip 2008, 8, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Polk, B.J.; Stelzenmuller, A.; Mijares, G.; MacCrehan, W.; Gaitan, M. Ag/AgCl Microelectrodes with Improved Stability for Microfluidics. Sens. Actuators B Chem. 2006, 114, 239–247. [Google Scholar] [CrossRef]
- Reid, S.W.; Reid, T.A.; Clarke, J.A.; White, S.P.; Sanghera, G.S. Formation of Layers of Amphiphilic Molecules. US 2009,0167,288 A3, 2 July 2009. [Google Scholar]
- Gonzalez-Perez, A.; Stibius, K.B.; Vissing, T.; Nielsen, C.H.; Mouritsen, O.G. Biomimetic Triblock Copolymer Membrane Arrays: A Stable Template for Functional Membrane Proteins. Langmuir 2009, 25, 10447–10450. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Bithi, S.S.; Vanapalli, S.A. Microfluidic Static Droplet Arrays with Tuneable Gradients in Material Composition. Lab Chip. 2011, 11, 3949–3952. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.R.; Bahamon, P.M.O.; Brown, C.G.; Heron, A.J.; Mackett, P.R. Formation of Array of Membranes and Apparatus Therefor. US 2015,0265,994 A1, 24 September 2015. [Google Scholar]
- Bayley, J.H.P.; Holden, M.; Heron, A.J.; Needham, D. Formation of Bilayers of Amphipathic Molecules. US 2010,0032,627 A1, 18 September 2012. [Google Scholar]
- Wallace, M.I.; Heron, A.J.; Holden, M.A. Bilayers. US 2013,0129,910 A1, 21 August 2007. [Google Scholar]
- Laub, J.H. Low Cost Flip Chip Technologies for DCA, WLCSP, and PBGA Assemblies; McGraw-Hill: New York, NY, USA, 2000. [Google Scholar]
- Ebefors, T.; Fredlund, J.; Perttu, D.; van Dijk, R.; Cifola, L.; Kaunisto, M.; Rantakari, P.; Vähä-Heikkilä, T. The Development and Evaluation of RF TSV for 3D IPD Applications. In Proceedings of the 2013 IEEE International 3D Systems Integration Conference (3DIC), San Francisco, CA, USA, 2–4 October 2013; pp. 1–8.
- Kalvesten, E.; Ebefors, T.; Svedin, N.; Rangsten, P.; Schonberg, T. Electrical Connections in Substrates. US 2007,0020,926, 21 March 2003. [Google Scholar]
- Bauer, T. First High Volume Via Process for Packaging and Integration of MEMS/CMOS. Technical report. Silex Microsystems, 2011. [Google Scholar]
- Kalvesten, E.; Ebefors, T.; Svedin, N.; Eriksson, A. Bonding Process and Bonded Structures. US 8,729,685 B2, 16 July 2013. [Google Scholar]
- Rosenstein, J.K.; Wanunu, M.; Merchant, C.A.; Drndic, M.; Shepard, K.L. Integrated nanopore sensing platform with sub-microsecond temporal resolution. Nat. Methods 2012, 9, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Uddin, A.; Yemenicioglu, S.; Chen, C.H.; Corgliano, E.; Milaninia, K.; Xia, F.; Plaxco, K.; Theogarajan, L. Biosensing with Integrated CMOS Nanopores. In Proceedings of the SPIE, San Diego, CA, USA, 12 August 2012; Volume 8460, pp. 1–6.
- Uddin, A.; Yemenicioglu, S.; Chen, C.H.; Corgliano, E.; Milaninia, K.; Theogarajan, L. Integration of Solid-State Nanopores in a 0.5 μm CMOS Foundry Process. Nanotechnology 2013, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Gozzini, F.; Molari, A.; Sampietro, M. Transimpedance Amplifier for High Sensitivity Current Measurements on Nanodevices. IEEE J. Solid-State Circuits 2009, 44, 1609–1616. [Google Scholar] [CrossRef]
- Kim, J.; Maitra, R.; Pedrotti, K.D.; Dunbar, W.B. A Patch-Clamp ASIC for Nanopore-Based DNA Analysis. IEEE Trans. Biomed. Circuits Syst. 2013, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Sigworth, F.J. Electronic Design of the Patch Clamp. In Single-Channel Recording, 2nd ed.; Sakmann, B., Neher, E., Eds.; Springer: New York, NY, USA, 1995. [Google Scholar]
- Huang, Y.; Magierowski, S.; Ghafar-Zadeh, E. CMOS for High-Speed Nanopore DNA Basecalling. 2016. Available online: http://waset.org/publications/10001103/cmos-solid-statenanopore-dna-system-level-sequencing-techniques-enhancement (accessed on 5 August 2016).
