Sensing Protein Structural Transitions with Microfluidic Modulation Infrared Spectroscopy

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Modeling of ⍺-Helical and β-Sheet Proteins
2.2. Purification of Recombinant Tau and pTau Proteins
2.3. Sample Preparation
2.4. Microfluidic Modulation Spectroscopy (MMS)
2.5. IR Spectrum Analysis
2.6. Statistical Analysis
3. Results
3.1. Microfluidic Modulation Spectroscopy-Infrared (MMS) Enables Quantitative Analysis of Protein Secondary Structure
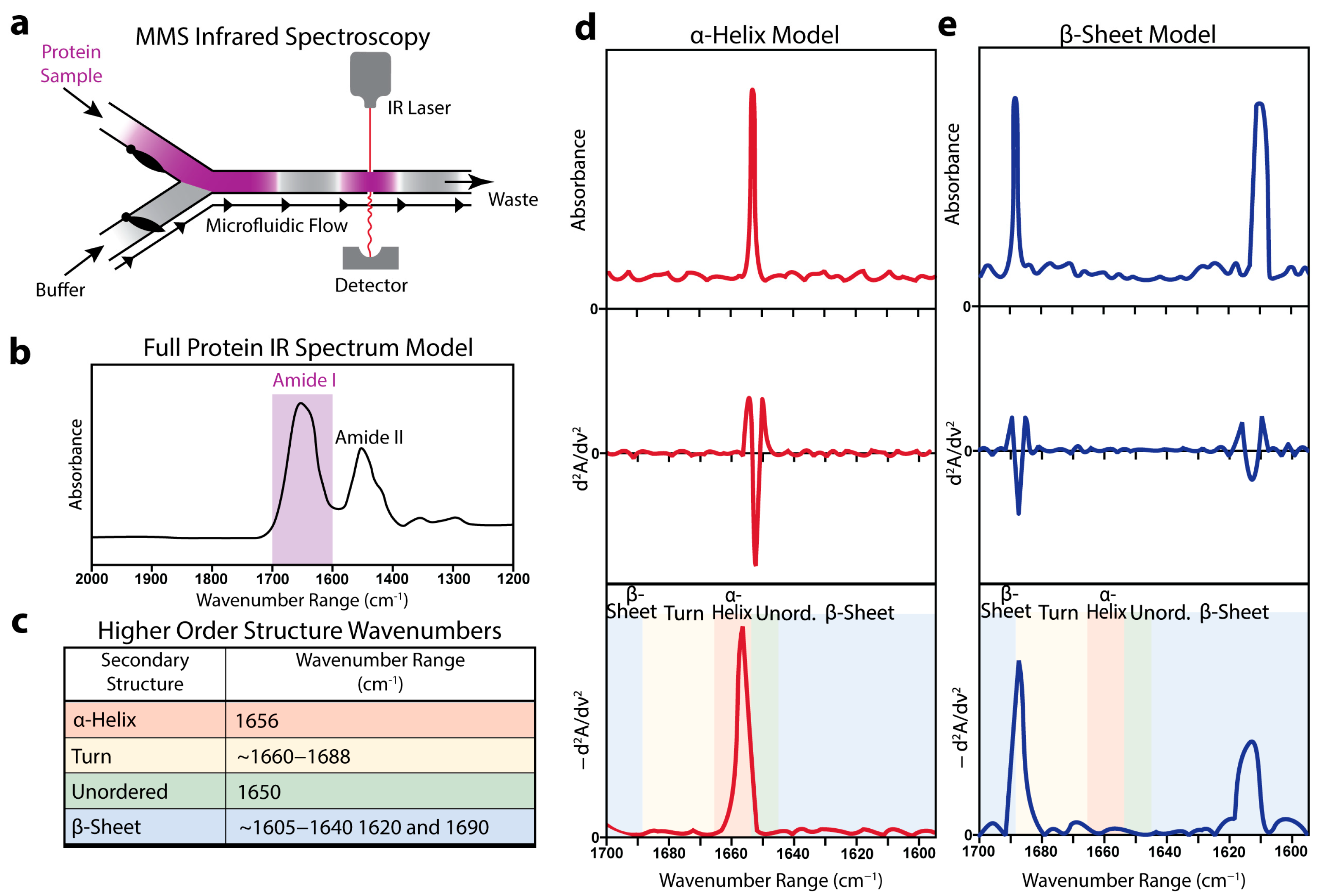
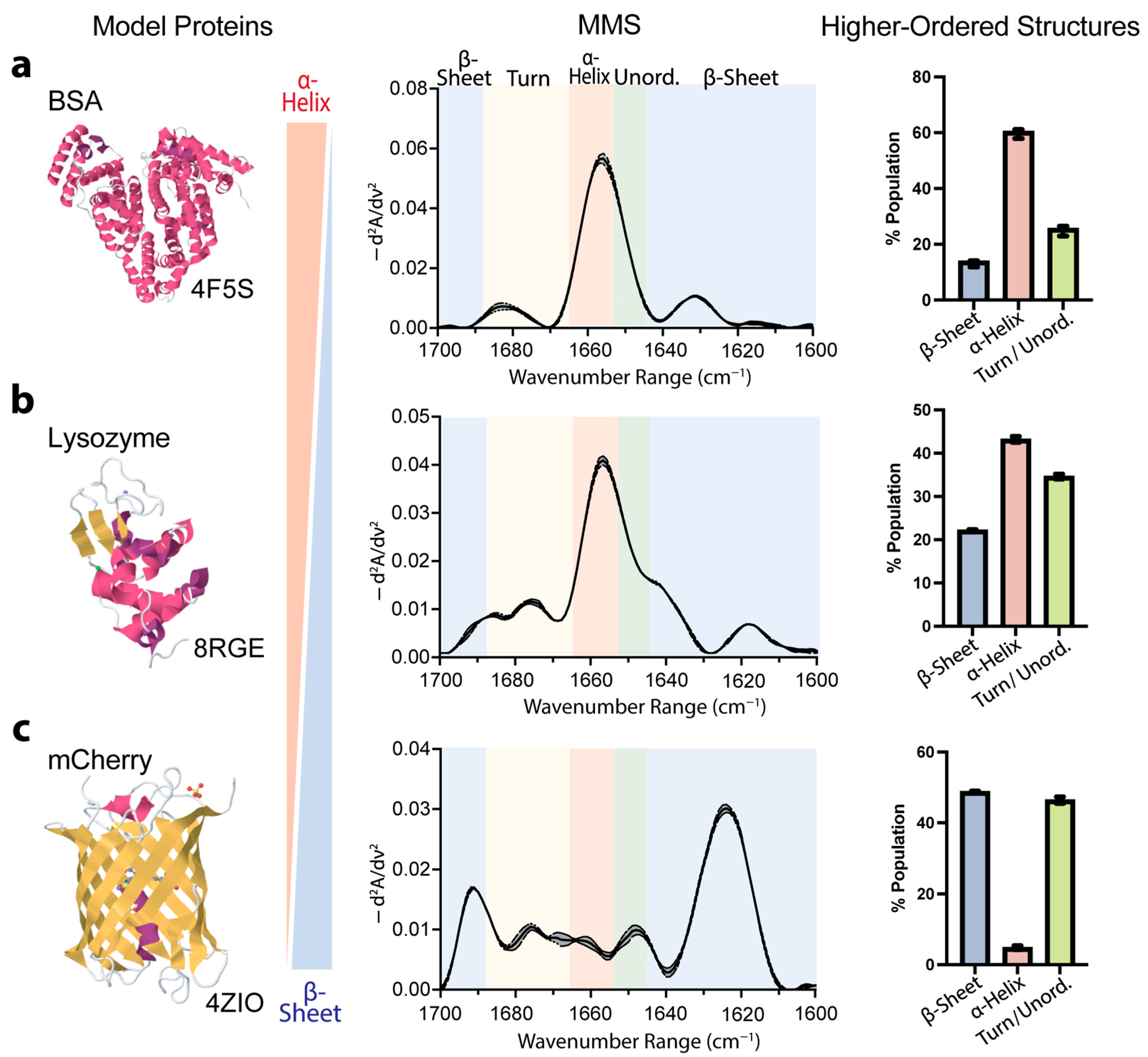
3.2. Validation of MMS-Based Secondary-Structure Analysis Using Model Proteins
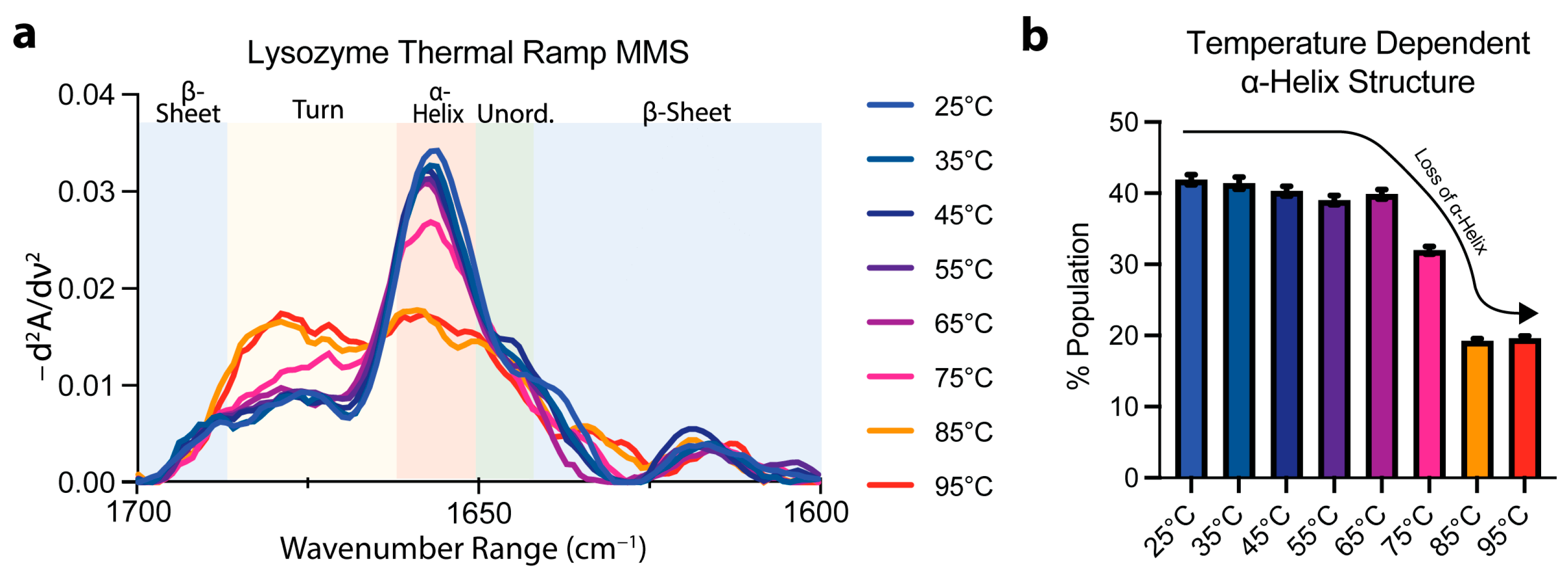
3.3. Protein Thermal Unfolding Can Be Monitored by MMS
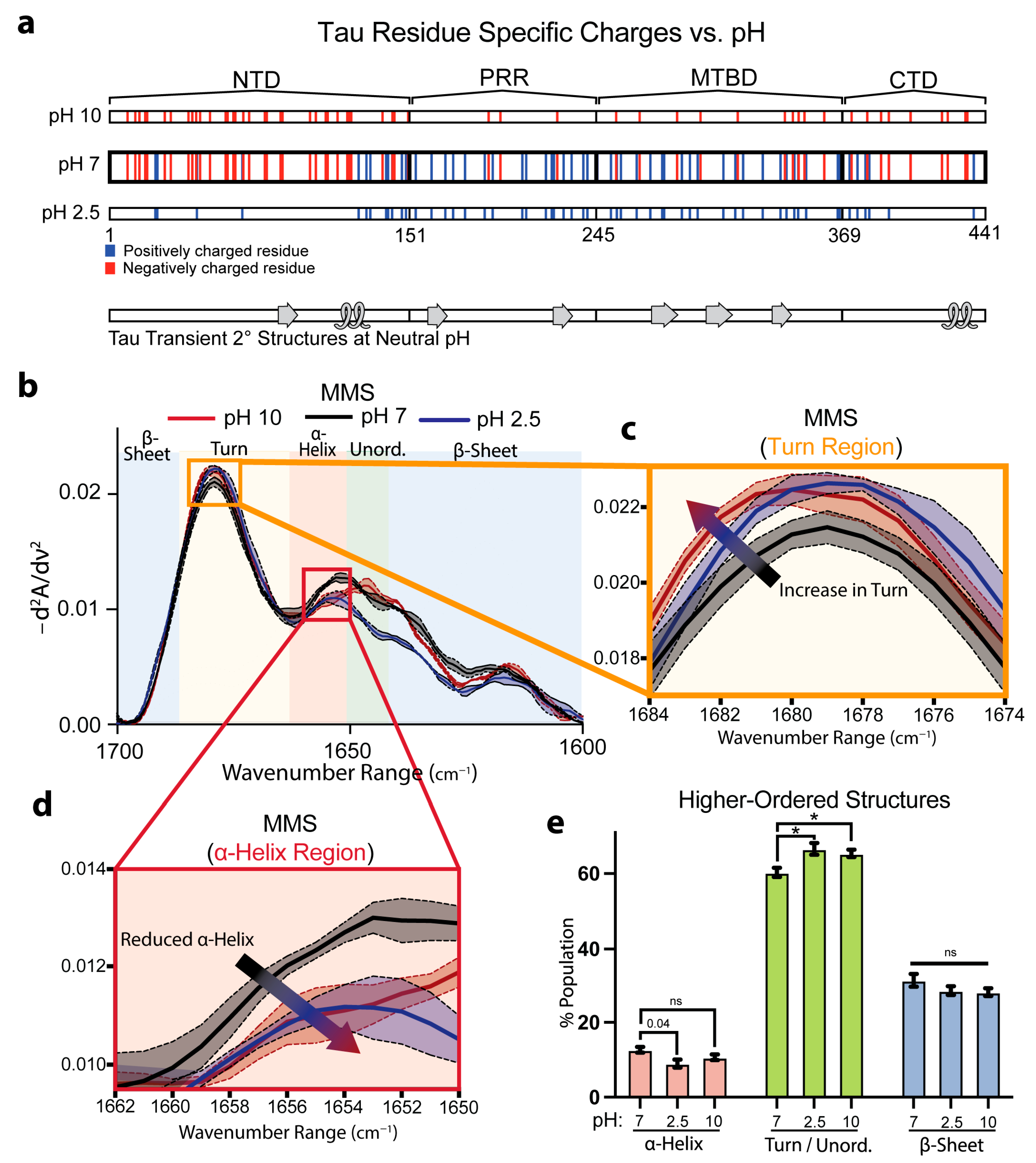
3.4. Tau Exhibits Secondary-Structure Changes Under Variable pH
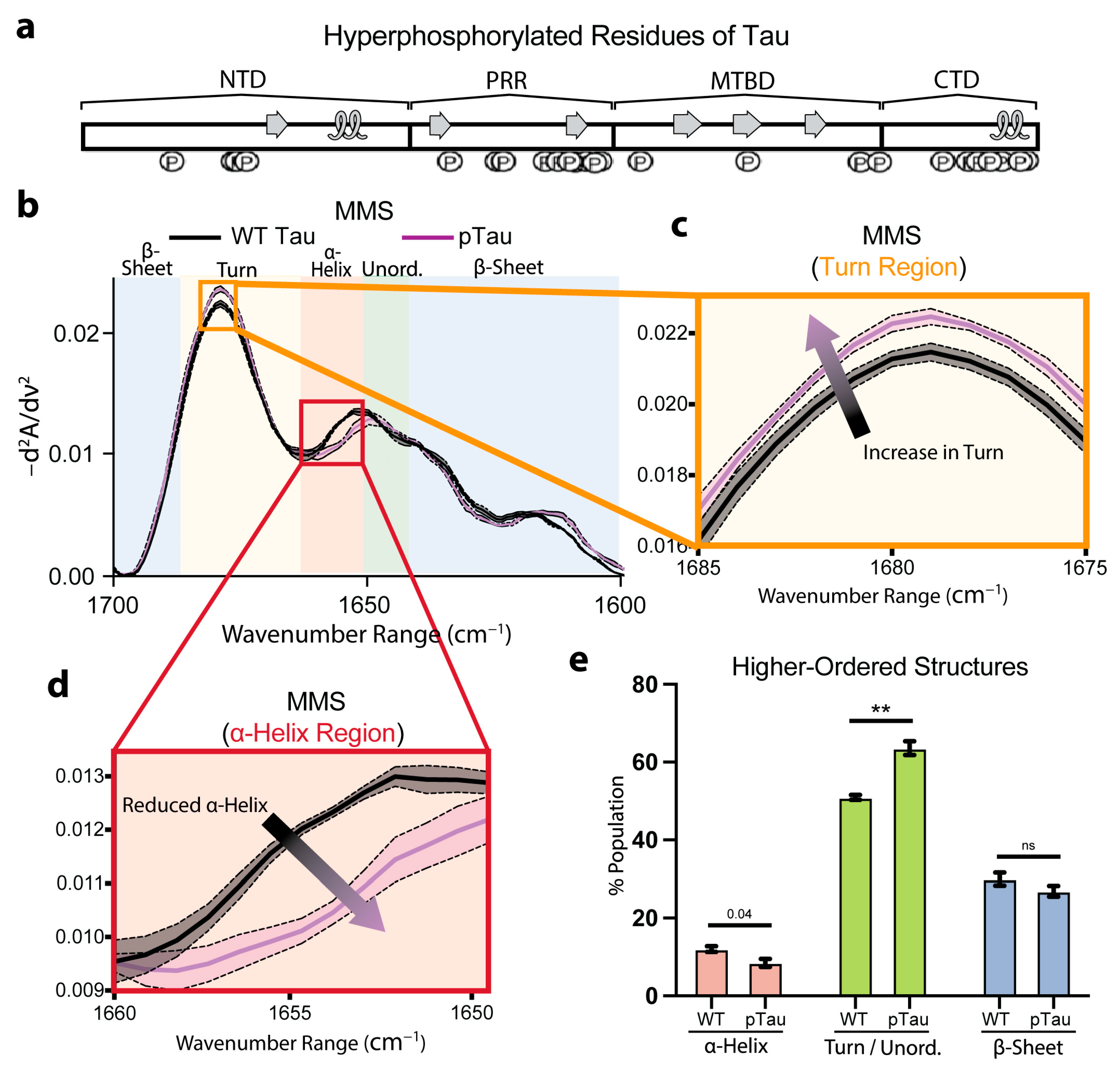
3.5. Hyperphosphorylation Moderately Affects Tau Secondary Structures
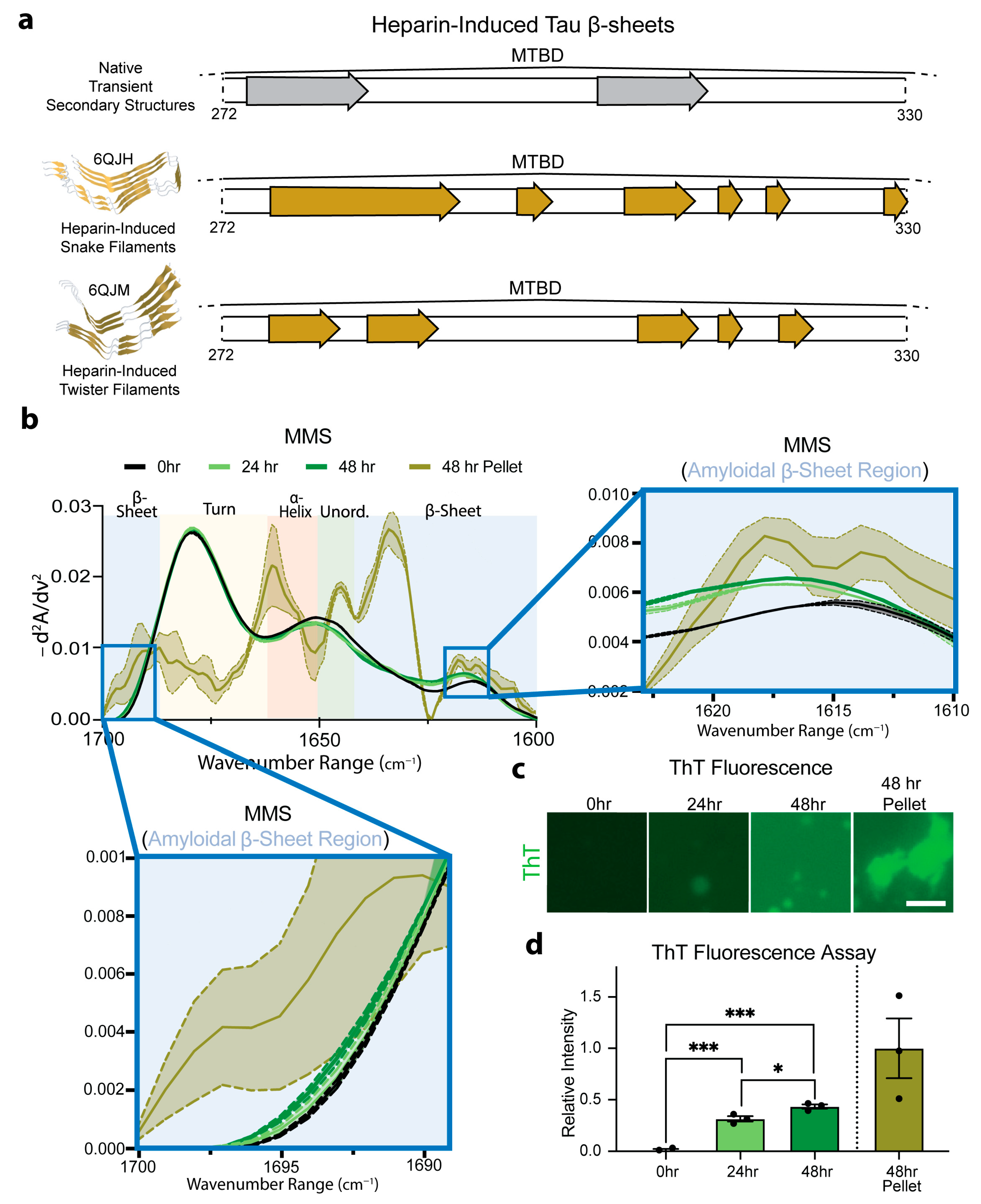
3.6. MMS Detects pTau Aggregation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- White, H.E.; Ignatiou, A.; Clare, D.K.; Orlova, E.V. Structural study of heterogeneous biological samples by cryoelectron microscopy and image processing. BioMed Res. Int. 2017, 2017, 1032432. [Google Scholar] [CrossRef]
- Martin, A.C.; Orengo, C.A.; Hutchinson, E.G.; Jones, S.; Karmirantzou, M.; Laskowski, R.A.; Mitchell, J.B.; Taroni, C.; Thornton, J.M. Protein folds and functions. Structure 1998, 6, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Analyzing protein structure and function. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Louros, N.; Schymkowitz, J.; Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 2023, 24, 912–933. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.R. Protein misfolding and aggregation in proteinopathies: Causes, mechanism and cellular response. Diseases 2023, 11, 30. [Google Scholar] [CrossRef]
