Fiber Optic Localized Surface Plasmon Resonance Sensor Based on Carboxymethylated Dextran Modified Gold Nanoparticles Surface for High Mobility Group Box 1 (HMGB1) Analysis


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
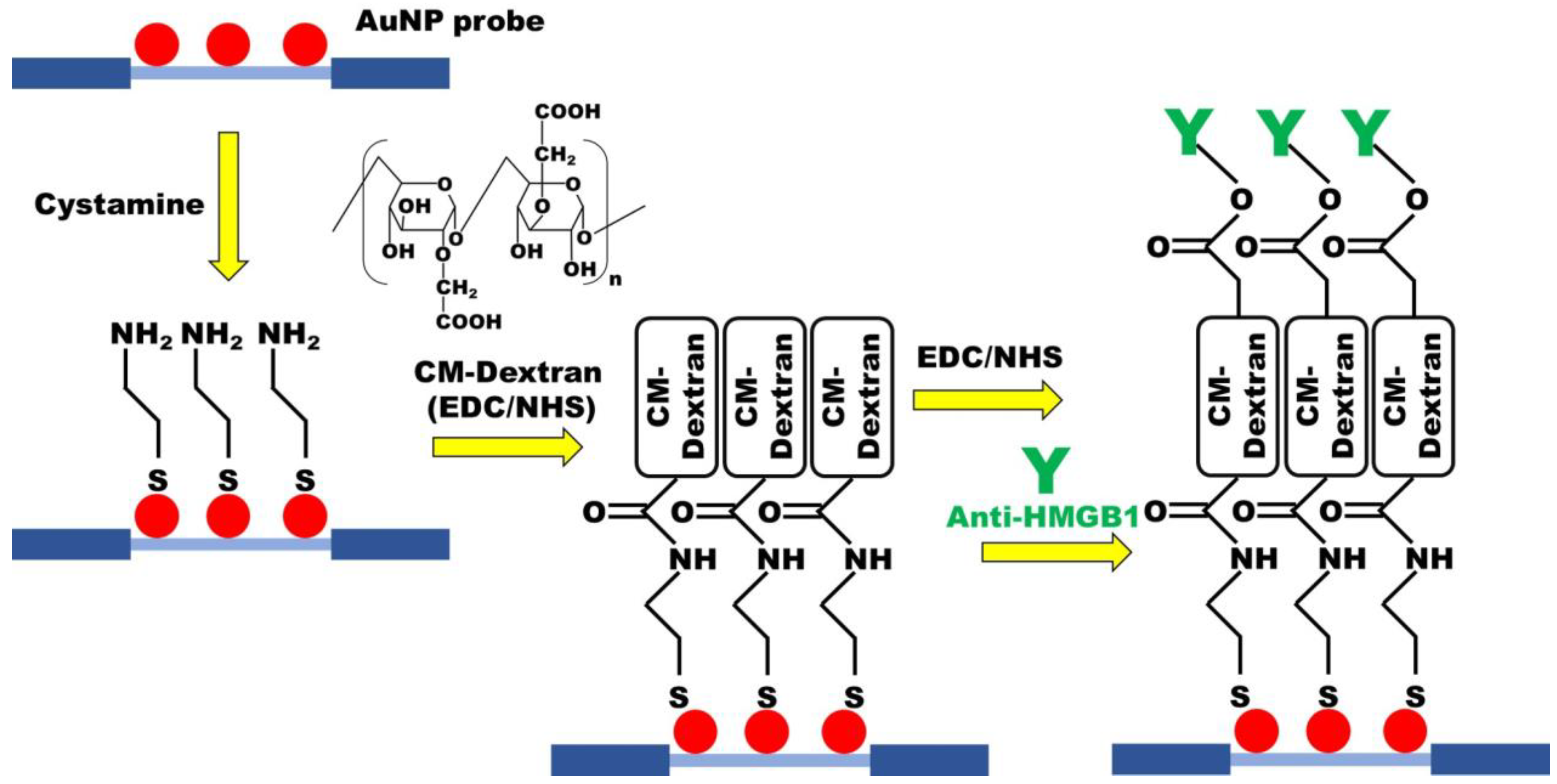
2.2. Preparation of AuNP Probe
2.3. Microfluidic Sensing Chip and FOLSPR Sensing System
2.4. Preparation of CM-Dextran Solution and Functionalization of CM-Dextran Sensing Chips
2.5. Preparation of Functional MUA/MCH Sensing Chip
2.6. Sample Preparation
3. Results
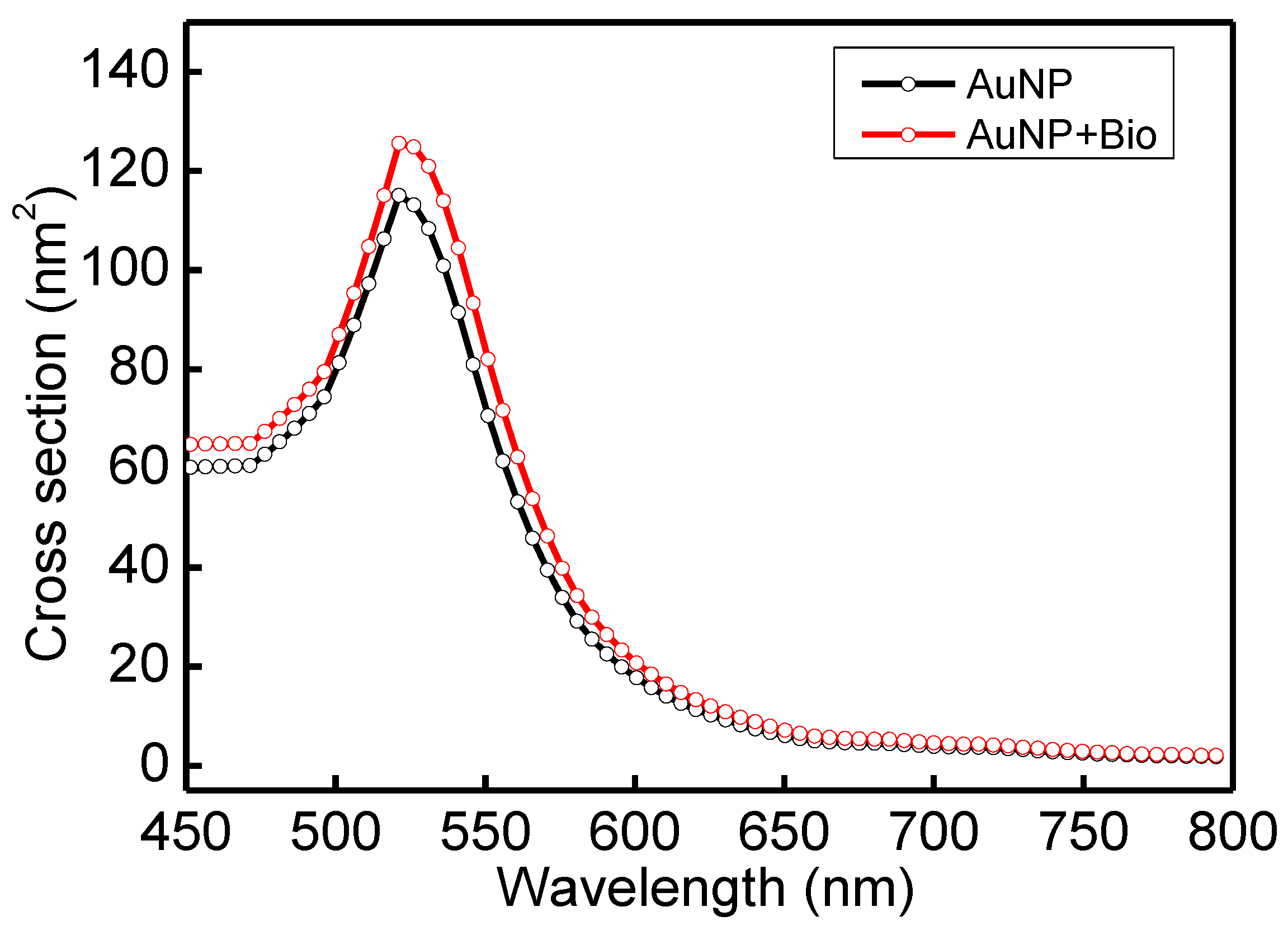
3.1. Material Analysis
3.2. Principle of FOLSPR Sensors
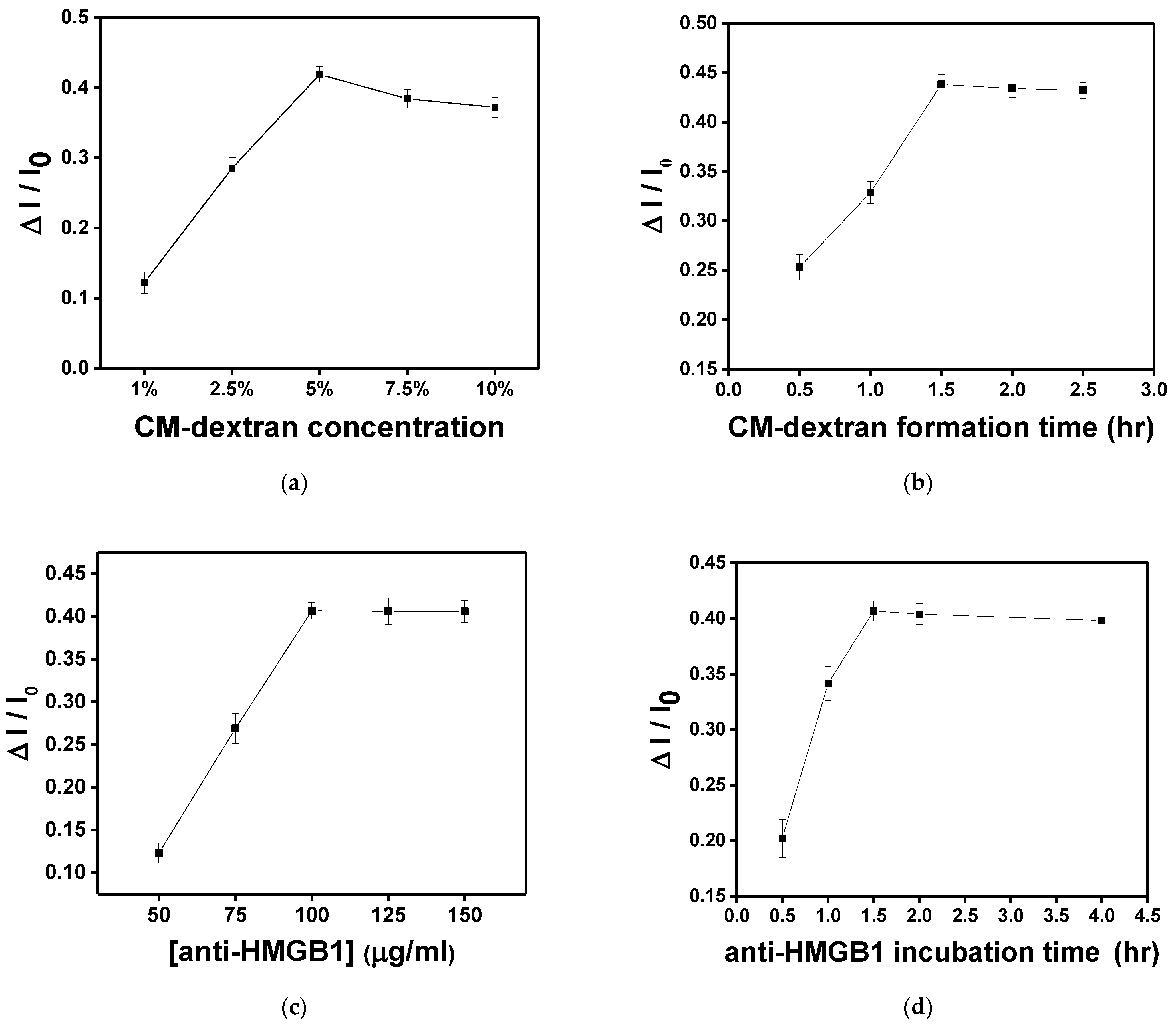
3.3. Optimization of the Sensor
3.4. Comparison of CM-Dextran and MUA/MCH Sensing Chip Modification Anti-HMGB1 Responses
3.5. Nonspecific Adsorption Test
3.6. Sensitivity of the Biosensor
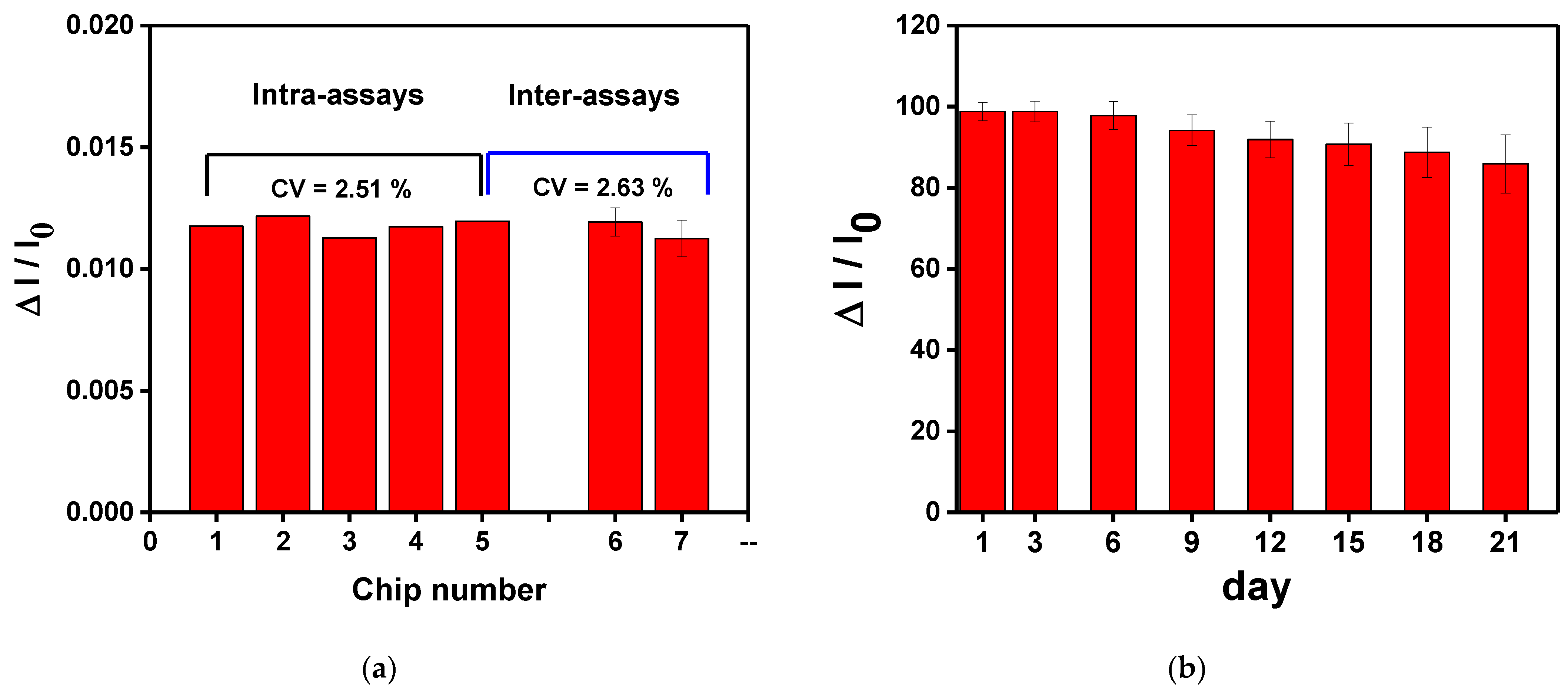
3.7. Sensing Chip Repeatability and Stability Tests
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Aki, H.S.; Fujita, M.; Yamashita, S.; Fujimoto, K.; Kumagai, K.; Tsuruta, R.; Kasaoka, S.; Aoki, T.; Nanba, M.; Murata, H.; et al. Elevation of jugular venous superoxide anion radical is associated with early inflammation, oxidative stress, and endothelial injury in forebrain ischemia–reperfusion rats. Brain Res. 2009, 1292, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Agresti, A.; Bianchi, M.E. HMGB proteins and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of High Mobility Group Box 1 by Dendritic Cells Controls T Cell Activation via the Receptor for Advanced Glycation End Products1. J. Immunol. 2005, 174, 7506–7515. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Czura, C.J.; Tracey, K.J. The cytokine activity of HMGB1. J. Leukoc. Biol. 2005, 78, 1–8. [Google Scholar] [CrossRef]
- Venereau, E.; De Leo, F.; Mezzapelle, R.; Careccia, G.; Musco, G.; Bianchi, M.E. HMGB1 as biomarker and drug target. Pharmacol. Res. 2016, 111, 534–544. [Google Scholar] [CrossRef]
