1. Introduction
Norovirus (NoV) is a leading cause of acute gastroenteritis, with millions of cases reported annually worldwide. NoV can spread rapidly through contaminated food, water, surfaces, and person-to-person contact [
1]. Given its ability to survive on surfaces for several days and to resist disinfectants, NoV can cause outbreaks, especially in close-contact environments, such as schools, cruise ships, and healthcare facilities. Furthermore, only a few viral particles are required for infection, underscoring the ease of its spread [
2]. Exposure to the virus induces vomiting, diarrhea, nausea, and abdominal pain, which lasts 1–3 days. Although most cases are mild, dehydration can be a significant risk factor for disease severity, particularly for young children, older adults, and immunocompromised individuals. NoV genogroup II (GII) is the most common genogroup linked to human infections, with the GII.4 genotype being the major cause of outbreaks and sporadic cases worldwide [
1,
3].
Accurate diagnosis of NoV infection is crucial for effective outbreak management and epidemiological tracking. NoV cannot be cultivated in conventional cell culture systems; thus, molecular and immunological methods are required to detect genetic material or viral antigen in clinical samples for laboratory diagnosis. Reverse transcription polymerase chain reaction (RT-PCR) is the gold standard for diagnosing NoV. It detects and quantifies viral RNA with high sensitivity and specificity, often providing differentiation between genogroup and strains [
4,
5]. However, RT-PCR has certain limitations that may reduce its practicality in low-resource settings, as it requires specialized equipment, specific reagents, and trained personnel. Moreover, the process involves multiple steps, including RNA extraction, reverse transcription, amplification, and analysis, which can be time-consuming. In contrast, immunological methods have lower sensitivity and specificity than molecular techniques. These methods often fail to detect low viral loads, resulting in an increased risk of false-negative results, particularly in early-stage or asymptomatic infections. Furthermore, the antigenic diversity of norovirus strains can compromise the binding efficiency of the antibodies used in the immunoassays [
6,
7]. The limitations of RT-PCR and immunological methods make them impractical in urgent diagnostic situations. Therefore, alternative, rapid, simplified, and cost-effective testing methods are needed for screening, identifying outbreaks, and supporting epidemiological studies.
Gold nanoparticles (AuNPs) are a versatile tool in rapid diagnostic technologies because of their unique physical, chemical, and optical properties. Specifically, their unique optical properties are attributed to surface plasmon resonance (SPR), a phenomenon in which free electrons on the nanoparticle surface resonate with incident light at specific wavelengths. The SPR characteristics of AuNPs vary with their size and shape. Spherical AuNPs generally exhibit a single SPR peak within the visible spectrum. Smaller AuNPs (5–20 nm) typically appear red because of their absorption properties, whereas larger particles tend to scatter light more and show color shifts tending toward purple (redshift) [
8,
9,
10]. AuNPs provide a high surface area for the immobilization of diagnostic probes, such as antibodies, aptamers, and nucleic acids, which amplify signal detection [
11,
12,
13].
Aptamers are short synthetic nucleic acid sequences that bind to specific molecular targets with high affinity and specificity. They are preferable to antibodies because of their greater stability, ease of synthesis, and the ability to target a wide range of analytes, including proteins, small molecules, and whole viruses [
14]. The functionalization of AuNPs with aptamers facilitates rapid diagnostic testing, which often produces simple colorimetric outputs that are visible to the naked eye, making them promising alternative probes for diagnostic assays [
15,
16,
17]. Aptamer-functionalized AuNPs have been developed for the colorimetric detection of various pathogens, such as SARS-CoV-2 [
18], Zika virus [
19], and
Pseudomonas aeruginosa [
20], using aptamers specific to pathogen-associated proteins. Furthermore, this detection system is applied in targeting specific cancer biomarkers, hormones, or small molecules in clinical samples with high precision [
11,
21,
22].