- Moses, D.; Guadalupe, K.; Yu, F.; Flores, E.; Perez, A.R.; McAnelly, R.; Shamoon, N.M.; Kaur, G.; Cuevas-Zepeda, E.; Merg, A.D.; et al. Structural biases in disordered proteins are prevalent in the cell. Nat. Struct. Mol. Biol. 2024, 31, 283–292. [Google Scholar] [CrossRef]
- Shrestha, U.R.; Smith, J.C.; Petridis, L. Full structural ensembles of intrinsically disordered proteins from unbiased molecular dynamics simulations. Commun. Biol. 2021, 4, 243. [Google Scholar] [CrossRef]
- Levine, Z.A.; Shea, J.E. Simulations of disordered proteins and systems with conformational heterogeneity. Curr. Opin. Struct. Biol. 2017, 43, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Han, K.H. Transient secondary structures as general target-binding motifs in intrinsically disordered proteins. Int. J. Mol. Sci. 2018, 19, 3614. [Google Scholar] [CrossRef]
- Trivedi, R.; Nagarajaram, H.A. Intrinsically disordered proteins: An overview. Int. J. Mol. Sci. 2022, 23, 14050. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Moran, C.R.; Gambin, Y.; Deniz, A.A. Single-molecule fluorescence studies of intrinsically disordered proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2010; Volume 472, pp. 179–204. [Google Scholar]
- Forman-Kay, J.D.; Mittag, T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure 2013, 21, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Ando, T. Functional implications of dynamic structures of intrinsically disordered proteins revealed by high-speed AFM imaging. Biomolecules 2022, 12, 1876. [Google Scholar] [CrossRef]
- Miles, A.J.; Drew, E.D.; Wallace, B.A. DichroIDP: A method for analyses of intrinsically disordered proteins using circular dichroism spectroscopy. Commun. Biol. 2023, 6, 823. [Google Scholar] [CrossRef]