- Rocha, D.C.P.; Souza, T.M.A.; Nunes, P.C.G.; Mohana-Borges, R.; Paes, M.V.; Guimarães, G.M.C.; Arcila, J.C.S.; Paiva, I.A.; Azeredo, E.L.d.; Damasco, P.V.; et al. Increased circulating levels of High Mobility Group Box 1 (HMGB1) in acute-phase Chikungunya virus infection: Potential disease biomarker. J. Clin. Virol. 2022, 146, 105054. [Google Scholar] [CrossRef]
- Sobajima, J.; Ozaki, S.; Uesugi, H.; Osakada, F.; Shirakawa, H.; Yoshida, M.; Nakao, K. Prevalence and characterization of perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA) directed against HMG1 and HMG2 in ulcerative colitis (UC). Clin. Exp. Immunol. 1998, 111, 402–407. [Google Scholar] [CrossRef]
- Urbonaviciute, V.; Fürnrohr, B.G.; Weber, C.; Haslbeck, M.; Wilhelm, S.; Herrmann, M.; Voll, R.E. Factors masking HMGB1 in human serum and plasma. J. Leukoc. Biol. 2007, 81, 67–74. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, L.; Li, J.; Zhu, S.; Yeung, M. Analysis of the Released Nuclear Cytokine HMGB1 in Human Serum. In Cytokine Bioassays: Methods and Protocols; Vancurova, I., Ed.; Springer: New York, NY, USA, 2014; pp. 13–25. [Google Scholar] [CrossRef]
- Arteaga, A.; Ranathunga, D.T.S.; Qu, J.; Biguetti, C.C.; Nielsen, S.O.; Rodrigues, D.C. Exogenous Protein Delivery of Ionic Liquid-Mediated HMGB1 Coating on Titanium Implants. Langmuir 2023, 39, 2204–2217. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Bianchi, M.E.; Coleman, T.R.; Tracey, K.J.; Al-Abed, Y. Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol. Med. 2018, 24, 21. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, Y.; Takakusagi, Y.; Kusayanagi, T.; Kuramochi, K.; Imai, T.; Hirayama, T.; Ito, I.; Yoshida, M.; Sakaguchi, K.; Sugawara, F. Identification and Characterization of the Direct Interaction between Methotrexate (MTX) and High-Mobility Group Box 1 (HMGB1) Protein. PLoS ONE 2013, 8, e63073. [Google Scholar] [CrossRef]
- Gaillard, C.; Borde, C.; Gozlan, J.; Maréchal, V.; Strauss, F. A High-Sensitivity Method for Detection and Measurement of HMGB1 Protein Concentration by High-Affinity Binding to DNA Hemicatenanes. PLoS ONE 2008, 3, e2855. [Google Scholar] [CrossRef] [PubMed]
