A Gene Circuit Combining the Endogenous I-E Type CRISPR-Cas System and a Light Sensor to Produce Poly-β-Hydroxybutyric Acid Efficiently


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Strains and Plasmids
2.2. Characterization of the CcaS-CcaR System
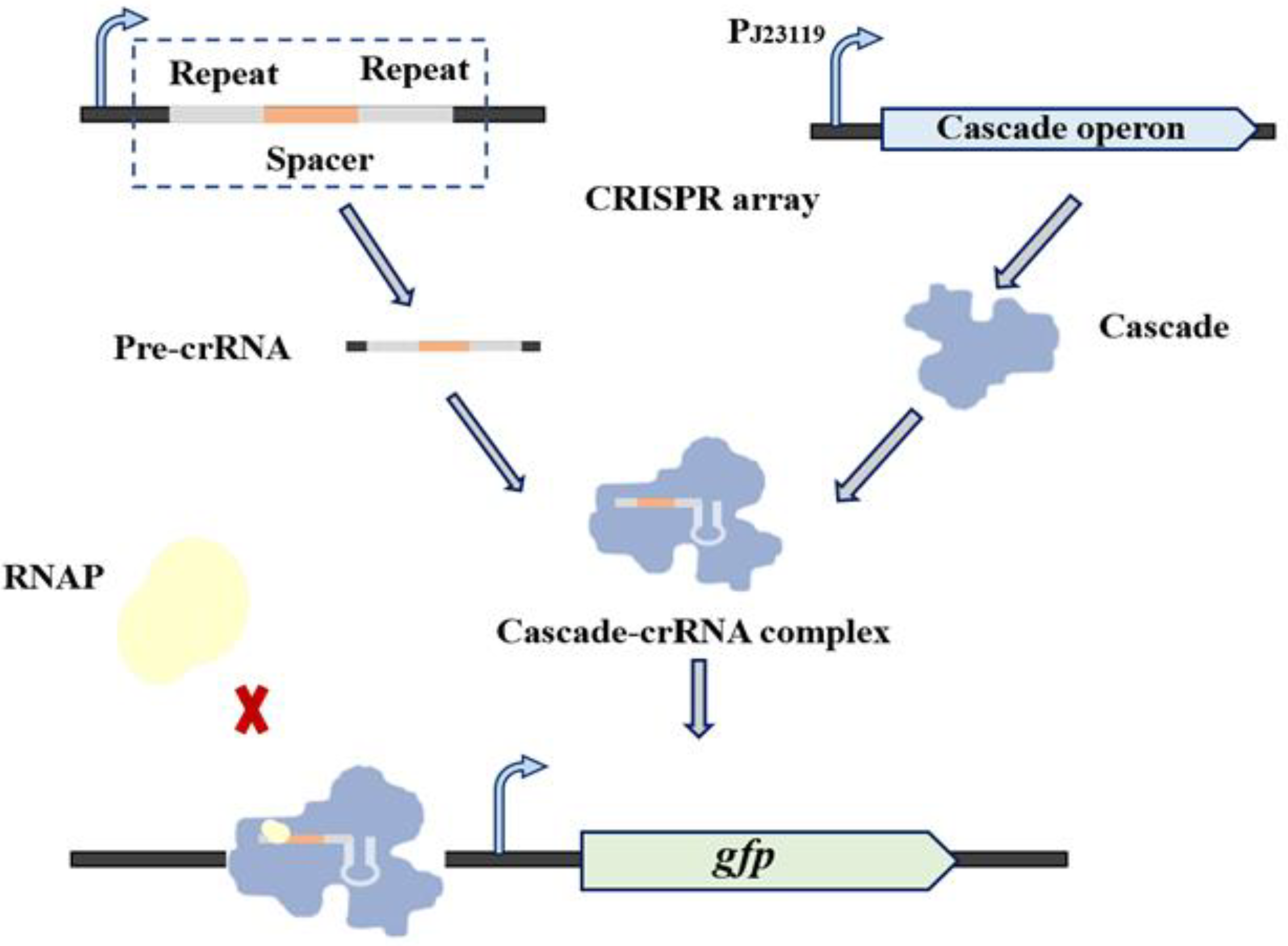
2.3. Characterization of crRNA
2.4. Fermentation of PHB
2.5. Measurement of PHB Production
3. Results and Discussion
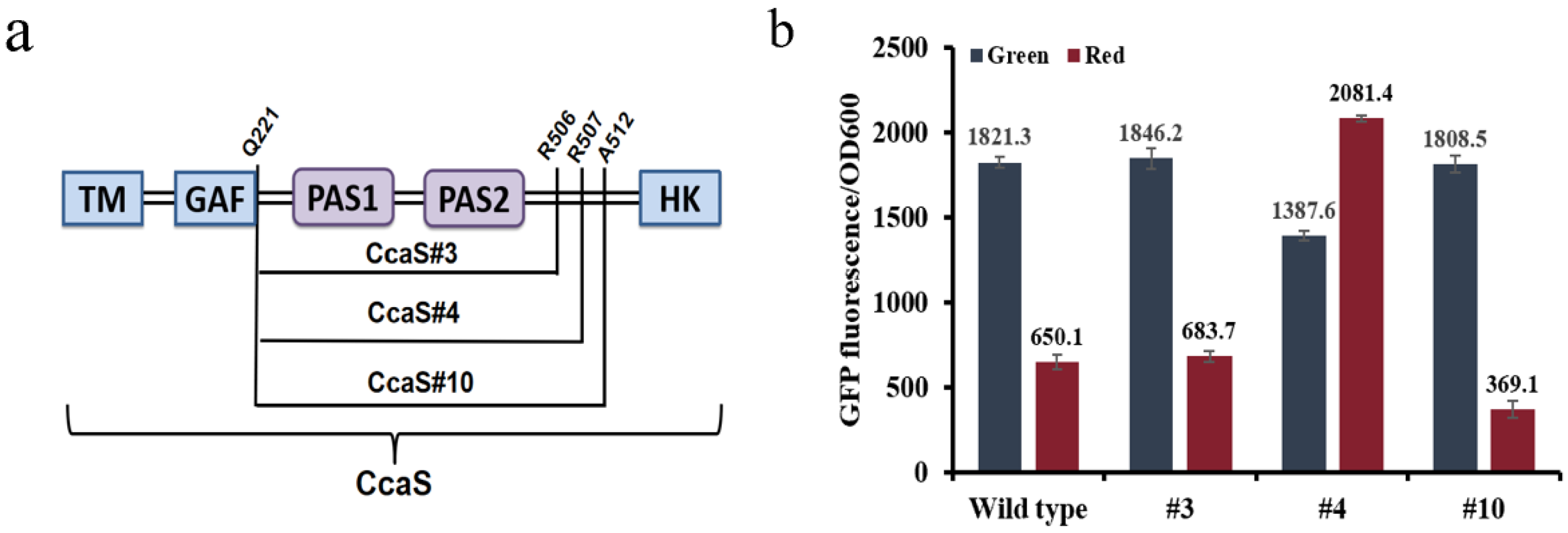
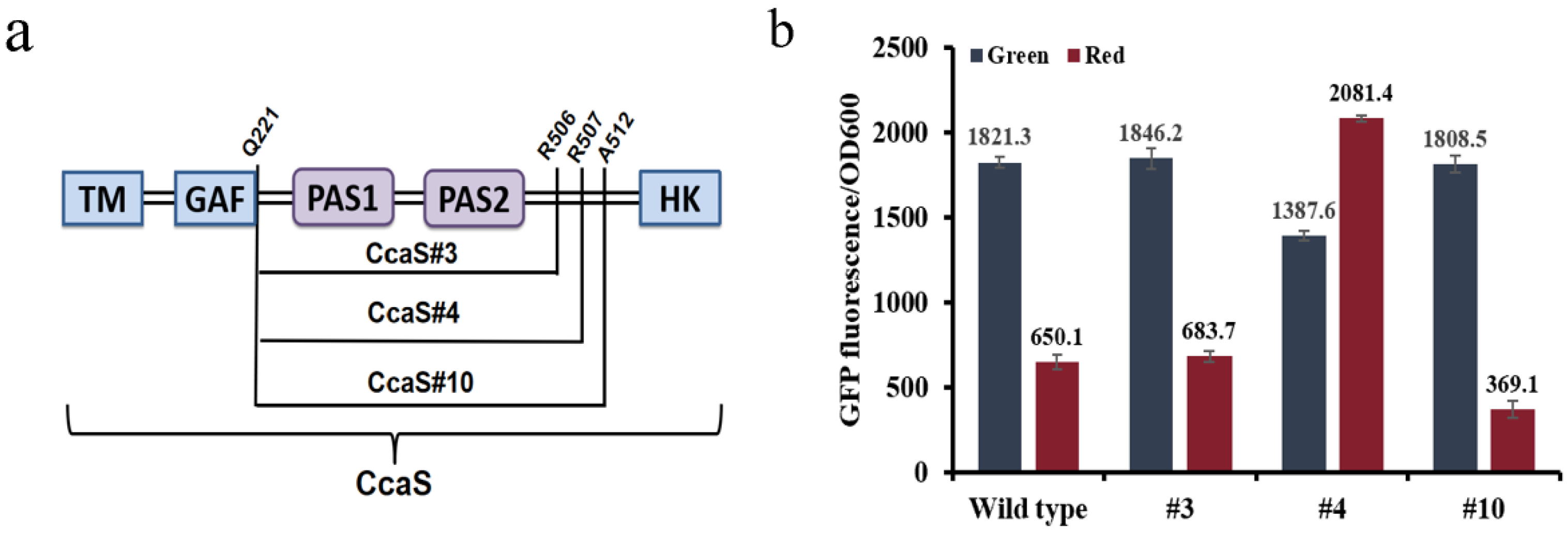
3.1. Optimization of the Optogenetic CcaS/CcaR Sensor
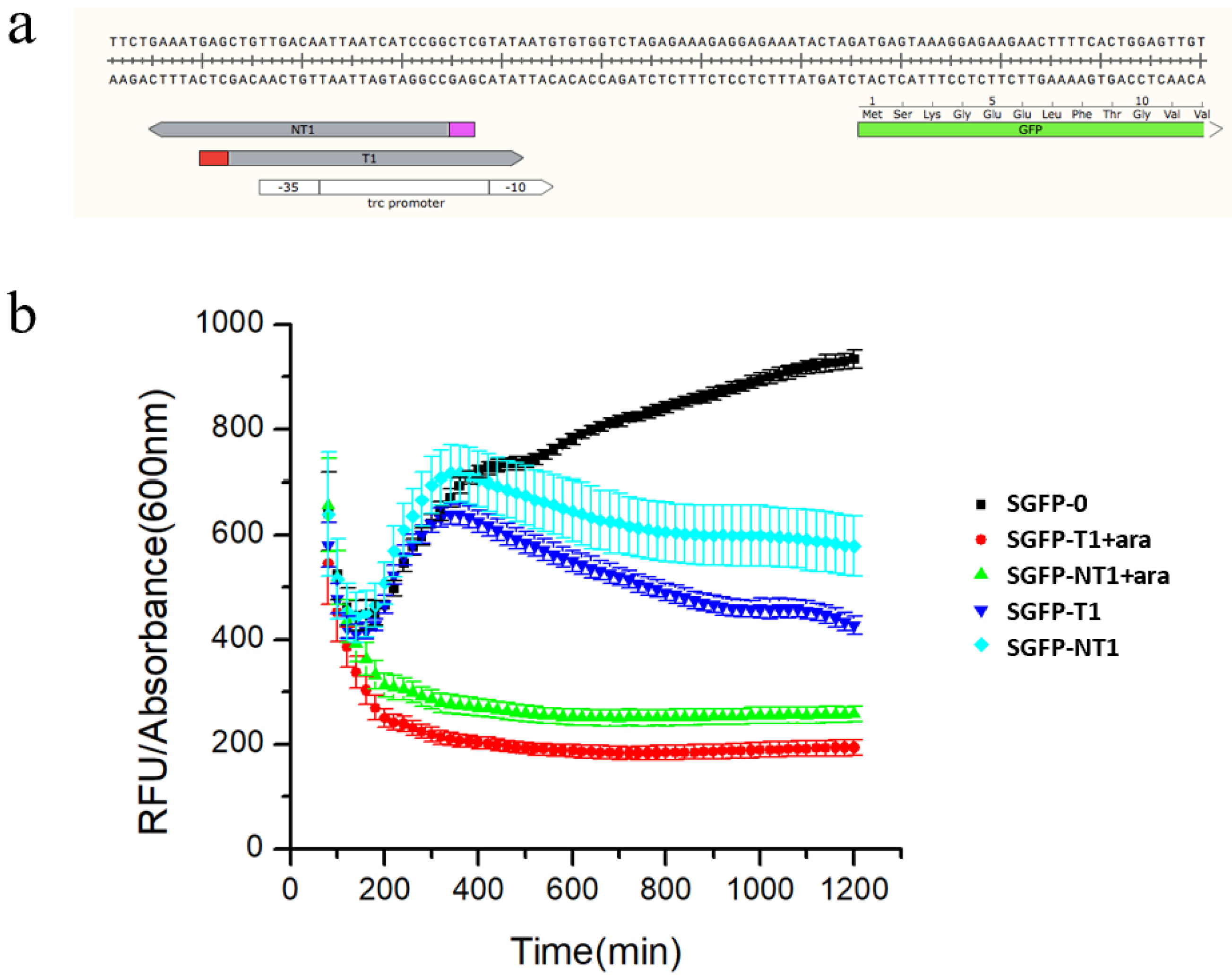
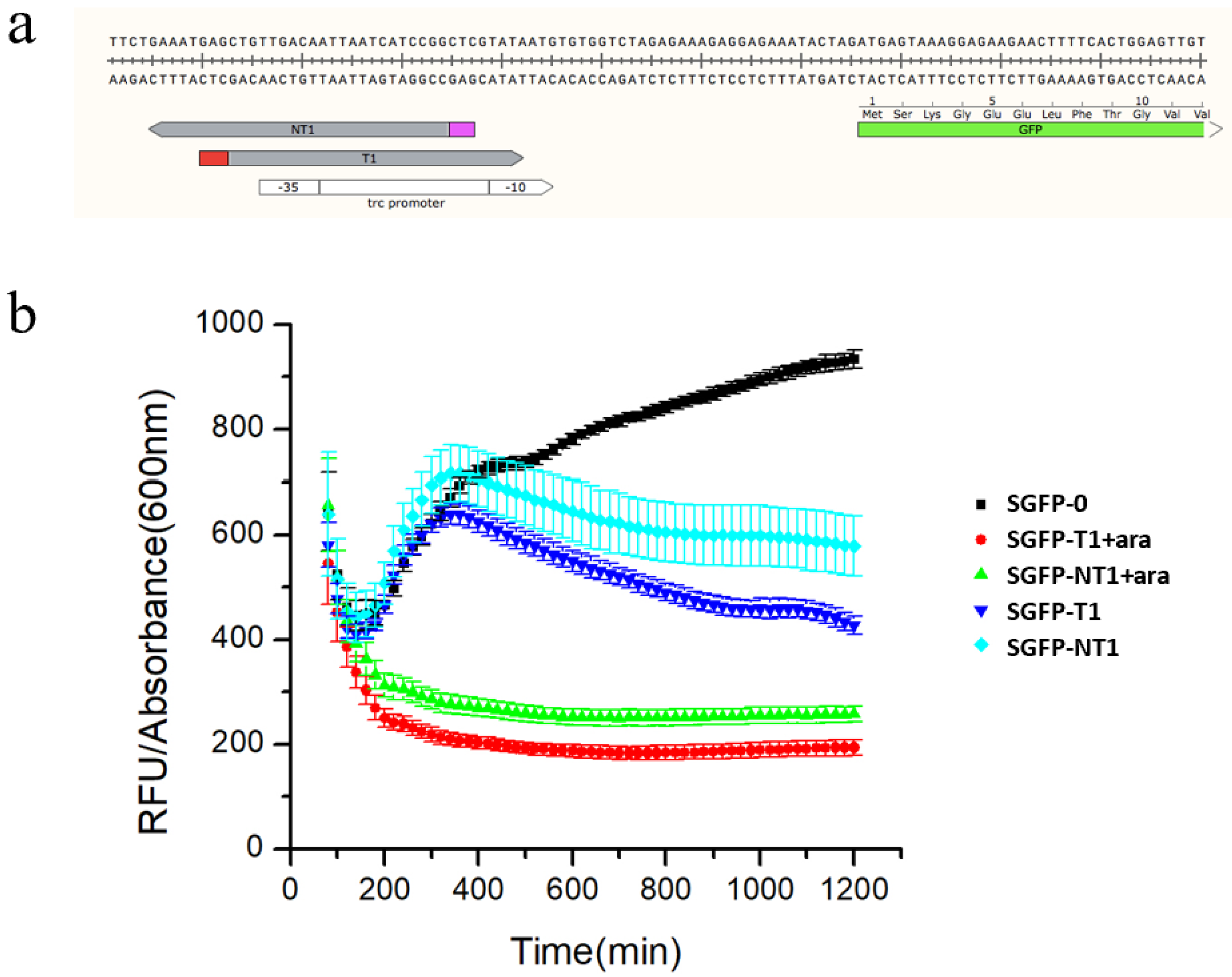
3.2. Characterization of the Type I-E CRISPRi System
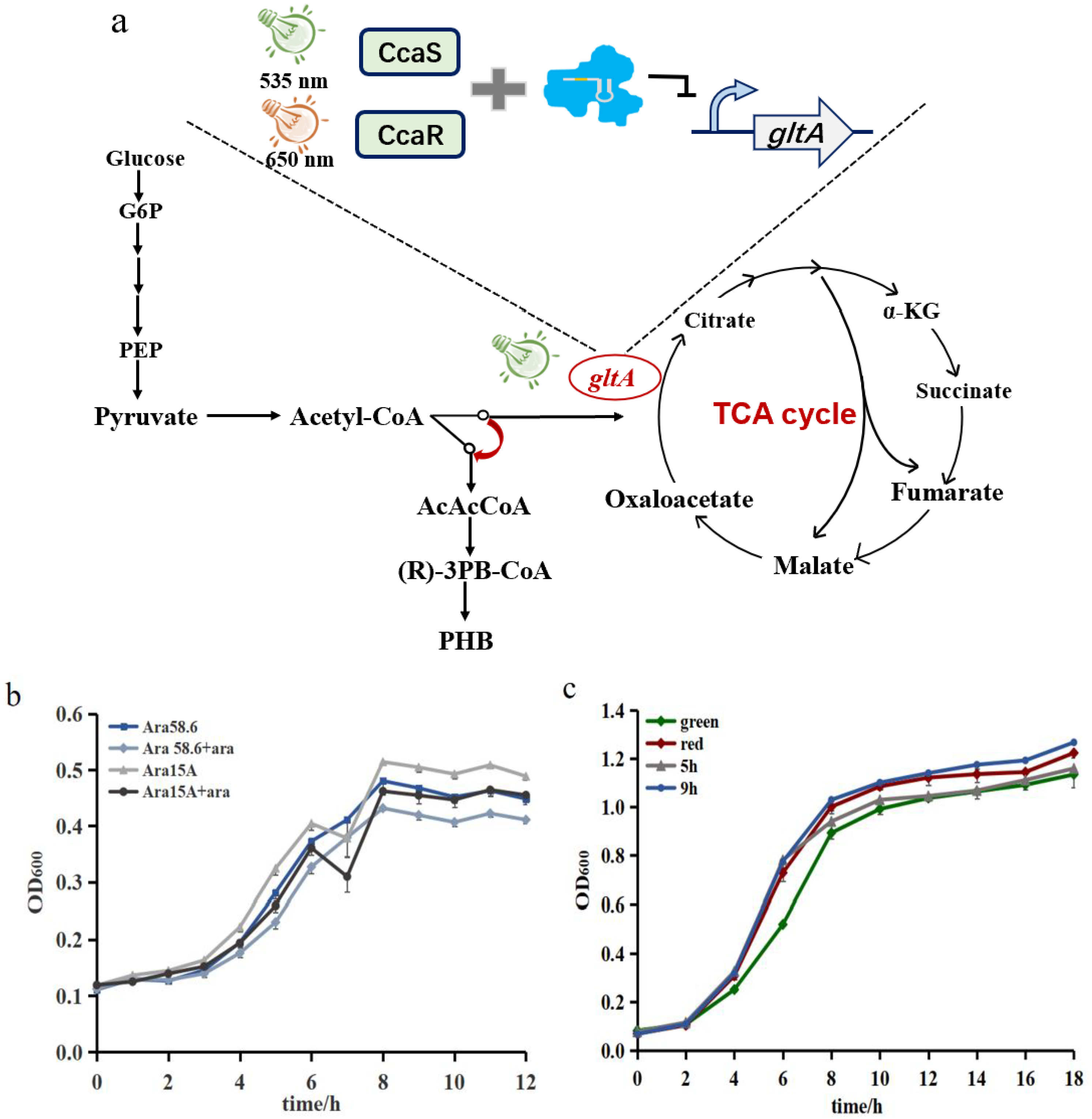
3.3. Construction of the Type-I-E CRISPR System for Regulating gltA Expression
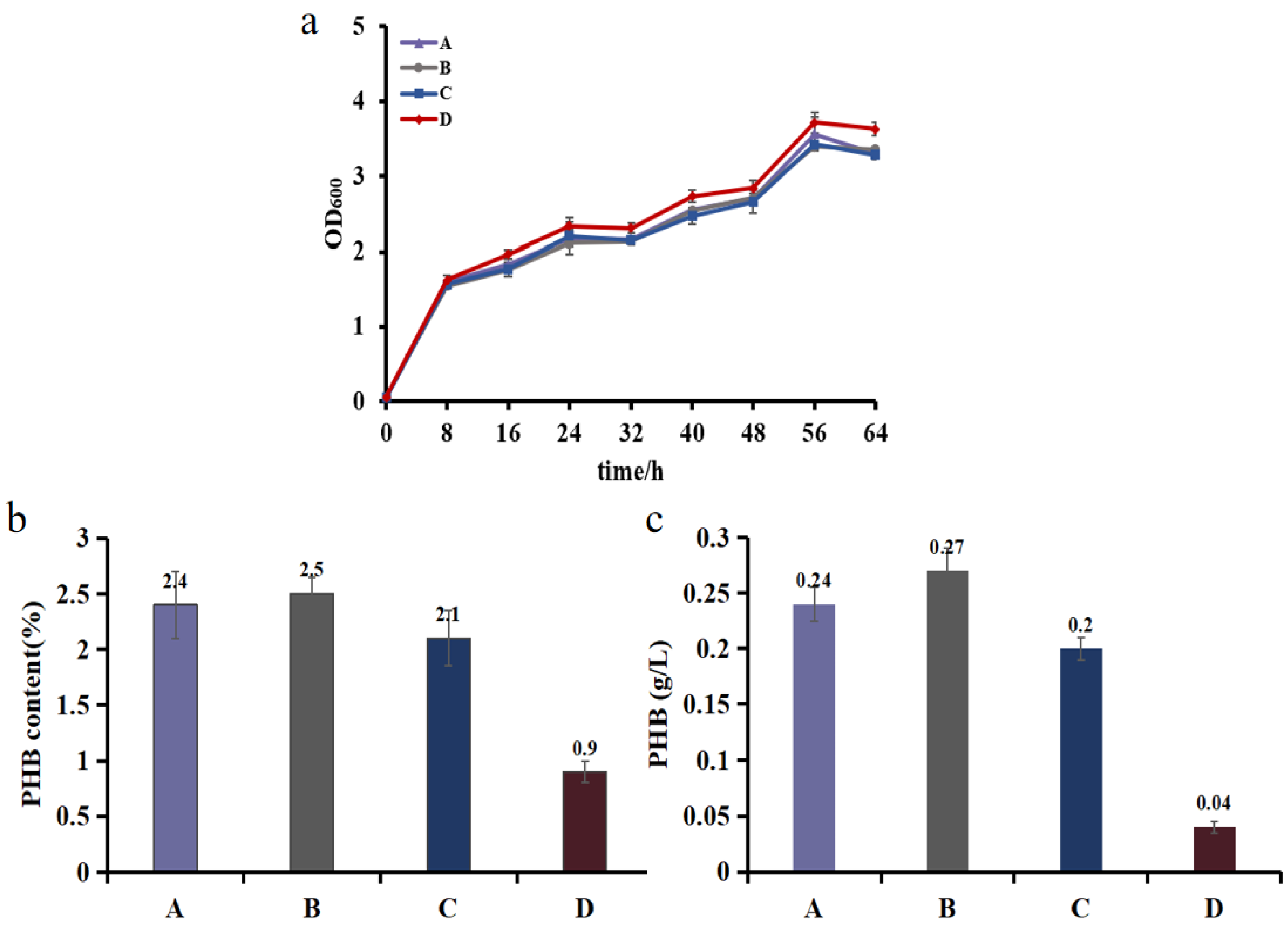
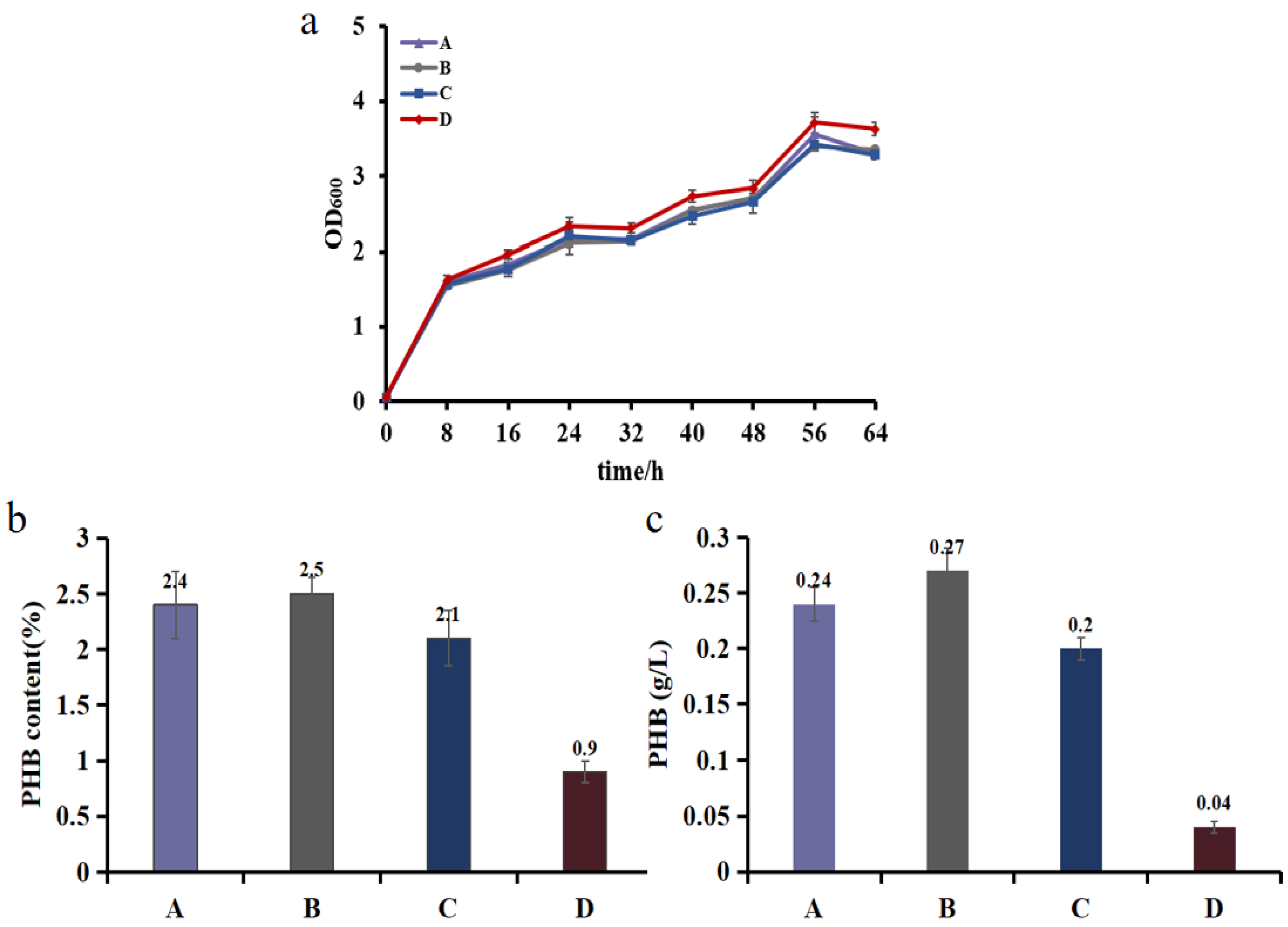
3.4. Demonstrating Light-Controlled Redirection of Metabolic Flux
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, K.R.; Jang, W.D.; Yang, D.; Cho, J.S.; Park, D.; Lee, S.Y. Systems metabolic engineering strategies: Integrating systems and synthetic biology with metabolic engineering. Trends Biotechnol. 2019, 37, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Keasling, J.D. Manufacturing Molecules Through Metabolic Engineering. Science 2010, 330, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch-Deere, C.A.; Oliver, J.W.K.; Rodriguez, G.M.; Atsumi, S. Synthetic Biology and Metabolic Engineering Approaches To Produce Biofuels. Chem. Rev. 2013, 113, 4611–4632. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, M.A.; Ip, S.S.; Carrasco-López, C.; Day, C.; Zhao, E.M.; Kawabe, H.; Avalos, J.L. Optogenetic control of the lac operon for bacterial chemical and protein production. Nat. Chem. Biol. 2020, 17, 71–79. [Google Scholar] [CrossRef]
- Wu, G.; Yan, Q.; Jones, J.A.; Tang, Y.J.; Fong, S.S.; Koffas, M.A. Metabolic Burden: Cornerstones in Synthetic Biology and Metabolic Engineering Applications. Trends Biotechnol. 2016, 34, 652–664. [Google Scholar] [CrossRef]
- Lalwani, M.A.; Zhao, E.M.; Avalos, J.L. Current and future modalities of dynamic control in metabolic engineering. Curr. Opin. Biotechnol. 2018, 52, 56–65. [Google Scholar] [CrossRef]
- Pham, V.D.; Somasundaram, S.; Lee, S.H.; Park, S.J.; Hong, S.H. Redirection of Metabolic Flux into Novel Gamma-Aminobutyric Acid Production Pathway by Introduction of Synthetic Scaffolds Strategy in Escherichia Coli. Appl. Biochem. Biotechnol. 2015, 178, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Soma, Y.; Tsuruno, K.; Wada, M.; Yokota, A.; Hanai, T. Metabolic flux redirection from a central metabolic pathway toward a synthetic pathway using a metabolic toggle switch. Metab. Eng. 2014, 23, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lutz, R.; Bujard, H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997, 25, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.; Ge, X.; Tian, P. High Production of 3-Hydroxypropionic Acid in Klebsiella pneumoniae by Systematic Optimization of Glycerol Metabolism. Sci. Rep. 2016, 6, 26932. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Lee, S.-M.; Um, Y.; Sim, S.J.; Woo, H.M. Development of SyneBrick Vectors As a Synthetic Biology Platform for Gene Expression in Synechococcus elongatus PCC 7942. Front. Plant Sci. 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Nair, A.; Hermiston, T.W. A comparative study examining the cytotoxicity of inducible gene expression system ligands in different cell types. Toxicol. Vitr. 2008, 22, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Pang, W.L.; Ostroff, N.A.; Baumgartner, B.L.; Nayak, S.; Tsimring, L.S.; Hasty, J. Metabolic gene regulation in a dynamically changing environment. Nature 2008, 454, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Sato, S.; Huq, E.; Tepperman, J.M.; Quail, P.H. A light-switchable gene promoter system. Nat. Biotechnol. 2002, 20, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.-M.; Chng, S.; Teo, W.; Cho, H.-S.; Chang, M. A Two-Layer Gene Circuit for Decoupling Cell Growth from Metabolite Production. Cell Syst. 2016, 3, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Rinaldi, N.J.; Robert, F.; Odom, D.T.; Bar-Joseph, Z.; Gerber, G.K.; Hannett, N.M.; Harbison, C.T.; Thompson, C.M.; Simon, I.; et al. Transcriptional Regulatory Networks in Saccharomyces cerevisiae. Science 2002, 298, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-P.; Lei, Q.