Fluorogenic Aptamer-Based Hybridization Chain Reaction for Signal-Amplified Imaging of Apurinic/Apyrimidinic Endonuclease 1 in Living Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Design and Preparation of Probes
2.3. HCR
2.4. In Vitro APE1 Detection
2.5. Construction and Characterization of Carrier
2.6. Cell Culture
2.7. APE1 Imaging in Living Cells
3. Results
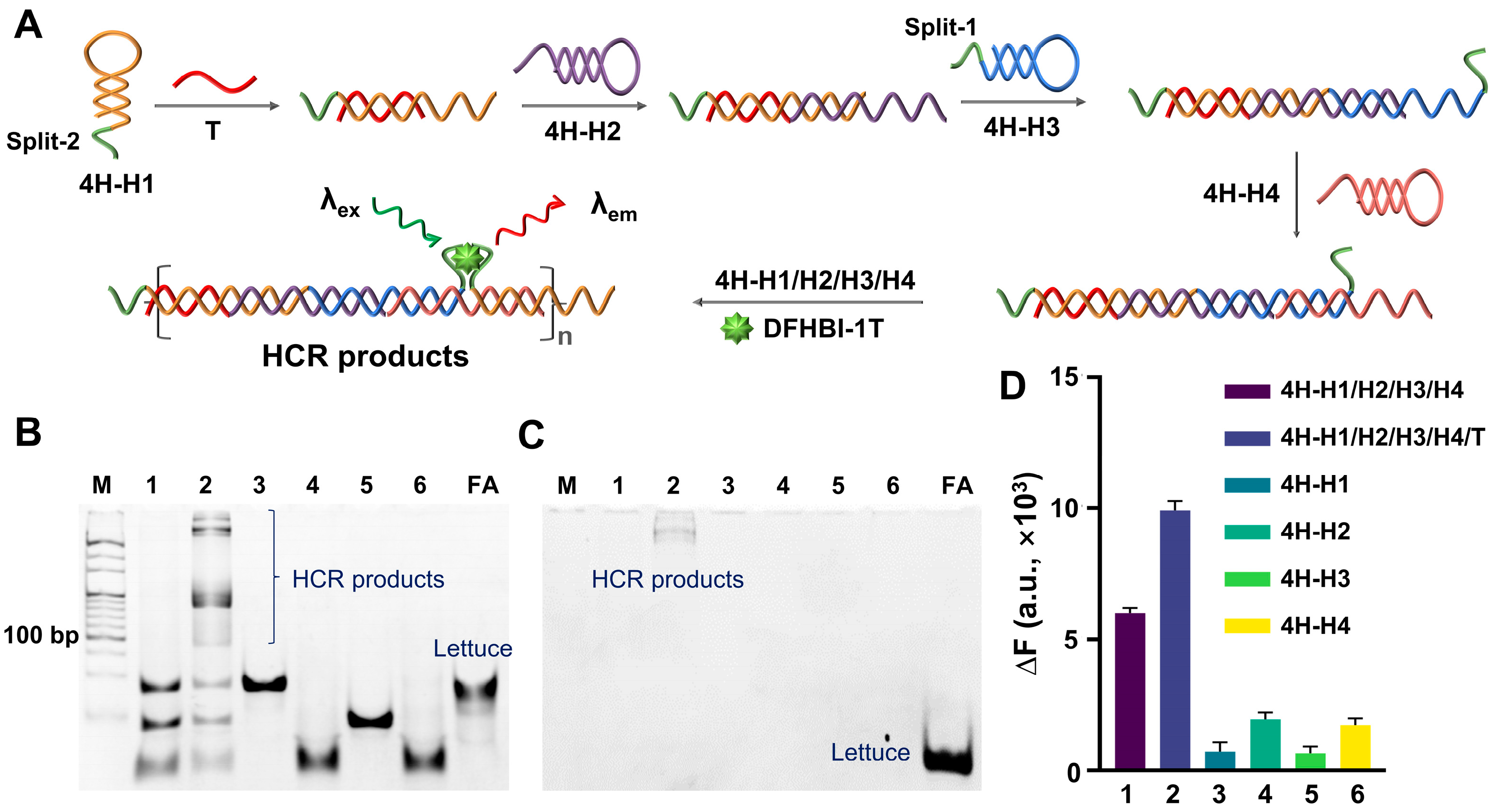
3.1. Four-HP-Mediated HCR (4H-HCR)
3.2. Two-HP-Mediated HCR (2H-HCR)
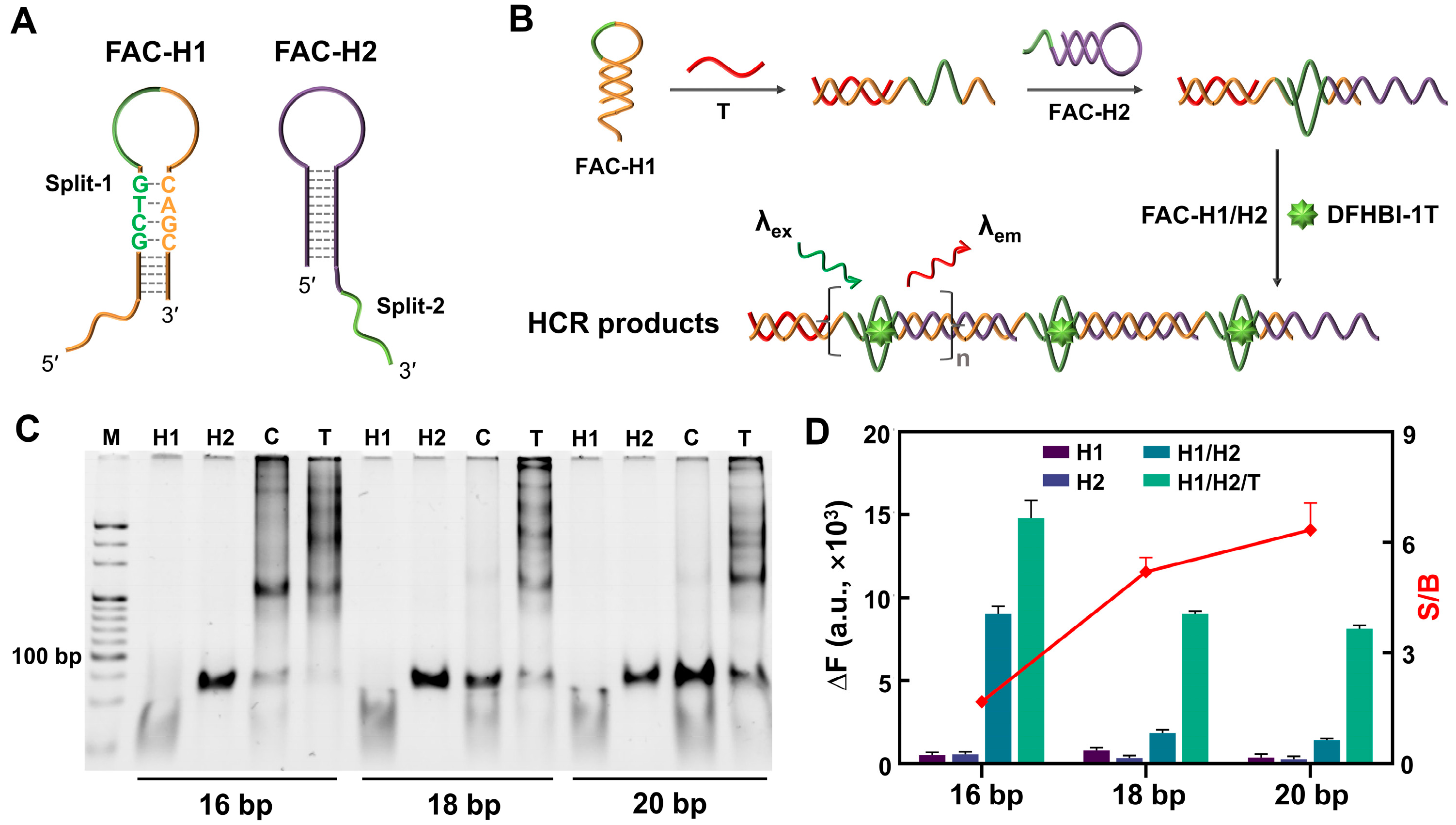
3.3. FA-Centered HCR (FAC-HCR)
3.4. Performance of FAC-HCR
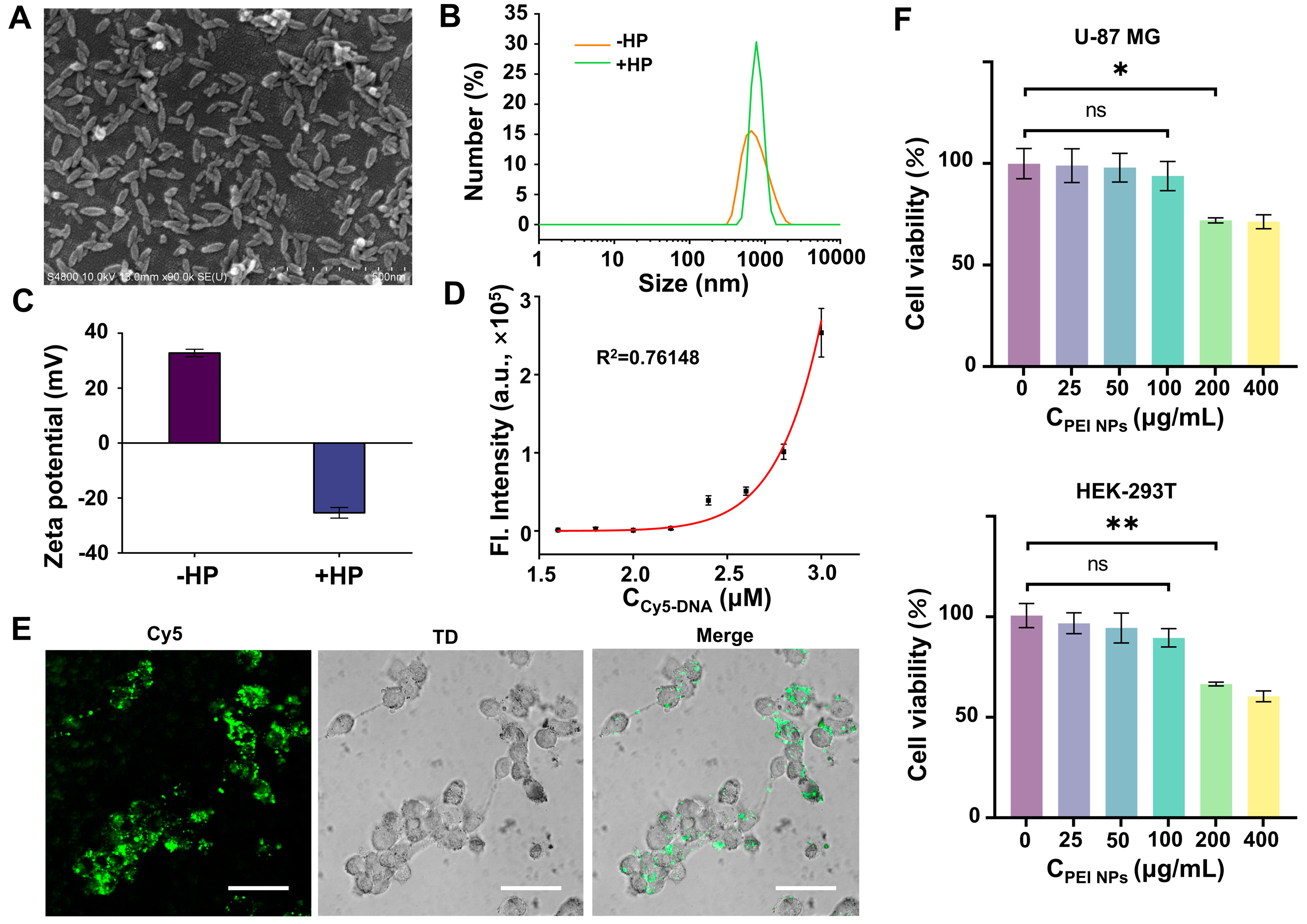
3.5. Delivery of FAC-HCR
3.6. APE1 Imaging
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.; Li, A.Z.; Liu, J. Surfactant-assisted label-free fluorescent aptamer biosensors and binding assays. Biosensors 2023, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- Neubacher, S.; Hennig, S. RNA structure and cellular applications of fluorescent light-up aptamers. Angew. Chem. Int. Ed. 2019, 58, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Paige, J.S.; Wu, K.Y.; Jaffrey, S.R. RNA mimics of green fluorescent protein. Science 2011, 333, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Filonov, G.S.; Moon, J.D.; Svensen, N.; Jaffrey, S.R. Broccoli: Rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014, 136, 16299–16308. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, Y.; Kobayashi, M.; Maruyama, R.; Sato, Y.; Makino, K.; Michiue, T.; Yui, H.; Nishizawa, S.; Yoshimoto, K. Programmable RNA detection with a fluorescent RNA aptamer using optimized three-way junction formation. RNA 2019, 25, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, H.; Litke, J.L.; Wu, J.; Jaffrey, S.R. Fluorophore-promoted RNA folding and photostability enables imaging of single broccoli-tagged mRNAs in live mammalian cells. Angew. Chem. Int. Ed. 2020, 59, 4511–4518. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Wu, R.; Karunanayake Mudiyanselage, A.P.K.K.; Yu, Q.; Zhao, B.; Xie, Y.; Bagheri, Y.; Tian, Q.; You, M. In situ genetically cascaded amplification for imaging RNA subcellular locations. J. Am. Chem. Soc. 2020, 142, 2968–2974. [Google Scholar] [CrossRef]