- Madhurantakam, S.; Muthukumar, S.; Prasad, S. Emerging Electrochemical Biosensing Trends for Rapid Diagnosis of COVID-19 Biomarkers as Point-of-Care Platforms: A Critical Review. ACS Omega 2022, 7, 12467–12473. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.M.; Hafner, J.H. Localized Surface Plasmon Resonance Sensors. Chem. Rev. 2011, 111, 3828–3857. [Google Scholar] [CrossRef]
- Xu, T.; Geng, Z. Strategies to improve performances of LSPR biosensing: Structure, materials, and interface modification. Biosens. Bioelectron. 2021, 174, 112850. [Google Scholar] [CrossRef]
- Behrouzi, K.; Lin, L. Gold nanoparticle based plasmonic sensing for the detection of SARS-CoV-2 nucleocapsid proteins. Biosens. Bioelectron. 2022, 195, 113669. [Google Scholar] [CrossRef]
- Miller, M.M.; Lazarides, A.A. Sensitivity of Metal Nanoparticle Surface Plasmon Resonance to the Dielectric Environment. J. Phys. Chem. B 2005, 109, 21556–21565. [Google Scholar] [CrossRef]
- Meira, D.I.; Proença, M.; Rebelo, R.; Barbosa, A.I.; Rodrigues, M.S.; Borges, J.; Vaz, F.; Reis, R.L.; Correlo, V.M. Chitosan Micro-Membranes with Integrated Gold Nanoparticles as an LSPR-Based Sensing Platform. Biosensors 2022, 12, 951. [Google Scholar] [CrossRef]
- Nath, N.; Chilkoti, A. A Colorimetric Gold Nanoparticle Sensor To Interrogate Biomolecular Interactions in Real Time on a Surface. Anal. Chem. 2002, 74, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Uthaman, S.; Lee, J.; Hwang, H.; Kim, G.; Yoo, P.J.; Hammock, B.D.; Kim, C.S.; Park, Y.-S.; Park, I.-K. In-direct localized surface plasmon resonance (LSPR)-based nanosensors for highly sensitive and rapid detection of cortisol. Sens. Actuators B: Chem. 2018, 266, 710–716. [Google Scholar] [CrossRef]