SMV25 is a single-stranded DNA aptamer that targets the NoV capsid protein [
23]. It was developed through the systematic evolution of ligands by exponential enrichment (SELEX) using Snow Mountain virus (SMV), the prototype GII.2 human norovirus (HuNoV), as the selection target. Secondary structural prediction revealed a dominant loop and two protruding hairpins (
Figure S1). The binding capacity of SMV25 was evaluated against various NoV-like particles (VLPs) and HuNoV in outbreak-derived stool specimens. Its signal intensity ratio (T/N ratio) indicated that SMV25 exhibited a strong binding affinity for GII.2 and GII.4, followed by GII.1, GII.7, GII.6, GI.8, and GII.3 VLPs. Similarly, SMV25 displayed high T/N ratios for GII.2 (SMV), GII.4, and GII.1 in diluted, partially purified stool samples from outbreaks. Furthermore, SMV25 demonstrated high capture efficiency during virus preconcentration before performing RT-PCR analysis of artificially contaminated lettuce samples. To date, the SMV aptamer has not been exploited in combination with AuNPs as a biosensing material for the development of simple colorimetric assays for NoV detection.
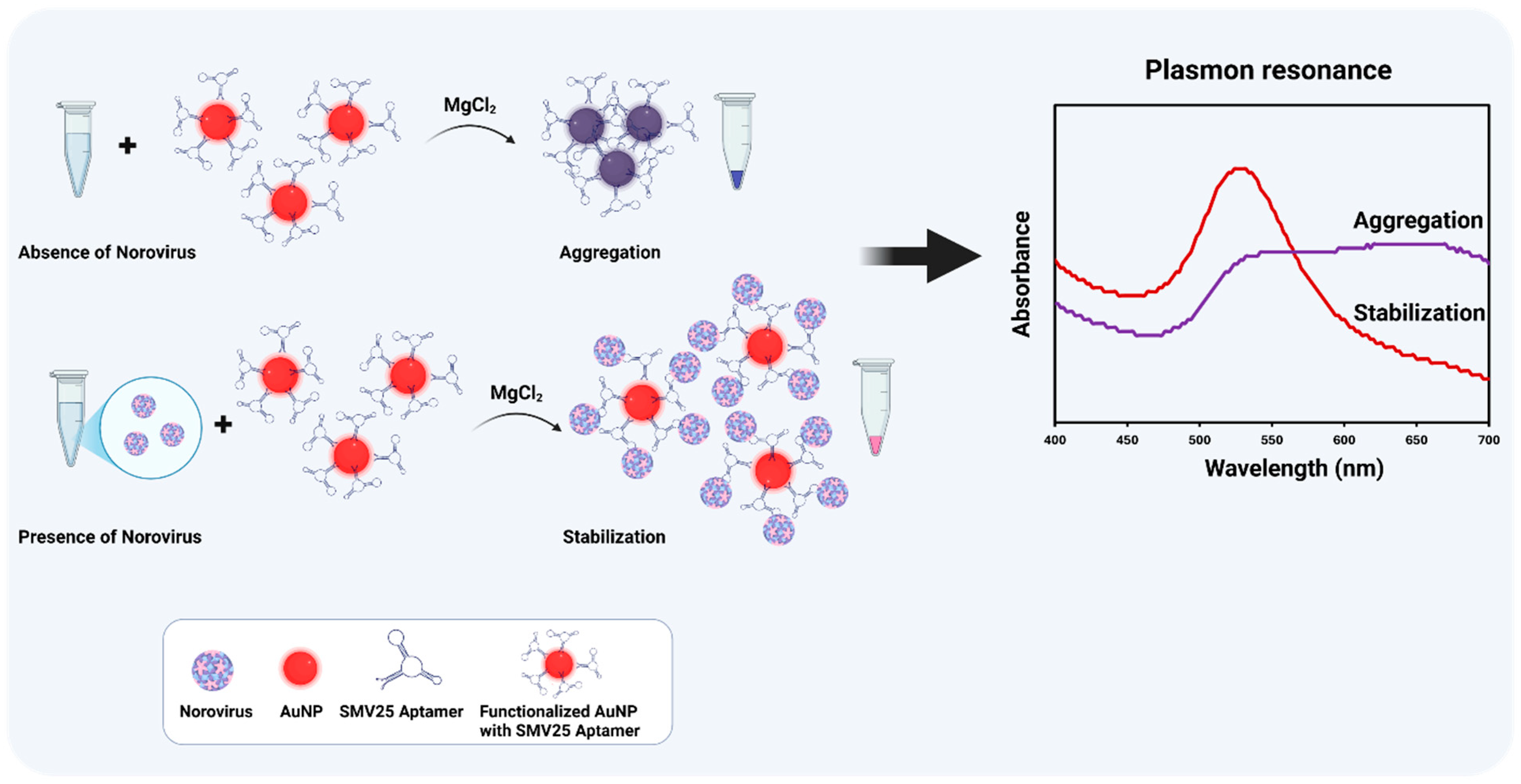
Addressing this gap and highlighting a significant step forward in NoV diagnostics, this study introduces an innovative approach: we have developed and validated a simple, user-friendly, and cost-effective colorimetric method for the direct detection of NoV in stool samples by employing SMV25 aptamer-functionalized AuNPs. The novelty of our assay lies not only in this first-time combination but also in its specific design for direct application to complex matrices and its operational simplicity. Detection is based on the visible red color obtained in the reaction following MgCl
2 induction after aptamer–NoV binding. The red color arises from enhanced steric stabilization, which reduces AuNP aggregation and minimizes the wavelength shift in the surface plasmon absorbance. In contrast, in the absence of NoV, the net charge of the functionalized AuNPs decreases due to neutralization by magnesium ions (Mg
2+), promoting particle aggregation and causing a redshift in the surface plasmon absorbance toward higher wavelengths. In addition to visible colorimetric detection, the absorbance spectra were measured between 400 and 700 nm using spectral scanning spectrophotometers (
Figure 1). The absorbance ratio at 630 nm to 520 nm indicates the aggregation levels; a higher ratio signifies greater aggregation (with increased absorbance at 630 nm), whereas a lower ratio indicates dispersed nanoparticles, the absorbance of which is stronger at 520 nm. The significance of this work is underscored by its potential as a screening tool; this functionalized AuNP-based colorimetric assay provides crucial proof-of-concept for detecting NoV without requiring specialized equipment or highly trained personnel. This makes the assay highly useful for rapid, on-site diagnostics in resource-limited environments, thereby facilitating timely outbreak response and improving public health surveillance capabilities for norovirus.