-Y. Metabolite sensing and signaling in cell metabolism. Signal Transduct. Target. Ther. 2018, 3, 9. [Google Scholar] [CrossRef]
- Meng, W.; Sijin, L.; Huimin, Z. Design and engineering of intracellular-metabolite-sensing/regulation gene circuits in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2016, 113, 206–215. [Google Scholar]
- Liu, D.; Evans, T.; Zhang, F. Applications and advances of metabolite biosensors for metabolic engineering. Metab. Eng. 2015, 31, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-Z.; Yu, H.-B.; Han, M.; Liu, L.-M.; Zhang, W.-G. Metabolic engineering of glucose uptake systems in Corynebacterium glutamicum for improving the efficiency of l-lysine production. J. Ind. Microbiol. Biotechnol. 2019, 46, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Jin, J.; Geng, Z.; Qi, Q.; Liang, Q. Programming Bacteria With Light—Sensors and Applications in Synthetic Biology. Front. Microbiol. 2018, 9, 2692. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.M.; Zhang, Y.; Mehl, J.; Park, H.; Lalwani, M.A.; Toettcher, J.E.; Avalos, J.L. Optogenetic regulation of engineered cellular metabolism for microbial chemical production. Nature 2018, 555, 683–687. [Google Scholar] [CrossRef]
