Sandwich Hybridization Assay for In Situ Real-Time Cyanobacterial Detection and Monitoring: A Review


Abstract
:1. Introduction
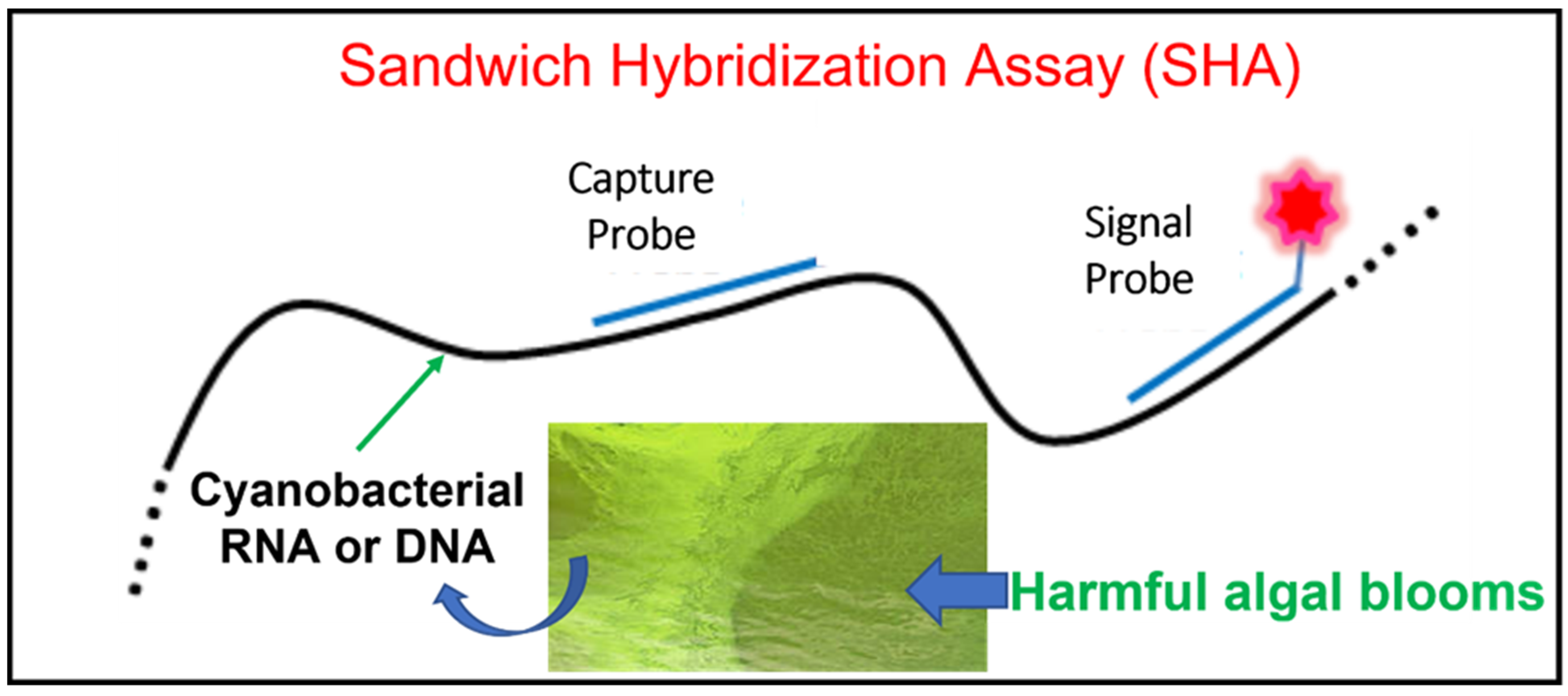
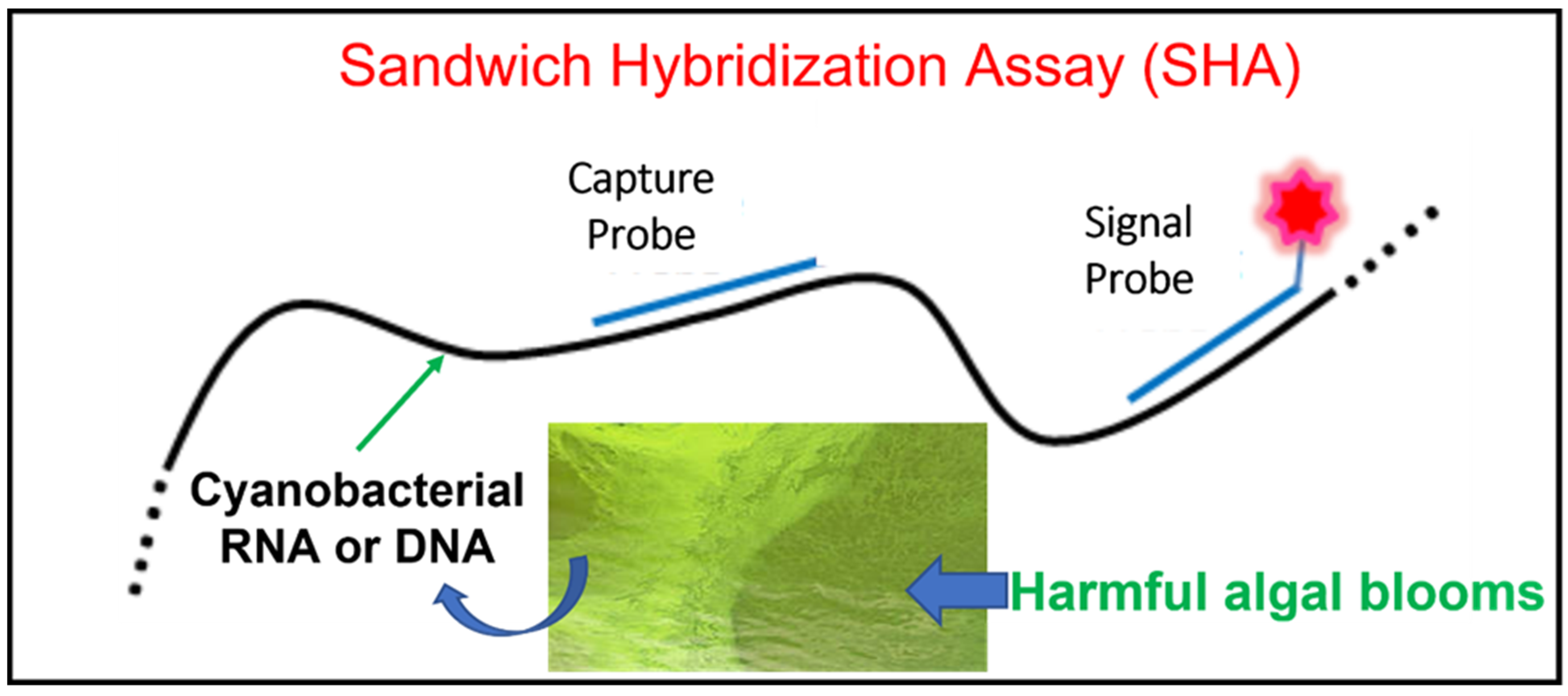
2. Sandwich Hybridization Assay (SHA)
2.1. What Is a SHA?
2.2. SHA Development and Application
2.3. SHA Application to Cyanobacterial Detection and Monitoring
2.4. Advantages of SHA
2.5. Technical Limitations of SHA
3. Future Perspectives
3.1. Does SHA Have an Established Niche in Field Work for cHABs?
3.2. Improving SHA for More Convenient and Broader Applications for In-Situ cHAB Monitoring
3.3. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Capture/signal probes and detection method | |
| AP | alkaline phosphatase |
| BBTP | 2'-(2-benzothiazolyl)-6'-hydroxybenzothiazole phosphate |
| DIG | digoxigenine |
| ELISA | enzyme-linked immunosorbent assay |
| FITC | fluorescein isothiocyanate |
| HRP | horseradish peroxidase |
| PNA | peptide nucleic acid |
| TMB | 3,3’,5,5’-tetra-methylbenzidine |
| Washing and hybridization buffers | |
| BSA | bovine serum albumin |
| DEPC | diethylpyrocarbonate |
| EDTA | ethylenediaminetetraacetic acid |
| GuSCN | guanidine thiocyanate |
| PBS | phosphate buffered saline |
| PVP | polyvinyl pyrrolidone |
| SDS | sodium dodecyl sulfate |
| SSC | saline-sodium citrate |
References
- Merel, S.; Walker, D.; Chicana, R.; Snyder, A.; Baurès, E.; Thomas, O. State of knowledge and concerns on cyanobacterial blooms and cyanotoxins. Environ. Inter. 2013, 59, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Harke, M.J.; Steffen, M.M.; Gobler, C.J.; Otten, T.G.; Wilhelm, S.W.; Wood, S.A.; Paerl, H.W. A review of the global ecology, genomics, and biogeography of the toxic cyanobacterium, Microcystis spp. Harmful Algae 2016, 54, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Hoagland, P.; Kaoru, Y.; White, A.W. Estimated Annual Economic Impacts from Harmful Algal Blooms (HABs) in the United States. Technical Report WHOI-2000-11. 2000. Available online: https://www.whoi.edu/cms/files/Economics_report_18564_23050.pdf (accessed on 20 July 2022).