2. Materials and Methods
2.1. Aptamer
The SMV25 aptamer (5′-AGTATACGTATTACCTGCAGCCATCTGTGTGAAGACTATATGGCGCTCACATATTTCTTTCCGATATCTCGGAGATCTTGC-3′) [
23] was obtained from Bio Basic Inc., Markham, ON, Canada. The aptamer was modified with thiols at its 3′ end using high-performance liquid chromatography for purification and verification. The thiolated end group would facilitate the attachment of the aptamer onto the AuNP surfaces. Prior to immobilization, the disulfide bonds in the thiolated end of the aptamer were reduced by treatment with Tris (2-carboxyethyl) phosphine hydrochloride (TCEP, Sigma Aldrich, Oakville, ON, Canada) at a molar ratio of 10:1 (TCEP:aptamer), following the manufacturer’s recommendations. Briefly, 10 μM of aptamer solution was incubated with 0.1 mM TCEP for 1 h in the dark. The reduced aptamer required no additional purification or dialysis because TCEP does not interfere with thiol–gold binding. To restore the active conformation of the aptamer, it was refolded by heating at 85 °C for 5 min in a water bath, followed by rapid cooling in an ice bath.
2.2. The Synthesis and Characterization of the AuNPs
Citrate-capped AuNPs were synthesized by sodium citrate reduction of HAuCl
4, as described by Zhang et al. [
24], with some modifications. Briefly, an aqueous solution of 1 mM HAuCl
4 (100 mL) (Sigma-Aldrich, St. Louis, MO, USA) was reduced with a 38.8 mM trisodium citrate solution (10 mL) (RCI Labscan Ltd., Bangkok, Thailand) at 90 °C under vigorous stirring for 15 min, resulting in a color change in the mixture from pale yellow to a reddish wine-like color, indicating nanoparticle formation. The solution was boiled, stirred for 10 min, and cooled to room temperature (RT). The solution was then filtered through a 0.45-mm membrane filter and stored in dark glass bottles at 4 °C. The particle size and zeta potential (ZP) of the AuNPs were measured by dynamic light scattering with a Zetasizer (Malvern Instruments, Worcestershire, UK). The AuNPs were then visualized by scanning transmission electron microscopy (STEM). A 10 μL portion of the sample was dropped onto a piece of a carbon-coated copper grid and allowed to air-dry. STEM images were acquired using a JSM-IT800 Ultrahigh Resolution Field Emission SEM (JEOL, Peabody, MA, USA). UV–visible (UV–Vis) spectroscopy was acquired, and spectral analysis was performed at wavelengths between 400 and 700 nm.