- Toettcher, J.E.; Voigt, C.A.; Weiner, O.; Lim, W.A. The promise of optogenetics in cell biology: Interrogating molecular circuits in space and time. Nat. Methods 2010, 8, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Audrey, O.; Laila, S.; Natalie, K.; Dirk-Jan, S. Site-Directed Mutagenesis to Improve Sensitivity of a Synthetic Two-Component Signaling System. PLoS ONE 2016, 11, e0147494. [Google Scholar]
- Baumschlager, A.; Aoki, S.K.; Khammash, M. Dynamic Blue Light-Inducible T7 RNA Polymerases (Opto-T7RNAPs) for Precise Spatiotemporal Gene Expression Control. ACS Synth. Biol. 2017, 6, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Yuu, H.; Rei, N.; Mitsunori, K.; Masahiko, I. Cyanobacteriochrome CcaS regulates phycoerythrin accumulation in Nostoc punctiforme, a group II chromatic adapter. Proc. Natl. Acad. Sci. USA 2010, 107, 8854–8859. [Google Scholar]
- Burgie, E.S.; Vierstra, R.D. Phytochromes: An Atomic Perspective on Photoactivation and Signaling. Plant Cell 2014, 26, 4568–4583. [Google Scholar] [CrossRef]
- Herrou, J.; Crosson, S. Function, structure and mechanism of bacterial photosensory LOV proteins. Nat. Rev. Micro. 2011, 9, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.S.; Zhang, F.; Bamberg, E.; Nagel, G.; Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 2005, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rodriguez, J.; Moser, F.; Song, M.; Voigt, C.A. Engineering RGB color vision into Escherichia coli. Nat. Chem. Biol. 2017, 13, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, J.; Yin, P.; Ortiz, M.E.; Subsoontorn, P.; Endy, D. Amplifying Genetic Logic Gates. Science 2013, 340, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.M.; Lalwani, M.A.; Chen, J.