- Chen, S.; Svedendahl, M.; Van Duyne, R.P.; Käll, M. Plasmon-Enhanced Colorimetric ELISA with Single Molecule Sensitivity. Nano Lett. 2011, 11, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, H.; Zhu, W.; Agrawal, A.; Lezec, H.; Li, L.; Peng, W.; Zou, Y.; Lu, Y.; Xu, T. Subradiant Dipolar Interactions in Plasmonic Nanoring Resonator Array for Integrated Label-Free Biosensing. ACS Sens. 2017, 2, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, X.; Hu, Y.; Chen, P.; Xu, H.; Hu, W.; Zhao, Y.; Wu, P.; Liu, G.L. Dual AuNPs detecting probe enhanced the NanoSPR effect for the high-throughput detection of the cancer microRNA21 biomarker. Biosens. Bioelectron. 2023, 225, 115084. [Google Scholar] [CrossRef]
- Chang, T.-C.; Wu, C.-C.; Wang, S.-C.; Chau, L.-K.; Hsieh, W.-H. Using A Fiber Optic Particle Plasmon Resonance Biosensor To Determine Kinetic Constants of Antigen–Antibody Binding Reaction. Anal. Chem. 2013, 85, 245–250. [Google Scholar] [CrossRef]
- Cheng, S.-F.; Chau, L.-K. Colloidal Gold-Modified Optical Fiber for Chemical and Biochemical Sensing. Anal. Chem. 2003, 75, 16–21. [Google Scholar] [CrossRef]
- Chang, T.-C.; Sun, A.Y.; Huang, Y.-C.; Wang, C.-H.; Wang, S.-C.; Chau, L.-K. Integration of Power-Free and Self-Contained Microfluidic Chip with Fiber Optic Particle Plasmon Resonance Aptasensor for Rapid Detection of SARS-CoV-2 Nucleocapsid Protein. Biosensors 2022, 12, 785. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Hsieh, M.-L.; Huang, K.-W.; Chau, L.-K.; Chang, C.-M.; Lyu, S.-R. Fiber-optic particle plasmon resonance sensor for detection of interleukin-1β in synovial fluids. Biosens. Bioelectron. 2010, 26, 1036–1042. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Ning, W.; Yang, E.; Li, Y.; Luo, Z.; Duan, Y. Sandwich method-based sensitivity enhancement of Ω-shaped fiber optic LSPR for time-flexible bacterial detection. Biosens. Bioelectron. 2022, 201, 113911. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, X.; Li, X.; Gong, P.; Zhang, Y.; Zhao, Y. Recent Advancements of LSPR Fiber-Optic Biosensing: Combination Methods, Structure, and Prospects. Biosensors 2023, 13, 405. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Chiang, C.-Y.; Li, C.-H.; Chang, T.-C.; Chiang, C.