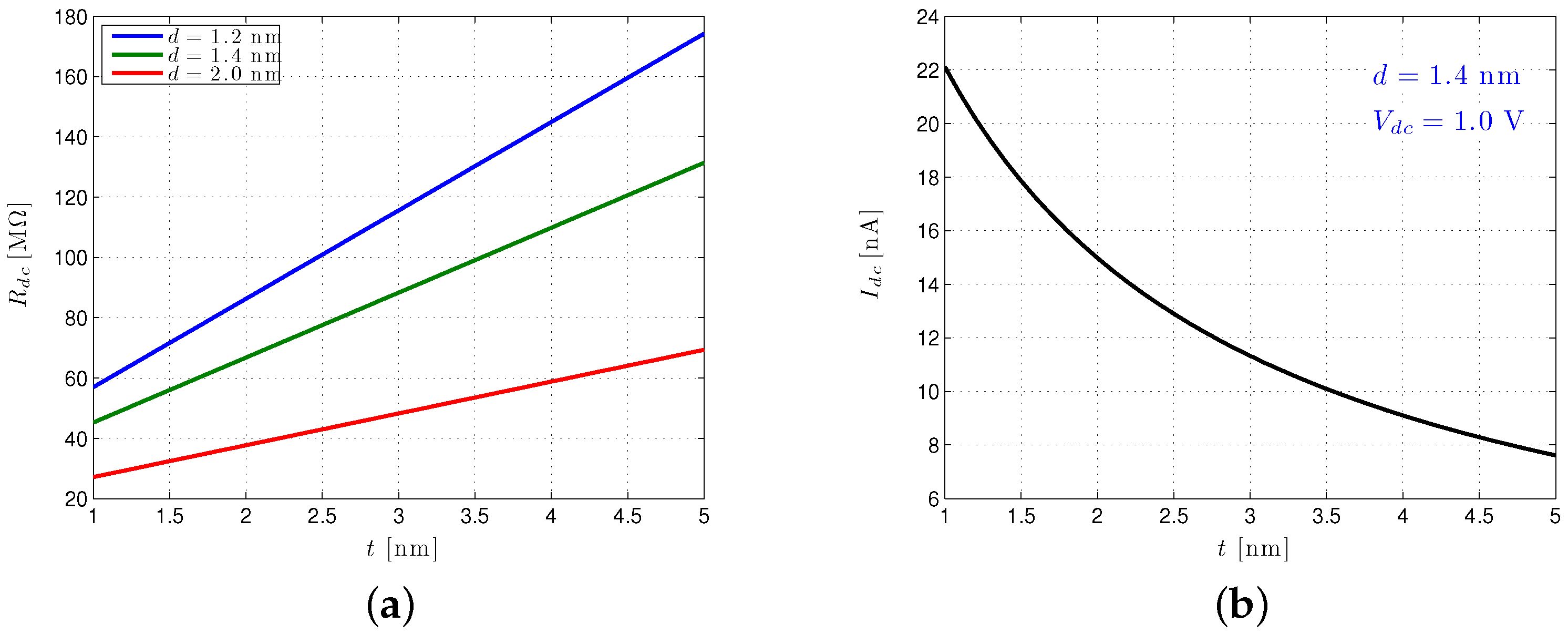




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
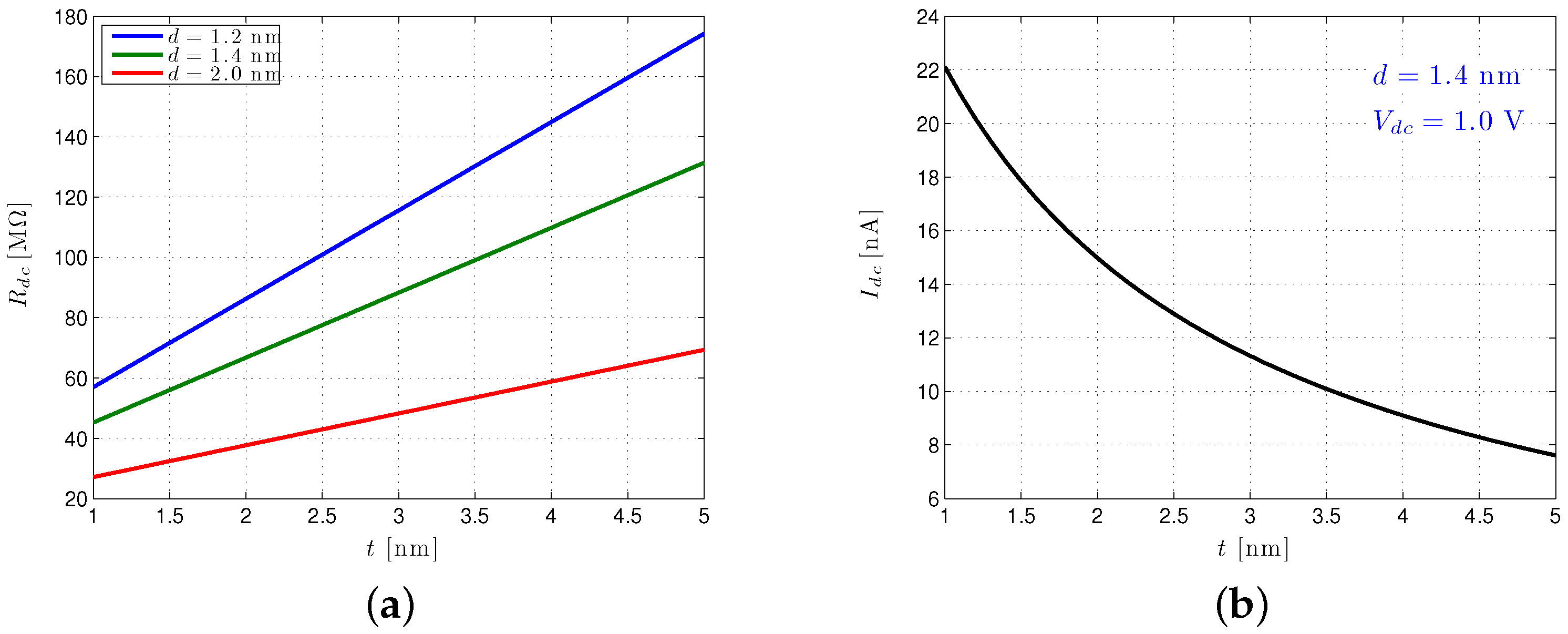{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magierowski, S.; Huang, Y.; Wang, C.; Ghafar-Zadeh, E. Nanopore-CMOS Interfaces for DNA Sequencing. Biosensors 2016, 6, 42. https://doi.org/10.3390/bios6030042
Magierowski S, Huang Y, Wang C, Ghafar-Zadeh E. Nanopore-CMOS Interfaces for DNA Sequencing. Biosensors. 2016; 6(3):42. https://doi.org/10.3390/bios6030042
Chicago/Turabian StyleMagierowski, Sebastian, Yiyun Huang, Chengjie Wang, and Ebrahim Ghafar-Zadeh. 2016. "Nanopore-CMOS Interfaces for DNA Sequencing" Biosensors 6, no. 3: 42. https://doi.org/10.3390/bios6030042
APA StyleMagierowski, S., Huang, Y., Wang, C., & Ghafar-Zadeh, E. (2016). Nanopore-CMOS Interfaces for DNA Sequencing. Biosensors, 6(3), 42. https://doi.org/10.3390/bios6030042