-M.; Orillac, P.; Toettcher, J.E.; Avalos, J.L. Optogenetic Amplification Circuits for Light-Induced Metabolic Control. ACS Synth. Biol. 2021, 10, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Chen, Y.; Liu, M.; Xiao, G.; Yuan, J. Engineering an Optogenetic CRISPRi Platform for Improved Chemical Production. ACS Synth. Biol. 2020, 10, 125–131. [Google Scholar] [CrossRef]
- Polstein, L.R.; Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Su, T.; Qi, Q.; Liang, Q. Easy regulation of metabolic flux in Escherichia coli using an endogenous type I-E CRISPR-Cas system. Microb. Cell Factories 2016, 15, 195. [Google Scholar] [CrossRef]
- Nakajima, M.; Ferri, S.; Rögner, M.; Sode, K. Construction of a Miniaturized Chromatic Acclimation Sensor from Cyanobacteria with Reversed Response to a Light Signal. Sci. Rep. 2016, 6, 37595. [Google Scholar] [CrossRef]
- Hirose, Y.; Shimada, T.; Narikawa, R.; Katayama, M.; Ikeuchi, M. Cyanobacteriochrome CcaS is the green light receptor that induces the expression of phycobilisome linker protein. Proc. Natl. Acad. Sci. USA 2008, 105, 9528–9533. [Google Scholar] [CrossRef]
- Ong, N.T.X.; Tabor, J.J. A Miniaturized Escherichia coli Green Light Sensor with High Dynamic Range. ChemBioChem 2018, 19, 1255–1258. [Google Scholar] [CrossRef]