- Miles, A.J.; Janes, R.W.; Wallace, B.A. Tools and methods for circular dichroism spectroscopy of proteins: A tutorial review. Chem. Soc. Rev. 2021, 50, 8400–8413. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolym. Orig. Res. Biomol. 2008, 89, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Khrapunov, S. Circular dichroism spectroscopy has intrinsic limitations for protein secondary structure analysis. Anal. Biochem. 2009, 389, 174–176. [Google Scholar] [CrossRef]
- Morris, G.A.; Barjat, H. Reference deconvolution. In Analytical Spectroscopy Library; Elsevier: Amsterdam, The Netherlands, 1997; Volume 8, pp. 303–316. [Google Scholar]
- De Meutter, J.; Goormaghtigh, E. Evaluation of protein secondary structure from FTIR spectra improved after partial deuteration. Eur. Biophys. J. 2021, 50, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Zuber, G.; Prestrelski, S.J.; Benedek, K. Application of Fourier transform infrared spectroscopy to studies of aqueous protein solutions. Anal. Biochem. 1992, 207, 150–156. [Google Scholar] [CrossRef]
- Kong, J.; Yu, S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. [Google Scholar] [CrossRef]
- Borcherds, W.M.; Daughdrill, G.W. Using NMR Chemical Shifts to Determine Residue-Specific Secondary Structure Populations for Intrinsically Disordered Proteins; Academic Press: Cambridge, MA, USA, 2018; Volume 611, pp. 101–136. [Google Scholar]
- Zhang, Z.; Zhao, Q.; Gong, Z.; Du, R.; Liu, M.; Zhang, Y.; Zhang, L.; Li, C. Progress, challenges and opportunities of NMR and XL-MS for cellular structural biology. JACS Au 2024, 4, 369–383. [Google Scholar] [CrossRef]
- Chalmers, G.R.; Eletsky, A.; Morris, L.C.; Yang, J.Y.; Tian, F.; Woods, R.J.; Moremen, K.W.; Prestegard, J.H. NMR resonance assignment methodology: Characterizing large sparsely labeled glycoproteins. J. Mol. Biol. 2019, 431, 2369–2382. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Kim, P.S.; Dobson, C.M.; Redfield, C. A residue-specific NMR view of the non-cooperative unfolding of a molten globule. Nat. Struct. Biol. 1997, 4, 630–634. [Google Scholar] [CrossRef]