-S.; Chau, L.-K.; Huang, K.-W.; Wu, C.-W.; Wang, S.-C.; Lyu, S.-R. Quantification of tumor necrosis factor-α and matrix metalloproteinases-3 in synovial fluid by a fiber-optic particle plasmon resonance sensor. Analyst 2013, 138, 4599–4606. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-W.; Chiang, C.-Y.; Chen, C.-H.; Chiang, C.-S.; Wang, C.-T.; Chau, L.-K. Self-referencing fiber optic particle plasmon resonance sensing system for real-time biological monitoring. Talanta 2016, 146, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Chiang, C.-Y.; Wu, C.-W.; Wang, C.-T.; Chau, L.-K. Integrated Graphene Oxide with Noble Metal Nanoparticles to Develop High-Sensitivity Fiber Optic Particle Plasmon Resonance (FOPPR) Biosensor for Biomolecules Determination. Nanomaterials 2021, 11, 635. [Google Scholar] [CrossRef]
- Ambrosetti, E.; Conti, M.; Teixeira, A.I.; Zilio, S.D. Patterned Carboxymethyl-Dextran Functionalized Surfaces Using Organic Mixed Monolayers for Biosensing Applications. ACS Appl. Bio Mater. 2022, 5, 3310–3319. [Google Scholar] [CrossRef]
- Saftics, A.; Türk, B.; Sulyok, A.; Nagy, N.; Gerecsei, T.; Szekacs, I.; Kurunczi, S.; Horvath, R. Biomimetic Dextran-Based Hydrogel Layers for Cell Micropatterning over Large Areas Using the FluidFM BOT Technology. Langmuir 2019, 35, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Narouz, M.R.; Munro, K.; Hao, B.; Crudden, C.M.; Horton, J.H.; Hao, H. Carboxymethylated Dextran-Modified N-Heterocyclic Carbene Self-Assembled Monolayers on Gold for Use in Surface Plasmon Resonance Biosensing. ACS Appl. Mater. Interfaces 2017, 9, 39223–39234. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lu, Y.; Li, X.; Cao, S.; Pei, Y.; Aastrup, T.; Pei, Z. Optimization of 3D Surfaces of Dextran with Different Molecule Weights for Real-Time Detection of Biomolecular Interactions by a QCM Biosensor. Polymers 2017, 9, 409. [Google Scholar] [CrossRef]