- Schmidl, S.R.; Sheth, R.U.; Wu, A.; Tabor, J.J. Refactoring and Optimization of Light-Switchable Escherichia coli Two-Component Systems. ACS Synth. Biol. 2014, 3, 820–831. [Google Scholar] [CrossRef]
- Westra, E.R.; van Erp, P.B.; Künne, T.; Wong, S.P.; Staals, R.H.; Seegers, C.L.; Bollen, S.; Jore, M.M.; Semenova, E.; Severinov, K.; et al. CRISPR Immunity Relies on the Consecutive Binding and Degradation of Negatively Supercoiled Invader DNA by Cascade and Cas3. Mol. Cell 2012, 46, 595–605. [Google Scholar] [CrossRef]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Victor, K.; Rotem, S.; Philip, H. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome biology 2007, 8, 1–7. [Google Scholar]
- Gilvarg, C.; Davis, B.D. The role of the tricarboxylic acid cycle in acetate oxidation in Escherichia coli. J. Biol. Chem. 1956, 222, 307–319. [Google Scholar] [CrossRef]
- Wang, W.; Yu, X.; Wei, Y.; Ledesma-Amaro, R.; Ji, X.-J. Reprogramming the metabolism of Klebsiella pneumoniae for efficient 1,3-propanediol production. Chem. Eng. Sci. 2021, 236, 116539. [Google Scholar] [CrossRef]
- Shao, J.; Wang, M.; Yu, G.; Zhu, S.; Yu, Y.; Heng, B.C.; Wu, J.; Ye, H. Synthetic far-red light-mediated CRISPR-dCas9 device for inducing functional neuronal differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, E6722–E6730. [Google Scholar] [CrossRef]