- Nwanochie, E.; Uversky, V.N. Structure determination by single-particle cryo-electron microscopy: Only the sky (and intrinsic disorder) is the limit. Int. J. Mol. Sci. 2019, 20, 4186. [Google Scholar] [CrossRef]
- Srivastava, A.; Nagai, T.; Srivastava, A.; Miyashita, O.; Tama, F. Role of computational methods in going beyond X-ray crystallography to explore protein structure and dynamics. Int. J. Mol. Sci. 2018, 19, 3401. [Google Scholar] [CrossRef]
- Holcomb, J.; Spellmon, N.; Zhang, Y.; Doughan, M.; Li, C.; Yang, Z. Protein crystallization: Eluding the bottleneck of X-ray crystallography. AIMS Biophys. 2017, 4, 557. [Google Scholar] [CrossRef]
- Suder, D.S.; Gonen, S. Mitigating the Blurring Effect of CryoEM Averaging on a Flexible and Highly Symmetric Protein Complex through Sub-Particle Reconstruction. Int. J. Mol. Sci. 2024, 25, 5665. [Google Scholar] [CrossRef] [PubMed]
- Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 2019, 294, 5181–5197. [Google Scholar] [CrossRef] [PubMed]
- Ivancic, V.A.; Lombardo, H.L.; Ma, E.; Wikström, M.; Batabyal, D. Advancing secondary structure characterization of monoclonal antibodies using Microfluidic Modulation Spectroscopy. Anal. Biochem. 2022, 646, 114629. [Google Scholar] [CrossRef]
- Ma, T.M.; Huang, R.H.; Collins, V.I.; Wikström, M.; Batabyal, D. Advancement of microfluidic modulation spectroscopy as a highly sensitive protein characterization technique for the detection of small structural changes. Anal. Biochem. 2023, 683, 115350. [Google Scholar] [CrossRef]
- Naudi-Fabra, S.; Tengo, M.; Jensen, M.R.; Blackledge, M.; Milles, S. Quantitative description of intrinsically disordered proteins using single-molecule FRET, NMR, and SAXS. J. Am. Chem. Soc. 2021, 143, 20109–20121. [Google Scholar] [CrossRef]