- Huang, C.-F.; Yao, G.-H.; Liang, R.-P.; Qiu, J.-D. Graphene oxide and dextran capped gold nanoparticles based surface plasmon resonance sensor for sensitive detection of concanavalin A. Biosens. Bioelectron. 2013, 50, 305–310. [Google Scholar] [CrossRef]
- Mannelli, I.; Lecerf, L.; Guerrouache, M.; Goossens, M.; Millot, M.-C.; Canva, M. DNA immobilisation procedures for surface plasmon resonance imaging (SPRI) based microarray systems. Biosens. Bioelectron. 2007, 22, 803–809. [Google Scholar] [CrossRef]
- Polzius, R.; Schneider, T.; Biert, F.F.; Bilitewski, U.; Koschinski, W. Optimization of biosensing using grating couplers: Immobilization on tantalum oxide waveguides. Biosens. Bioelectron. 1996, 11, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Akkoyun, A.; Bilitewski, U. Optimisation of glass surfaces for optical immunosensors. Biosens. Bioelectron. 2002, 17, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Tang, M.; Bowyer, A.; Eisenthal, R.; Hubble, J. A novel pH- and ionic-strength-sensitive carboxy methyl dextran hydrogel. Biomaterials 2005, 26, 4677–4683. [Google Scholar] [CrossRef] [PubMed]
- Tabasi, O.; Falamaki, C.; Mahmoudi, M. A Detailed Study on the Fabrication of Surface Plasmon Sensor Chips: Optimization of Dextran Molecular Weight. Plasmonics 2019, 14, 1145–1159. [Google Scholar] [CrossRef]
- Taitt, C.R.; Anderson, G.P.; Lingerfelt, B.M.; Feldstein, M.J.; Ligler, F.S. Nine-Analyte Detection Using an Array-Based Biosensor. Anal. Chem. 2002, 74, 6114–6120. [Google Scholar] [CrossRef]
- Löfås, S.; Johnsson, B. A novel hydrogel matrix on gold surfaces in surface plasmon resonance sensors for fast and efficient covalent immobilization of ligands. J. Chem. Soc. Chem. Commun. 1990, 21, 1526–1528. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Chen, C.-H.; Wang, C.-T. Fiber Optic Particle Plasmon Resonance-Based Immunoassay Using a Novel Multi-Microchannel Biochip. Sensors 2020, 20, 3086. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Huang, C.-H.; Lu, S.-H.; Kuo, I.T.; Chau, L.-K. Direct detection of orchid viruses using nanorod-based fiber optic particle plasmon resonance immunosensor. Biosens. Bioelectron. 2014, 51, 371–378. [Google Scholar] [CrossRef]