- Luo, M.L.; Mullis, A.S.; Leenay, R.T.; Beisel, C.L. Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 2014, 43, 674–681. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, H.; Xia, Y.; Kang, Z.; Qi, Q. Complete PHB mobilization in Escherichia coli enhances the stress tolerance: A potential biotechnological application. Microb. Cell Factories 2009, 8, 47. [Google Scholar] [CrossRef]
- Mainguet, S.E.; Gronenberg, L.S.; Wong, S.S.; Liao, J.C. A reverse glyoxylate shunt to build a non-native route from C4 to C2 in Escherichia coli. Metab. Eng. 2013, 19, 116–127. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Jiang, W.; Qi, Q.; Liang, Q. A Gene Circuit Combining the Endogenous I-E Type CRISPR-Cas System and a Light Sensor to Produce Poly-β-Hydroxybutyric Acid Efficiently. Biosensors 2022, 12, 642. https://doi.org/10.3390/bios12080642
Li X, Jiang W, Qi Q, Liang Q. A Gene Circuit Combining the Endogenous I-E Type CRISPR-Cas System and a Light Sensor to Produce Poly-β-Hydroxybutyric Acid Efficiently. Biosensors. 2022; 12(8):642. https://doi.org/10.3390/bios12080642
Chicago/Turabian StyleLi, Xiaomeng, Wei Jiang, Qingsheng Qi, and Quanfeng Liang. 2022. "A Gene Circuit Combining the Endogenous I-E Type CRISPR-Cas System and a Light Sensor to Produce Poly-β-Hydroxybutyric Acid Efficiently" Biosensors 12, no. 8: 642. https://doi.org/10.3390/bios12080642
APA StyleLi, X., Jiang, W., Qi, Q., & Liang, Q. (2022). A Gene Circuit Combining the Endogenous I-E Type CRISPR-Cas System and a Light Sensor to Produce Poly-β-Hydroxybutyric Acid Efficiently. Biosensors, 12(8), 642. https://doi.org/10.3390/bios12080642