- VarnBuhler, B.S.; Moon, J.; Dey, S.K.; Wu, J.; Jaffrey, S.R. Detection of SARS-CoV-2 RNA using a DNA aptamer mimic of green fluorescent protein. ACS Chem. Biol. 2022, 17, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, D.; Su, N.; Bao, B.; Xie, X.; Zuo, F.; Yang, L.; Wang, H.; Jiang, L.; Lin, Q.; et al. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 2019, 37, 1287–1293. [Google Scholar] [CrossRef]
- Passalacqua, L.F.M.; Banco, M.T.; Moon, J.D.; Li, X.; Jaffrey, S.R.; Ferré-D’Amaré, A.R. Intricate 3D architecture of a DNA mimic of GFP. Nature 2023, 618, 1078–1084. [Google Scholar] [CrossRef]
- Kolpashchikov, D.M.; Spelkov, A.A. Binary (split) light-up aptameric sensors. Angew. Chem. Int. Ed. 2021, 60, 4988–4999. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Huang, L.-J.; Zhao, W.; Zhang, T.-T.; Cui, M.-R.; Yang, X.-J.; Zhao, X.-L.; Chen, H.-Y.; Xu, J.-J. Living-cell microRNA imaging with self-assembling fragments of fluorescent protein-mimic RNA aptamer. ACS Sens. 2021, 6, 2339–2347. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, P.; Ge, M.; Wang, H.; Cheng, Y.; Li, G.; Huang, Q.; He, H.; Cao, C.; Lin, D.; et al. Elucidation of leak-resistance DNA hybridization chain reaction with universality and extensibility. Nucleic Acids Res. 2020, 48, 2220–2231. [Google Scholar] [CrossRef]
- Chen, Y.; Nagao, R.; Murayama, K.; Asanuma, H. Orthogonal amplification circuits composed of acyclic nucleic acids enable RNA detection. J. Am. Chem. Soc. 2022, 144, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, D.; Cao, X.; Guo, Y. The amplified DNA logic gates based on aptamer-receptor recognition for cell detection and bioimaging. Biosensors 2023, 13, 968. [Google Scholar] [CrossRef] [PubMed]