- LeBlanc, S.J.; Kulkarni, P.; Weninger, K.R. Single molecule FRET: A powerful tool to study intrinsically disordered proteins. Biomolecules 2018, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Metskas, L.A.; Rhoades, E. Single-molecule FRET of intrinsically disordered proteins. Annu. Rev. Phys. Chem. 2020, 71, 391–414. [Google Scholar] [CrossRef] [PubMed]
- Moosa, M.M.; Ferreon, J.C.; Ferreon, A.C.M. Single-molecule fret detection of early-stage conformations in α-synuclein aggregation. In Alpha-Synuclein: Methods and Protocols; Springer: New York, NY, USA, 2019; pp. 221–233. [Google Scholar]
- Quan, M.D.; Liao, S.C.J.; Ferreon, J.C.; Ferreon, A.C.M. Fluorescence lifetime imaging microscopy of biomolecular condensates. In Phase-Separated Biomolecular Condensates: Methods and Protocols; Springer: New York, NY, USA, 2022; pp. 135–148. [Google Scholar]
- Yoo, T.Y.; Meisburger, S.P.; Hinshaw, J.; Pollack, L.; Haran, G.; Sosnick, T.R.; Plaxco, K. Small-angle X-ray scattering and single-molecule FRET spectroscopy produce highly divergent views of the low-denaturant unfolded state. J. Mol. Biol. 2012, 418, 226–236. [Google Scholar] [CrossRef]
- Narang, D.; Lento, C.; Wilson, D.J. HDX-MS: An analytical tool to capture protein motion in action. Biomedicines 2020, 8, 224. [Google Scholar] [CrossRef]
- Seetaloo, N.; Zacharopoulou, M.; Stephens, A.D.; Kaminski Schierle, G.S.; Phillips, J.J. Millisecond hydrogen/deuterium-exchange mass spectrometry approach to correlate local structure and aggregation in α-synuclein. Anal. Chem. 2022, 94, 16711–16719. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Deniz, A.A. α-Synuclein multistate folding thermodynamics: Implications for protein misfolding and aggregation. Biochemistry 2007, 46, 4499–4509. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef]
- Konoplev, G.; Agafonova, D.; Bakhchova, L.; Mukhin, N.; Kurachkina, M.; Schmidt, M.P.; Verlov, N.; Sidorov, A.; Oseev, A.; Stepanova, O.; et al. Label-free physical techniques and methodologies for proteins detection in microfluidic biosensor structures. Biomedicines 2022, 10, 207. [Google Scholar] [CrossRef]