- Pearson, L.; Dittmann, E.; Mazmouz, R.; Ongley, S.E.; D’Agostino, P.M.; Neilan, B.A. The genetics, biosynthesis and regulation of toxic specialized metabolites of cyanobacteria. Harmful Algae 2016, 54, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zurawell, R.W.; Chen, H.; Burke, J.M.; Prepas, E.E. Hepatotoxic cyanobacteria: A review of the biological importance of microcystins in freshwater environments. Toxicol. Environ. Health Part B 2005, 8, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Pearson, L.; Mihali, T.; Moffitt, M.; Kellmann, R.; Neilan, B. On the chemistry, toxicology and genetics of the cyanobacterial toxins, microcystin, nodularin, saxitoxin and cylindrospermopsin. Mar. Drugs 2010, 8, 1650–1680. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef]
- Méjean, A.; Paci, G.; Gautier, V.; Ploux, O. Biosynthesis of anatoxin-a and analogues (anatoxins) in cyanobacteria. Toxicon 2014, 91, 15–22. [Google Scholar] [CrossRef]
- Beversdorf, L.J.; Chaston, S.D.; Miller, T.R.; McMahon, K.D. Microcystin mcyA and mcyE gene abundances are not appropriate indicators of microcystin concentrations in lakes. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef]
- Almuhtaram, H.; Kibuye, F.A.; Ajjampur, S.; Glover, C.M.; Hofmann, R.; Gaget, V.; Owen, C.; Wert, E.C.; Zamyadi, A. State of knowledge on early warning tools for cyanobacteria detection. Ecol. Indic. 2021, 133, 108442. [Google Scholar] [CrossRef]
- Li, X.; Qiu, D.; Chen, S.; Li, J.; Luo, C.; Hu, D.; Li, J.; Zhu, J.; Chen, H.; Li, S.; et al. Evaluation of RNA degradation in pure culture and field Microcystis samples preserved with various treatments. J. Microbiol. Methods 2019, 164, 105684. [Google Scholar] [CrossRef]
- Rautio, J.; Barken, K.B.; Lahdenperä, J.; Breitenstein, A.; Molin, S.; Neubauer, P. Sandwich hybridisation assay for quantitative detection of yeast RNAs in crude cell lysates. Microb. Cell Fact. 2003, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P.J.; Malcolm, A.D.B. Nucleic acid analysis by sandwich hybridization. J. Clin. Lab. Anal. 1989, 3, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Goffredi, S.K.; Jones, W.J.; Scholin, C.A.; Marin, R., III.; Vrijenhoek, R.C. Molecular detection of marine invertebrate larvae. Mar. Biotechnol. 2006, 8, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.M.; Zhang, L.A.; Manna, A.; Armitage, B.A.; Ly, D.H.; Schneider, J.W. High affinity γPNA sandwich hybridization assay for rapid detection of short nucleic acid targets with single mismatch discrimination. Biomacromolecules 2013, 14, 2253–2261. [Google Scholar] [CrossRef]