2.3. The Immobilization of the SMV25 Aptamer onto AuNPs
The SMV25 aptamer was immobilized onto AuNPs, first by preparing 2.5 μM of the SMV25 aptamer by dilution of a 10 μM stock in phosphate-buffered saline (PBS; HyClone, Logan, UT, USA) containing 0.55 mM MgCl2 (Sigma Aldrich, Canada). Subsequently, 5 μL of the 2.5 μM aptamer solution was combined with 25 μL of AuNPs and incubated at RT for 16 h. This process utilized the strong affinity of the thiol groups for the gold surfaces to establish the direct immobilization of the thiolated aptamer onto the AuNPs by adsorption.
2.4. Optimization of MgCl2 Concentration for SMV25-Functionalized AuNP Aggregation
To develop the colorimetric detection method, the effect of MgCl2 concentration on the aggregation of AuNPs was investigated. A mixture was prepared by combining 30 μL AuNPs, 5 μL PBS, and 20 μL MgCl2 at varying concentrations (8, 10, 12, 14, 16, and 18 mM) in a 0.2 mL PCR tube, yielding a total volume of 55 μL. The color change was observed with the naked eye immediately after the addition of MgCl2; the reddish color indicates dispersed AuNPs, which was interpreted as positive, while the purple indicates aggregated AuNPs, which was interpreted as negative. Furthermore, absorbance intensity changes were recorded using a UV–Vis spectrophotometer (BioTek, Agilent, Winooski, VT, USA) within the wavelength range of 400–700 nm. The absorbance ratio at 630 nm to 520 nm (A630/520) was analyzed. An A630/520 value ≥ 1 indicated aggregation, which was interpreted as a negative result, whereas A630/520 < 1 indicated dispersed AuNPs, which was interpreted as a positive result.
2.5. The Detection of the NoV Capsid Protein
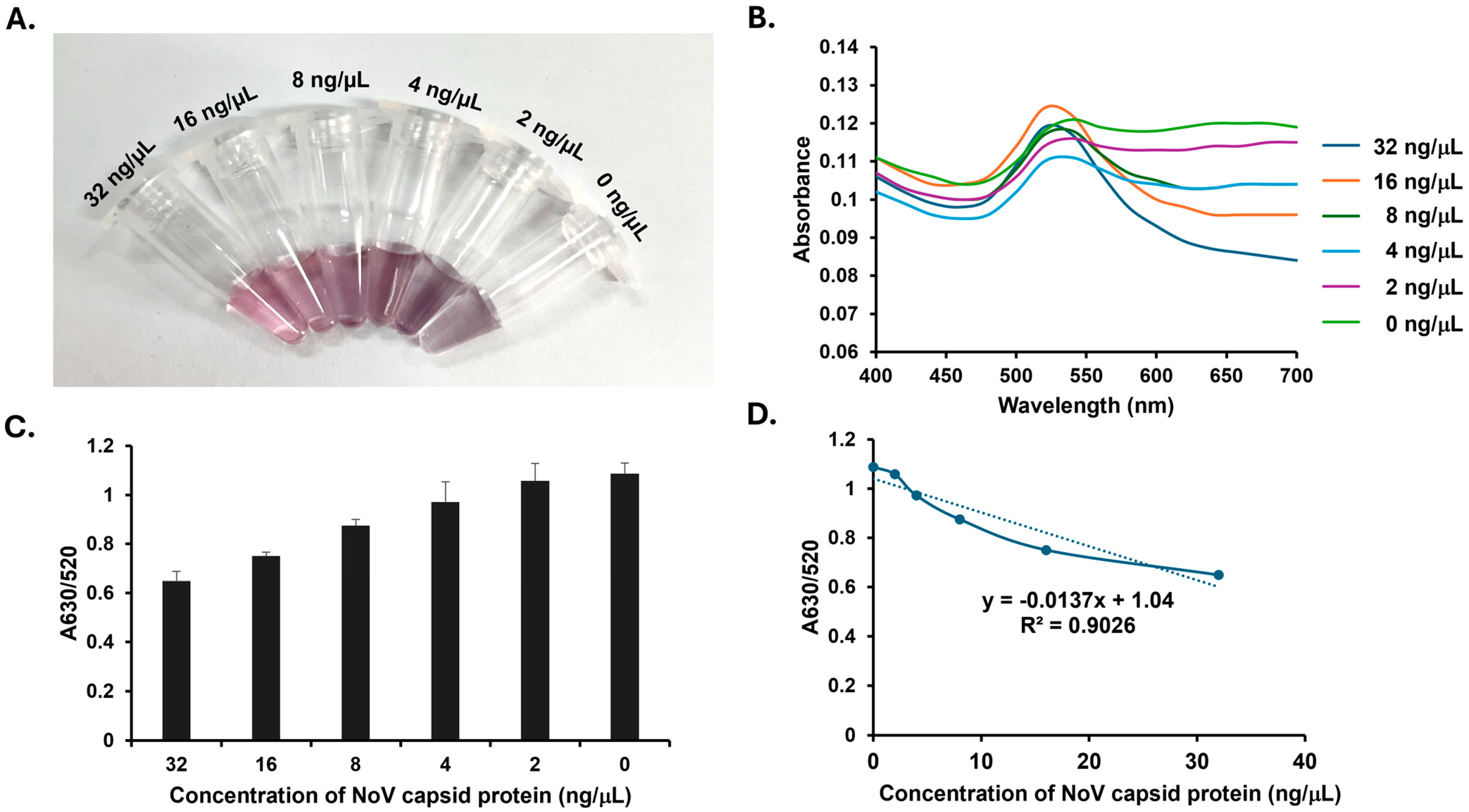
The recombinant NoV group-2 capsid protein was obtained from ProSpec, Rehovot, Israel. Various concentrations (0, 2, 4, 8, 16, and 32 ng/μL) of the protein were prepared by dilution in PBS, after which 5 μL of each concentration was mixed with 30 μL of SMV25-modified AuNP solution in PCR tubes. The mixtures were incubated at RT for 30 min. Following incubation, 20 μL of 14 mM MgCl2 was added to each sample. The resulting color changes were visually inspected. As mentioned above, a red solution indicates a positive result, whereas a purple solution indicates a negative result. The absorbance intensities were measured using a UV–visible spectrophotometer at the 400–700 nm wavelength. The absorbance ratio at 630 nm to 520 nm (A630/520) was calculated and interpreted as above.