- Rutten, I.; Daems, D.; Leirs, K.; Lammertyn, J. Highly sensitive multiplex detection of molecular biomarkers using hybridization chain reaction in an encoded particle microfluidic platform. Biosensors 2023, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Marras, S.A.E.; Bushkin, Y.; Tyagi, S. High-fidelity amplified FISH for the detection and allelic discrimination of single mRNA molecules. Proc. Natl. Acad. Sci. USA 2019, 116, 13921–13926. [Google Scholar] [CrossRef]
- Liu, X.; Mao, D.; Song, Y.; Zhu, L.; Isak, A.N.; Lu, C.; Deng, G.; Chen, F.; Sun, F.; Yang, Y.; et al. Computer-aided design of reversible hybridization chain reaction (CAD-HCR) enables multiplexed single-cell spatial proteomics imaging. Sci. Adv. 2022, 8, eabk0133. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, L.; Xie, L.; Shang, J.; He, S.; Liu, J.; Wang, F. Modular assembly of a concatenated DNA circuit for in vivo amplified aptasensing. Small 2022, 18, 2200983. [Google Scholar] [CrossRef]
- Karunanayake Mudiyanselage, A.P.K.K.; Yu, Q.; Leon-Duque, M.A.; Zhao, B.; Wu, R.; You, M. Genetically encoded catalytic hairpin assembly for sensitive RNA imaging in live cells. J. Am. Chem. Soc. 2018, 140, 8739–8745. [Google Scholar] [CrossRef]
- Sun, J.; Jiang, W.; Zhu, J.; Li, W.; Wang, L. Label-free fluorescence dual-amplified detection of adenosine based on exonuclease III-assisted DNA cycling and hybridization chain reaction. Biosens. Bioelectron. 2015, 70, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Genot, A.J.; Zhang, D.Y.; Bath, J.; Turberfield, A.J. Remote toehold: A mechanism for flexible control of DNA hybridization kinetics. J. Am. Chem. Soc. 2011, 133, 2177–2182. [Google Scholar] [CrossRef] [PubMed]
- McNeill, D.R.; Whitaker, A.M.; Stark, W.J.; Illuzzi, J.L.; McKinnon, P.J.; Freudenthal, B.D.; Wilson, D.M., III. Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 2020, 35, 27–38. [Google Scholar] [CrossRef]
- Chaim, I.A.; Nagel, Z.D.; Jordan, J.J.; Mazzucato, P.; Ngo, L.P.; Samson, L.D. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc. Natl. Acad. Sci. USA 2017, 114, E10379–E10388. [Google Scholar] [CrossRef] [PubMed]