- Clancy, E.; Burke, M.; Arabkari, V.; Barry, T.; Kelly, H.; Dwyer, R.M.; Kerin, M.J.; Smith, T.J. Amplification-free detection of microRNAs via a rapid microarray-based sandwich assay. Anal. Bioanal. Chem. 2017, 409, 3497–3505. [Google Scholar] [CrossRef] [PubMed]
- Thieme, D.; Neubauer, P.; Nies, D.H.; Grass, G. Sandwich hybridization assay for sensitive detection of dynamic changes in mRNA transcript levels in crude Escherichia coli cell extracts in response to copper ions. Appl. Environ. Microbiol. 2008, 74, 7463–7470. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.R.; Hassell, J.A. A novel method to map transcripts: Evidence for homology between an adenovirus mRNA and discrete multiple regions of the viral genome. Cell 1977, 12, 23–36. [Google Scholar] [CrossRef]
- Ishii, J.K.; Ghosh, S. Non-Isotopic Detection of Nucleic Acids Using A Polystyrene Support-Based Sandwich Hybridization Assay and Compositions Useful Therefor. U.S. Patent No. 5474895, 12 December 1995. [Google Scholar]
- Tyagi, S.; Kramer, F.R.; Lizardi, P.M.; Landegren, U.D.; Blok, H.J. Sensitive Nucleic Acid Sandwich Hybridization Assay. U.S. Patent No. 5759773, 2 June 1998. [Google Scholar]
- Zammatteo, N.; Alexandre, I.; Ernest, I.; Le, L.; Brancart, F.; Remacle, J. Comparison between microwell and bead supports for the detection of human cytomegalovirus amplicons by sandwich hybridization. Anal. Biochem. 1997, 253, 180–189. [Google Scholar] [CrossRef]
- Huhtamella, S.; Leinonen, M.; Nieminen, T.; Fahnert, B.; Myllykoski, L.; Breitenstein, A.; Neubauer, P. RNA-based sandwich hybridisation method for detection of lactic acid bacteria in brewery samples. J. Microbiol. Methods 2007, 68, 543–553. [Google Scholar] [CrossRef]
- Namimatsu, T.; Tsuna, M.; Imai, Y.; Futo, S.; Mitsuse, S.; Sakano, T.; Sato, S. Detection of Salmonella by using the colorimetric DNA/rRNA sandwich hybridization in microtiter wells. J. Vet. Med. Sci. 2000, 62, 615–619. [Google Scholar] [CrossRef]
- Leskelä, T.; Tilsala-Timisjärvi, A.; Kusnetsov, J.; Neubauer, P.; Breitenstein, A. Sensitive genus-specific detection of Legionella by a 16S rRNA based sandwich hybridization assay. J. Microbiol. Methods 2005, 62, 167–179. [Google Scholar] [CrossRef]