For the spiked assay, a 10% stool suspension was prepared from a healthy donor by weighing 1 g of stool and homogenizing it in 9 mL of PBS (pH 7.4) using a vortex mixer. The suspension was then aliquoted and stored at −20 °C until use. Prior to testing, the NoV capsid protein was spiked into the 10% stool suspension at final concentrations of 12.5, 25, 50, and 100 ng/μL. Subsequently, 5 μL of each spiked sample was added to 30 μL of SMV25-modified AuNP solution and incubated at RT for 30 min. As described in the abovementioned protocol, 20 μL of 14 mM MgCl2 was added to the mixture, and changes in color and absorbance intensities were recorded and analyzed.
2.6. Detection of NoV in Stool Samples
Ninety stool samples from patients with diarrhea were kindly provided by Professor Niwat Maneekarn of the Center of Excellence (Emerging and Re-emerging Diarrheal Viruses), Chiang Mai University. All samples had been previously tested by real-time RT-PCR, with the genogroup and genotypes determined for the positive cases.
For analysis, 5 μL of each stool suspension was directly added to the SMV25-functionalized AuNP solution. In cases where the samples appeared turbid or dark brown, the suspensions were diluted 1:1 with PBS before being mixed with the AuNPs. The mixtures were incubated at RT for 30 min, followed by the addition of 20 μL of 14 mM MgCl
2. The color changes were observed visually, and the absorbance intensities were measured. Quantitative assessment was performed using the calibration equation derived from the NoV capsid protein spiked stool suspension experiment. Finally, the assay’s sensitivity and specificity were analyzed using the MedCalc diagnostic test evaluation version 23.2.6, an online statistical calculator [
25].
2.7. The STEM of the SMV25-Functionalized AuNPs
STEM was performed to visualize the aggregation and dispersion patterns of various reactions involving the SMV25-functionalized AuNPs. The test conditions were as follows: (1) SMV25-functionalized AuNPs alone; (2) SMV25-functionalized AuNPs in the presence of 32 ng/µL of NoV capsid protein, representing a positive reaction; (3) SMV25-functionalized AuNPs with PBS as the negative control; (4) SMV25-functionalized AuNPs with a NoV-positive stool sample; and (5) SMV25-functionalized AuNPs with a NoV-negative stool sample. Following the addition of 14 mM MgCl2 and the observation of the resulting colorimetric reaction, a 10 µL portion of each sample was placed onto a carbon-coated copper grid and air-dried. STEM images were then captured using the JSM-IT800 Ultrahigh Resolution Field Emission SEM (JEOL, Tokyo, Japan).