- Lin, J.C.; Glover, Z.K.; Sreedhara, A. Assessing the utility of circular dichroism and FTIR spectroscopy in monoclonal-antibody comparability studies. J. Pharm. Sci. 2015, 104, 4459–4466. [Google Scholar] [CrossRef]
- Wei, Y.; Thyparambil, A.A.; Latour, R.A. Protein helical structure determination using CD spectroscopy for solutions with strong background absorbance from 190 to 230 nm. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2014, 1844, 2331–2337. [Google Scholar] [CrossRef]
- Watson, M.J.; Harkewicz, R.; Hodge, E.A.; Vorauer, C.; Palmer, J.; Lee, K.K.; Guttman, M. Simple platform for automating decoupled LC–MS analysis of hydrogen/deuterium exchange samples. J. Am. Soc. Mass Spectrom. 2020, 32, 597–600. [Google Scholar] [CrossRef] [PubMed]
- De Meutter, J.; Vandenameele, J.; Matagne, A.; Goormaghtigh, E. Infrared imaging of high density protein arrays. Analyst 2017, 142, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, C.; Duan, M.; Liu, C.; Li, M.; Wu, G.; Du, J.; Xu, G.; Yu, X.; Wang, L. Application of microfluidic modulation spectroscopy for simultaneous structural and thermal stability analysis of commercial mAbs under varying formulation conditions. Anal. Methods 2025, 17, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Savitzky, A.; Golay, M.J. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Quan, M.D.; Choi, K.J.; Dao, K.M.; Ferreon, J.C.; Ferreon, A.C.M. Electrostatic modulation of hnRNPA1 low-complexity domain liquid–liquid phase separation and aggregation. Protein Sci. 2021, 30, 1408–1417. [Google Scholar] [CrossRef]
- Edelhoch, H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 1967, 6, 1948–1954. [Google Scholar] [CrossRef]