- Zhang, N.; Appella, D.H. Colorimetric detection of anthrax DNA with a peptide nucleic acid sandwich-hybridization assay. J. Am. Chem. Soc. 2007, 129, 8424–8425. [Google Scholar] [CrossRef] [PubMed]
- Feuillie, C.; Merheb, M.M.; Gillet, B.; Montagnac, G.; Daniel, I.; Hänni, C. A novel SERRS sandwich-hybridization assay to detect specific DNA target. PLoS ONE 2011, 6, e17847. [Google Scholar] [CrossRef] [PubMed]
- Scholin, C.A.; Buck, K.R.; Britschgi, T.; Cangelosi, G.; Chavez, E.P. Identification of Pseudo-nitzschia australis (Bacillariophyceae) using rRNA-targeted probes in whole cell and sandwich hybridization formats. Phycologia 1996, 35, 190–197. [Google Scholar] [CrossRef]
- Scholin, C.; Miller, P.; Buck, K.; Chavez, F.; Harris, P.; Haydock, P.; Howard, J.; Cangelosi, G. Detection and quantification of Pseudo-nitzschia australis in cultured and natural populations using LSU rRNA-targeted probes. Limnol. Oceanogr. Methods 1997, 42, 1265–1272. [Google Scholar] [CrossRef]
- Greenfield, D.I.; Marin, R., III.; Jensen, S.; Massion, E.; Roman, B.; Feldman, J.; Scholin, C.A. Application of environmental sample processor (ESP) methodology for quantifying Pseudo-nitzschia australis using ribosomal RNA-targeted probes in sandwich and fluorescent in situ hybridization formats. Limnol. Oceanogr. Methods 2006, 4, 426–435. [Google Scholar] [CrossRef]
- Greenfield, D.I.; Marin, R., III.; Doucette, G.J.; Mikulski, C.; Jones, K.; Jensen, S.; Roman, B.; Alvarado, N.; Feldman, J.; Scholin, C. Field applications of the second-generation Environmental Sample Processor (ESP) for remote detection of harmful algae: 2006-2007. Limnol. Oceanogr. Methods 2008, 6, 667–679. [Google Scholar] [CrossRef]
- Doucette, G.J.; Mikulski, C.M.; Jones, K.L.; King, K.L.; Greenfield, D.I.; Marin, R.; Jensen, S.; Roman, B.; Elliott, C.T.; Scholin, C.A. Remote, subsurface detection of the algal toxin domoic acid onboard the Environmental Sample Processor: Assay development and field trials. Harmful Algae 2009, 8, 880–888. [Google Scholar] [CrossRef]
- Tyrrell, J.V.; Bergquist, P.R.; Bergquist, P.R.; Scholin, C.A. Detection and enumeration of Heterosigma akashiwo and Fibrocapsa japonica (Raphidophayceae) using rRNA-targeted oligonucleotide probes. Phycologia 2001, 40, 457–467. [Google Scholar] [CrossRef]