- Casper, J.; Schenk, S.H.; Parhizkar, E.; Detampel, P.; Dehshahri, A.; Huwyler, J. Polyethylenimine (PEI) in gene therapy: Current status and clinical applications. J. Control. Release 2023, 362, 667–691. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Wu, C. Progress and perspectives in developing polymeric vectors for in vitro gene delivery. Biomater. Sci. 2013, 1, 152–170. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Tong, H.; Shi, Q.; Fernandes, J.C.; Jin, T.; Dai, K.; Zhang, X. Uptake mechanisms of non-viral gene delivery. J. Control. Release 2012, 158, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.E.; Keswani, R.K.; Pack, D.W. Dependence of PEI and PAMAM gene delivery on clathrin- and caveolin-dependent trafficking pathways. Pharm. Res. 2015, 32, 2051–2059. [Google Scholar] [CrossRef]
- Fan, Z.; Zhao, J.; Chai, X.; Li, L. A cooperatively activatable, DNA-based fluorescent reporter for imaging of correlated enzymatic activities. Angew. Chem. Int. Ed. 2021, 60, 14887–14891. [Google Scholar] [CrossRef]
- Yu, C.; Wang, Y.; Wu, R.; Zhu, Z.; Li, B. Study on the functionalization and signaling efficiency of the hybridization chain reaction using traditional and single molecular characterizations. ACS Appl. Bio Mater. 2021, 4, 3649–3657. [Google Scholar] [CrossRef]
- Yang, C.; Shi, Y.; Zhang, Y.; He, J.; Li, M.; Huang, W.; Yuan, R.; Xu, W. Modular DNA tetrahedron nanomachine-guided dual-responsive hybridization chain reactions for discernible bivariate assay and cell imaging. Anal. Chem. 2023, 95, 10337–10345. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Blank, A.; Berger, M.S.; Stevens, B.A.; Silber, J.R. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin. Cancer Res. 2001, 7, 3510–3518. [Google Scholar] [PubMed]
- Silber, J.R.; Bobola, M.S.; Blank, A.; Schoeler, K.D.; Haroldson, P.D.; Huynh, M.B.; Kolstoe, D.D. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin. Cancer Res. 2002, 8, 3008–3018. [Google Scholar] [PubMed]