- Gill, S.C.; Von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Bolen, D.W. Thermodynamics of denaturant-induced unfolding of a protein that exhibits variable two-state denaturation. Biochemistry 2004, 43, 13357–13369. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Lorenz-Fonfria, V.A. Infrared difference spectroscopy of proteins: From bands to bonds. Chem. Rev. 2020, 120, 3466–3576. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta (BBA)-Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Sadat, A.; Joye, I.J. Peak fitting applied to fourier transform infrared and raman spectroscopic analysis of proteins. Appl. Sci. 2020, 10, 5918. [Google Scholar] [CrossRef]
- Schwaighofer, A.; Lendl, B. Infrared spectroscopy for structure analysis of protein inclusion bodies. In Inclusion Bodies: Methods and Protocols; Springer: New York, NY, USA, 2023; pp. 209–223. [Google Scholar]
- López-Lorente, Á.I.; Mizaikoff, B. Mid-infrared spectroscopy for protein analysis: Potential and challenges. Anal. Bioanal. Chem. 2016, 408, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.M.; Bourassa, M.W.; Smith, R.J. FTIR spectroscopic imaging of protein aggregation in living cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 2339–2346. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Sanchez-Weatherby, J.; Williams, L.J.; Mikolajek, H.; Sandy, J.; Worrall, J.A.; Hough, M.A. Efficient in situ screening of and data collection from microcrystals in crystallization plates. Biol. Crystallogr. 2024, 80, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Reddington, S.C.; Driezis, S.; Hartley, A.M.; Watson, P.D.; Rizkallah, P.J.; Jones, D.D. Genetically encoded phenyl azide photochemistry drives positive and negative functional modulation of a red fluorescent protein. RSC Adv. 2015, 5, 77734–77738. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e1000034. [Google Scholar] [CrossRef]
- Jebarupa, B.; Muralidharan, M.; Arun, A.; Mandal, A.K.; Mitra, G. Conformational heterogeneity of tau: Implication on intrinsic disorder, acid stability and fibrillation in Alzheimer’s disease. Biophys. Chem. 2018, 241, 27–37. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Lucas, L.; Rhoades, D.; Ferreon, J.C.; Ferreon, A.C.M. Electrostatic Effects on Tau Nanocondensates. Biomolecules 2025, 15, 406. [Google Scholar] [CrossRef]