- Doll, C.; Main, C.R.; Bianco, C.; Coyne, K.J.; Greenfield, D.I. Comparison of sandwich hybridization assay and quantitative PCR for the quantification of live and preserved cultures of Heterosigma akashiwo (Raphidophyceae). Limnol. Oceanogr. Methods 2014, 12, 232–245. [Google Scholar] [CrossRef]
- Haywood, A.J.; Scholin, C.A.; Marin, R., III.; Steidinger, K.A.; Heil, C.; Ray, J. Molecular detection of the brevetoxin-producing dinoflagellate Karenia brevis and closely related species using rRNA-targeted probes and a semiautomated sandwich hybridization assay. J. Phycol. 2007, 43, 1271–1286. [Google Scholar] [CrossRef]
- Anderson, D.M.; Kulis, D.M.; Keafer, B.A.; Gribble, K.E.; Marin, R.; Scholin, C.A. Identification and enumeration of Alexandrium spp. from the Gulf of Maine using molecular probes. Deep-Sea Res. II 2005, 52, 2467–2490. [Google Scholar] [CrossRef]
- Mikulski, C.M.; Park, Y.T.; Jones, K.L.; Lee, C.K.; Lim, W.A.; Lee, Y.; Scholin, C.A.; Doucette, G.J. Development and field application of rRNA-targeted probes for the detection of Cochlodinium polykrikoides Margalef in Korean coastal waters using whole cell and sandwich hybridization formats. Harmful Algae 2008, 7, 347–359. [Google Scholar] [CrossRef]
- Hull, R.; Al-Hakim, A. Nucleic acid hybridization in plant virus diagnosis and characterization. Trends Biotechnol. 1988, 6, 213–218. [Google Scholar] [CrossRef]
- Barken, K.B.; Gabig-Ciminska, M.; Holmgren, A.; Molin, S. Effect of unlabeled helper probes on detection of an RNA target by bead-based sandwich hybridization. Bio.Tech. 2004, 36, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Brandt, O.; Hoheisel, J.D. Peptide nucleic acids on microarrays and other biosensors. Trends Biotechnol. 2004, 22, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Van Ness, J.; Chen, L. The use of oligodeoxynucleotide probes in chaotrope-based hybridization solutions. Nucleic Acids Res. 1991, 19, 5143–5151. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Marin, R., III.; Jensen, S.D.; Feldman, J.; Birch, J.M.; Massion, E.I.; Delong, E.F.; Suzuki, M.; Wheeler, K.; Scholin, C.A. Near real-time, autonomous detection of marine bacterioplankton on a coastal mooring in Monterey Bay, California, using rRNA-targeted DNA probes. Environ. Microbiol. 2009, 11, 1168–1180. [Google Scholar] [CrossRef]