- Mulvaney, P. Surface Plasmon Spectroscopy of Nanosized Metal Particles. Langmuir 1996, 12, 788–800. [Google Scholar] [CrossRef]
- Nath, N.; Chilkoti, A. Label Free Colorimetric Biosensing Using Nanoparticles. J. Fluoresc. 2004, 14, 377–389. [Google Scholar] [CrossRef]
- Stewart, M.E.; Anderton, C.R.; Thompson, L.B.; Maria, J.; Gray, S.K.; Rogers, J.A.; Nuzzo, R.G. Nanostructured Plasmonic Sensors. Chem. Rev. 2008, 108, 494–521. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.A.; Rahim Ferhan, A.; Cho, N.-J. Nanoplasmonic sensors for biointerfacial science. Chem. Soc. Rev. 2017, 46, 3615–3660. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, A.; Zhang, L.; Hu, D.; Salmain, M.; Mazouzi, Y.; Flack, R.; Liedberg, B.; Boujday, S. Core–Shell Gold/Silver Nanoparticles for Localized Surface Plasmon Resonance-Based Naked-Eye Toxin Biosensing. ACS Appl. Mater. Interfaces 2019, 11, 46462–46471. [Google Scholar] [CrossRef] [PubMed]
- Ruemmele, J.A.; Hall, W.P.; Ruvuna, L.K.; Van Duyne, R.P. A Localized Surface Plasmon Resonance Imaging Instrument for Multiplexed Biosensing. Anal. Chem. 2013, 85, 4560–4566. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Yan, X.; Du, Y. An aptamer based wall-less LSPR array chip for label-free and high throughput detection of biomolecules. Biosens. Bioelectron. 2014, 53, 58–64. [Google Scholar] [CrossRef]
- Khijwania, S.K.; Gupta, B.D. Fiber optic evanescent field absorption sensor: Effect of fiber parameters and geometry of the probe. Opt. Quantum Electron. 1999, 31, 625–636. [Google Scholar] [CrossRef]
- Sital, S.; Baliyan, A.; Sharma, E.K.; Gupta, M. Optimization of Multimode Fibers for Surface Plasmon Resonance Based Sensors Under Spectral and Single Wavelength Intensity Interrogation. Plasmonics 2022, 17, 665–673. [Google Scholar] [CrossRef]
- Draine, B.T.; Flatau, P.J. Discrete-dipole approximation for periodic targets: Theory and tests. J. Opt. Soc. Am. A 2008, 25, 2693–2703. [Google Scholar] [CrossRef]