- Uversky, V.N.; Winter, S.; Galzitskaya, O.V.; Kittler, L.; Lober, G. Hyperphosphorylation induces structural modification of tau-protein. FEBS Lett. 1998, 439, 21–25. [Google Scholar] [CrossRef]
- Zhu, S.; Shala, A.; Bezginov, A.; Sljoka, A.; Audette, G.; Wilson, D.J. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS ONE 2015, 10, e0120416. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dexheimer, T.; Sui, D.; Hovde, S.; Deng, X.; Kwok, R.; Bochar, D.A.; Kuo, M.H. Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription drugs linked to Alzheimer’s disease and cognitive functions. Sci. Rep. 2020, 10, 16551. [Google Scholar] [CrossRef] [PubMed]
- Zandomeneghi, G.; Krebs, M.R.; McCammon, M.G.; Fändrich, M. FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 2004, 13, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, L.; Tsoi, P.S.; Nair, A.; Ferreon, A.C.M.; Ferreon, J.C. Sensing Protein Structural Transitions with Microfluidic Modulation Infrared Spectroscopy. Biosensors 2025, 15, 382. https://doi.org/10.3390/bios15060382
Lucas L, Tsoi PS, Nair A, Ferreon ACM, Ferreon JC. Sensing Protein Structural Transitions with Microfluidic Modulation Infrared Spectroscopy. Biosensors. 2025; 15(6):382. https://doi.org/10.3390/bios15060382
Chicago/Turabian StyleLucas, Lathan, Phoebe S. Tsoi, Ananya Nair, Allan Chris M. Ferreon, and Josephine C. Ferreon. 2025. "Sensing Protein Structural Transitions with Microfluidic Modulation Infrared Spectroscopy" Biosensors 15, no. 6: 382. https://doi.org/10.3390/bios15060382
APA StyleLucas, L., Tsoi, P. S., Nair, A., Ferreon, A. C. M., & Ferreon, J. C. (2025). Sensing Protein Structural Transitions with Microfluidic Modulation Infrared Spectroscopy. Biosensors, 15(6), 382. https://doi.org/10.3390/bios15060382