- Scholin, C.A.; Birch, J.; Jensen, S.; Marin, R., III.; Massion, E.; Pargett, D.; Preston, C.; Roman, B.; Ussler, W., III. The quest to develop ecogenomic sensors: A 25-Year history of the Environmental Sample Processor (ESP) as a case study. Oceanography 2017, 30, 100–113. [Google Scholar] [CrossRef]
- Matsunaga, T.; Takeyama, H.; Nakayama, H. 16S rRNA-targeted identification of cyanobacterial genera using oligonucleotide-proves immobilized on bacterial magnetic particles. J. Appl. Phycol. 2001, 13, 389–394. [Google Scholar] [CrossRef]
- Matsunaga, T.; Nakayama, H.; Okochi, M.; Takeyama, H. Fluorescent detection of cyanobacterial DNA using bacterial magnetic particles on a MAG-microarray. Biotechnol. Bioeng. 2001, 73, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Nubel, U.; Garcia-Pichel, F.; Muyzer, G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Appl. Environ. Microbiol. 1997, 63, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, B.; Rizzi, E.; Frosini, A.; Sivonen, K.; Rajaniemi, P.; Rantala, A.; Mugnai, M.A.; Ventura, S.; Wilmotte, A.; Boutte, C.; et al. Development of a universal microarray based on the ligation detection reaction and 16S rRNA gene polymorphism to target diversity of cyanobacteria. Appl. Environ. Microbiol. 2004, 70, 7161–7172. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.P.; Li, X.; Du, S. Identification and enumeration of Microcystis using a sandwich hybridization assay. J. Microbiol. 2012, 50, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.P.; Du, S.; Li, X. Detection of a microcystin-producing Microcystis in Guanqiao Lake using a sandwich hybridization assay. Can. J. Microbiol. 2012, 58, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Piccin-Santos, V.; Brandão, M.M.; Bittencourt-Oliveira, M.D.C. Phylogenetic study of Geitlerinema and Microcystis (Cyanobacteria) using PC-IGS and 16S-23S ITS as markers: Investigation of horizontal gene transfer. J. Phycol. 2014, 50, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Dearth, N.M. Development of A Sandwich Hybridization Assay for the Harmful Cyanobacteria Microcystis spp. Master’s Thesis, University of South Carolina, Columbia, SC, USA, 2017. Available online: https://scholarcommons.sc.edu/etd/4484 (accessed on 20 July 2022).