- Naidu, M.D.; Mason, J.M.; Pica, R.V.; Fung, H.; Peña, L.A. Radiation resistance in glioma cells determined by DNA damage repair activity of Ape1/Ref-1. J. Radiat. Res. 2010, 51, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, Y.; Li, Z.; Li, L.; Zhao, Y.; Li, M. An enzymatically gated catalytic hairpin assembly delivered by lipid nanoparticles for the tumor-specific activation of signal amplification in miRNA imaging. Angew. Chem. Int. Ed. 2022, 61, e202214230. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Chen, J.; Zeng, S.; Zhu, T.; Chen, J.; Qi, C.; Mao, G.; Liu, Y. Closed cyclic DNA machine for sensitive logic operation and APE1 detection. Small 2023, 19, 2207736. [Google Scholar] [CrossRef] [PubMed]
- Conole, D.; Cao, F.; am Ende, C.W.; Xue, L.; Kantesaria, S.; Kang, D.; Jin, J.; Owen, D.; Lohr, L.; Schenone, M.; et al. Discovery of a potent deubiquitinase (DUB) small-molecule activity-based probe enables broad spectrum DUB activity profiling in living cells. Angew. Chem. Int. Ed. 2023, 62, e202311190. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.W.; Ng, K.K.; Yam, B.H.; Ho, P.L.; Kao, R.Y.; Yang, D. Rapid broad spectrum detection of carbapenemases with a dual fluorogenic-colorimetric probe. J. Am. Chem. Soc. 2021, 143, 6886–6894. [Google Scholar] [CrossRef]
- He, S.; Li, J.; Lyu, Y.; Huang, J.; Pu, K. Near-Infrared fluorescent macromolecular reporters for real-time imaging and urinalysis of cancer immunotherapy. J. Am. Chem. Soc. 2020, 142, 7075–7082. [Google Scholar] [CrossRef]
- Zhao, S.J.; Zheng, P.; Wu, Z.; Jiang, J.H. DNA-templated bioorthogonal reactions via catalytic hairpin assembly for precise RNA imaging in live cells. Anal. Chem. 2022, 94, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zheng, Y.; Chen, D.; Cheng, Y.; Fang, X.; Zhong, C.; Huang, X.; Huang, Q.; Xu, J.; Xu, J.; et al. Branch-shaped trapping device regulates accelerated catalyzed hairpin assembly and its application for microRNA in situ imaging. Anal. Chem. 2023, 95, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, F.; Gan, C.; Yuan, R.; Xiang, Y. Sustainable and cascaded catalytic hairpin assembly for amplified sensing of microRNA biomarkers in living cells. Biosens. Bioelectron. 