4. Discussion
Several biosensor-based approaches have recently been developed as alternative methods for NoV detection to address the limitations of existing techniques. RT-PCR, the gold standard, while highly sensitive, is costly and time-consuming. Furthermore, immunological assays may fail to detect emerging NoV genotypes with high sensitivity. AuNP-based biosensors offer a promising alternative detection system that is portable and efficient. Despite these advantages, additional optimization is required to adapt these methods for routine use, including outbreak monitoring and public health surveillance.
This study explored the potential of aptamer-functionalized AuNPs as an innovative, rapid, and accessible platform for detecting NoV capsid proteins. The aptamer was modified by adding a thiol (-SH) group at the 3′ end, enabling its immobilization onto the AuNP surface via thiol–gold interactions. Thiolated aptamers bind to AuNPs by covalent bonding, ensuring strong and highly stable immobilization. This bonding enhances target recognition by reducing steric hindrance, thereby facilitating high sensitivity and long-term diagnostic assays. In contrast, non-thiolated aptamers offer a simpler and more cost-effective alternative but may exhibit reduced stability and reproducibility [
26,
27]. The salt-induced aggregation in the assay generated a detectable signal, manifesting as a shift in localized SPR visible by naked-eye visualization. NaCl and MgCl
2 are commonly used to induce the aggregation of AuNPs in colorimetric assays. The choice between the two depends on the specific biosensing application; NaCl is preferred for gradual and tunable aggregation, while MgCl
2 is favored for rapid and strong aggregation at lower concentrations [
28,
29]. In this study, strong aggregation was necessary for detecting the NoV capsid protein in stool samples. Therefore, MgCl
2, a divalent salt, was used as a coagulant because of its superior ability to neutralize surface charges, thereby inducing the robust aggregation of aptamer-functionalized AuNPs. Typically, AuNPs are stabilized in solution by a negative surface charge from the capping citrate ions. These charges create repulsive forces that prevent the nanoparticles from aggregating. When MgCl
2 is introduced, Mg
2+ neutralizes this surface charge by binding to negatively charged groups, weakening the electrostatic repulsion, thereby inducing aggregation. Similar studies have demonstrated the capability of nanoparticle systems for colorimetric detection. Aithal et al. [
18] used MgCl
2 to induce nanoparticle aggregation for SARS-CoV-2 spike protein detection, achieving sensitivities as low as 16 nM of spike protein in PBS and 3540 genome copies/μL of inactivated SARS-CoV-2 virus. Djisalov et al. [
30] demonstrated the rapid detection of
Trichoderma spp. via the MgCl
2-induced aggregation of AuNPs, which was followed by color change evaluation and absorbance spectra measurement. These examples highlight the potential of simple, equipment-free diagnostic testing using visual or absorbance measurements.