- Alterovitz, S.A.; Amirtharaj, P.M.; Apell, P.; Arakawa, E.T.; Ashok, J.; Barth, J.; Bezuidenhout, D.F.; Birch, J.R.; Birken, H.G.; Blessing, C.; et al. List of Contributors. In Handbook of Optical Constants of Solids; Palik, E.D., Ed.; Academic Press: Cambridge, MA, USA, 1998; pp. xv–xviii. [Google Scholar] [CrossRef]
- Štros, M.; Polanská, E.V.; Hlaváčová, T.; Skládal, P. Progress in Assays of HMGB1 Levels in Human Plasma-The Potential Prognostic Value in COVID-19. Biomolecules 2022, 12, 544. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gold Sphere Nanoparticle | Diameter (2r1) | 13.0 nm |
|---|---|---|
| Refractive Index (Nmetal) | Ref [60] | |
| Biomolecular | Radius (r2) | 3.2 nm |
| Refractive index (nB) | 1.45 | |
| Wavelength | 450~800 nm | |
| Cube size | 0.5 nm | |
| Surrounding medium (ns) | 1.42 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, C.-Y.; Chen, C.-H.; Wu, C.-W. Fiber Optic Localized Surface Plasmon Resonance Sensor Based on Carboxymethylated Dextran Modified Gold Nanoparticles Surface for High Mobility Group Box 1 (HMGB1) Analysis. Biosensors 2023, 13, 522. https://doi.org/10.3390/bios13050522
Chiang C-Y, Chen C-H, Wu C-W. Fiber Optic Localized Surface Plasmon Resonance Sensor Based on Carboxymethylated Dextran Modified Gold Nanoparticles Surface for High Mobility Group Box 1 (HMGB1) Analysis. Biosensors. 2023; 13(5):522. https://doi.org/10.3390/bios13050522
Chicago/Turabian StyleChiang, Chang-Yue, Chien-Hsing Chen, and Chin-Wei Wu. 2023. "Fiber Optic Localized Surface Plasmon Resonance Sensor Based on Carboxymethylated Dextran Modified Gold Nanoparticles Surface for High Mobility Group Box 1 (HMGB1) Analysis" Biosensors 13, no. 5: 522. https://doi.org/10.3390/bios13050522
APA StyleChiang, C.-Y., Chen, C.-H., & Wu, C.-W. (2023). Fiber Optic Localized Surface Plasmon Resonance Sensor Based on Carboxymethylated Dextran Modified Gold Nanoparticles Surface for High Mobility Group Box 1 (HMGB1) Analysis. Biosensors, 13(5), 522. https://doi.org/10.3390/bios13050522