2022, 197, 113809. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Qu, B.; Ying, Z.M.; Liu, J.W.; Wu, Z.; Yu, R.Q.; Jiang, J.H. Cascade circuits on self-assembled DNA polymers for targeted RNA imaging in vivo. Anal. Chem. 2020, 92, 15953–15958. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-Y.; Yates, M.V.; Mulchandani, A.; Chen, W. Visualizing the dynamics of viral replication in living cells via Tat peptide delivery of nuclease-resistant molecular beacons. Proc. Natl. Acad. Sci. USA 2008, 105, 17522–17525. [Google Scholar] [CrossRef]
- Kim, Y.; Yang, C.J.; Tan, W. Superior structure stability and selectivity of hairpin nucleic acid probes with an l-DNA stem. Nucleic Acids Res. 2007, 35, 7279–7287. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Tan, Y.; Zhou, C.; Fu, Z.; Huang, R.; Li, J.; Li, L. Fluorogenic Aptamer-Based Hybridization Chain Reaction for Signal-Amplified Imaging of Apurinic/Apyrimidinic Endonuclease 1 in Living Cells. Biosensors 2024, 14, 274. https://doi.org/10.3390/bios14060274
Liu M, Tan Y, Zhou C, Fu Z, Huang R, Li J, Li L. Fluorogenic Aptamer-Based Hybridization Chain Reaction for Signal-Amplified Imaging of Apurinic/Apyrimidinic Endonuclease 1 in Living Cells. Biosensors. 2024; 14(6):274. https://doi.org/10.3390/bios14060274
Chicago/Turabian StyleLiu, Meixi, Yunjie Tan, Chen Zhou, Zhaoming Fu, Ru Huang, Jin Li, and Le Li. 2024. "Fluorogenic Aptamer-Based Hybridization Chain Reaction for Signal-Amplified Imaging of Apurinic/Apyrimidinic Endonuclease 1 in Living Cells" Biosensors 14, no. 6: 274. https://doi.org/10.3390/bios14060274
APA StyleLiu, M., Tan, Y., Zhou, C., Fu, Z., Huang, R., Li, J., & Li, L. (2024). Fluorogenic Aptamer-Based Hybridization Chain Reaction for Signal-Amplified Imaging of Apurinic/Apyrimidinic Endonuclease 1 in Living Cells. Biosensors, 14(6), 274. https://doi.org/10.3390/bios14060274