Our study successfully demonstrated the detection of NoV capsid proteins in both colloidal and stool suspensions using aptamer–AuNP conjugation, as confirmed by naked-eye visualization and absorbance measurements. The method exhibited a consistent response in both matrices, with A630/A520 values showing a clear linear increase as the NoV capsid protein concentration decreased. Specifically, for the colloidal suspension, this linear relationship was observed over the 0–32 ng/µL range, yielding a regression correlation coefficient (R
2) of 0.9026. In the stool suspension, the linear trend extended across the 0–100 ng/µL range, with an R
2 value of 0.9170. These strong correlations, both approaching to 1, underscore the consistency and reliable response of the method, indicating its suitability for sensitive norovirus detection. STEM further confirmed the reliability of the system by demonstrating the well-defined dispersion and aggregation patterns, which are consistent with UV–vis spectroscopy findings. However, in real stool samples, the detection efficiency of our assay decreased, yielding 77% sensitivity and 65% specificity. This reduction can be primarily attributed to the highly complex and heterogeneous nature of stools. As a biological matrix, stool contains bacteria, undigested food, metabolites, and other diverse components that can interfere with the assay in multiple ways [
31]. For example, physical interference can arise from high turbidity or strong coloration in some samples, which may obscure the colorimetric readout. Additionally, various components within the stool matrix can engage in non-specific interactions with either the aptamers or the AuNPs, leading to unintended aggregation or stabilization events. The biochemical complexity of stool further impairs specific probe–target interactions; key contributing factors include potential enzymatic degradation of aptamers or the NoV capsid protein target, which diminishes available binding components, and variations in pH and ionic strength that can adversely alter AuNPs, aptamer conformation, and binding affinity. Collectively, these matrix-inherent factors can obscure signals and reduce overall assay performance [
32,
33]. Beyond these direct matrix effects, pre-analytical variables associated with sample handling and storage also significantly influence real-world reproducibility [
34]. Repeated freeze–thaw cycles, for example, can degrade target analytes, and inconsistencies in dilution protocols may introduce variability in the final analyte concentrations presented to the assay [
35,
36]. In our experiments, certain 10% stool suspension samples were further diluted 1:1 with PBS if they exhibited high turbidity or coloration that interfered with colorimetric detection. This pre-treatment proved critical for some highly turbid, RT-PCR-positive samples. Specifically, the dilution step resulted in a lower A630/A520 ratio compared to their undiluted counterparts, which enables a correct positive interpretation where the undiluted sample might have yielded false-negative results. These corrected interpretations were consistent with RT-PCR findings. This observation underscores the trade-off; while dilution effectively reduces matrix-induced interference, it unavoidably lowers the absolute concentration of viral particles, which can, in turn, decrease detection sensitivity. To address challenges stemming from both inherent matrix effects and sample management issues, future investigations could explore strategies such as optimizing pH and ionic strengths, establishing standardized sample handling/preparation procedures, and preparing matrix-matched calibrations to enhance detection robustness in complex samples. Furthermore, conducting a study to determine an optimal, standardized dilution factor by quantitatively assessing its impact on sensitivity would be valuable in refining the assay for routine field use.
Notably, the system detected multiple NoV genotypes with high sensitivity, particularly for GII.2 (100%) and GII.4 (78%). This result is largely due to the use of NoV GII.2 as the target for aptamer selection. The binding capacity of the selected aptamer to various VLPs had been previously investigated, and strong binding affinity was observed for GII.2 and GII.4, followed by GII.6 and GII.7, with slightly lower affinity for GII.3 and GII.17 [
23]. These results were consistent with our findings. During specificity testing in the present study, the assay showed no false-positive results in samples containing rotavirus or sapovirus, but did produce false positives for samples containing astrovirus, adenovirus, and enterovirus. However, definitive conclusions about the cross-reactivity with these viruses could not be made. Further validation using intact viruses or recombinant viral proteins is necessary to confirm the specificity of the assay for other gastroenteritis-causing viruses. Escudero-Abarca et al. [
23] reported that SMV25 exhibited significantly low T/N ratios for poliovirus and hepatitis A virus from cell culture lysates, indicating that it may not bind to non-target viruses. The false-positive results observed in the present study may have been caused by interfering substances in the stools that destabilized the AuNPs.
Various aptamer- and AuNP-based biosensors have been developed for NoV detection using fluorescence and electrochemical systems. Giamberardino et al. [
37] designed a biosensor that integrated a DNA aptamer with an AuNP-modified screen-printed carbon electrode to detect both HuNoV and murine norovirus in stool and environmental specimens using fluorescence anisotropy. Weng and Neethirajan [
38] developed a portable fluorometric paper-based microfluidic device that combined aptamers to detect norovirus spiked into food matrices. Similarly, Li et al. [
39] introduced a dual-mode aptasensor incorporating AuNPs that enabled both electrochemical and colorimetric NoV detection in fecal samples. Although these methods offered high sensitivity, they often involved complex procedures and required expensive instrumentation. Our approach used a simple mix-and-detect format, which could reduce the operational difficulty and costs of detection. However, the main limitation of this study is that we did not use live NoV or other gastroenteritis viruses to assess assay performance. The amount of recombinant capsid protein may not accurately reflect the actual virus levels in samples.
In summary, SMV25-functionalized AuNPs present a promising approach for NoV detection. The assay demonstrated high sensitivity in the buffer systems, although its performance in complex biological samples requires further optimization. Future research should focus on improving specificity and ensuring consistent applicability across genotypes to enhance practical implementation.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}