- Dearth, N.M.; Jones, W.J.; Espinosa, J.I.; Mortensen, R.A.; Pinckney, J.L.; Greenfield, D.I. A new sandwich hybridization assay method to identify and quantify Microcystis spp. Limnol. Oceanogr. Methods 2022, 20, 210–221. [Google Scholar] [CrossRef]
- Daims, H.; Brühl, A.; Amann, R.; Schleifer, K.H.; Wagner, M. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: Development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 1999, 22, 434–444. [Google Scholar] [CrossRef]
- Marin, R., III.; Scholin, C.A. Toxic algal detection using rRNA-targeted probes in a semi-automated sandwich hybridization format. In Microscopic and Molecular Methods for Quantitative Phytoplankton Analysis; Karlson, B., Cusack, C., Bresnan, E., Eds.; UNESCO: Paris, France, 2010; pp. 87–94. [Google Scholar]
- Scholin, C.; Doucette, G.; Jensen, S.; Roman, B.; Pargett, D.; Marin, R., III.; Preston, C.; Jones, W.; Feldman, J.; Everlove, C.; et al. Remote detection of marine microbes, small invertebrates, harmful algae, and biotoxins using the Environmental Sample Processor (ESP). Oceanography 2009, 22, 158–167. [Google Scholar] [CrossRef]
- Forootan, A.; Sjöback, R.; Björkman, J.; Sjögreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef]
- Okholm, A.H.; Nielsen, J.S.; Vinther, M.; Sørensen, R.S.; Schaffert, D.; Kjems, J. Quantification of cellular uptake of DNA nanostructures by qPCR. Methods 2014, 67, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.L.; Karlson, B.; Wulff, A.; Kudela, R.; Trick, C.; Asnaghi, V.; Berdalet, E.; Cochlan, W.; Davidson, K.; De Rijcke, M.; et al. Future HAB science: Directions and challenges in a changing climate. Harmful Algae 2020, 91, 101632. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Nakajima, N.; Okamoto, S.; Suzuki, I.; Tanabe, Y.; Tamaoki, M.; Makamura, Y.; Kasai, F.; Watanabe, A.; Kawashima, K.; et al. Complete genomic structure of the bloom-forming toxic cyanobacterium Microcystis aeruginosa NIES-843. DNA Res. 2007, 14, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.K.; Evitt, N.H.; Swartz, J.R. Chemical lysis of cyanobacteria. J. Biol. Eng. 2015, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Tillet, D.; Neilan, B.A. Xanthogenate nucleic acid isolation from cultured and environmental cyanobacteria. J. Phycol. 2000, 36, 251–258. [Google Scholar] [CrossRef]
- Devlin, S.; Meneely, J.P.; Greer, B.; Greef, C.; Lochgead, M.J.; Elliot, C.T. Next generation planar waveguide detection of microcystins in freshwater and cyanobacterial extracts, utilising a novel lysis method for portable sample preparation and analysis. Anal. Chim. Acta 2013, 769, 108–113. [Google Scholar] [CrossRef]
- Kim-Tiam, S.; Comte, K.; Dalle, C.; Duval, C.; Pancrace, C.; Gugger, M.; Marie, B.; Yéprémian, C.; Bernard, C. Development of a new extraction method based on high-intensity ultra-sonication to study RNA regulation of the filamentous cyanobacteria Planktothrix. PLoS ONE 2019, 14, e0222029. [Google Scholar] [CrossRef] [PubMed]
- Bickman, S.R.; Campbell, K.; Elliott, C.; Murphy, C.; O’Kennedy, R.; Papst, P.; Lochhead, M.J. An innovative portable biosensor system for the rapid detection of freshwater cyanobacterial algal bloom toxins. Environ. Sci. Technol. 2018, 52, 11691–11698. [Google Scholar] [CrossRef]

{kind=link}
| Reference | Solid Support | Capture Probe | Signal Probe | Detection Method | Washing Buffer | Hybridization Buffer | Incubation Time | Detection Limit | Detection Target |
|---|---|---|---|---|---|---|---|---|---|
| [12] Rautio et al. 2003 | Streptavidin-coated magnetic beads | 3′ Biotin labeled RNA | 3′ DIG-labeled RNA | Plate fluorescence reader; Anti-DIG Fab fragment-AP conjugate + BBTP | 50 mM Tris, 150 mM NaCl, 0.3% Tween 20 | 5 × SSC, 0.5% SDS, 0.02% Ficoll, 0.02% PVP, 0.02% BSA, 20% deionized formamide, 4% dextran sulfate | Denaturation: 65 °C 3–5 min | 2 fmol (1.2 × 109 molecules) | Yeast (Saccharomyces cerevisiae) SUC2 mRNA and 18S rRNA |
| Hybridization: 50 °C 30 min | |||||||||
| Bead immobilization: 37 °C 30 min | |||||||||
| Conjugation: 20 °C 30 min | |||||||||
| Substrate application: 37 °C 20 min | |||||||||
| [26] Feuillie et al. 2011 | Streptavidin-coated magnetic beads | 3′ Biotin labeled DNA | 5′ Rhodamine 6G labeled DNA | Surface Enhanced Resonance Raman Scattering (SERRS) | 0.25 × SSC, 0.5% Tween 20 | 4 × SSC, 0.05% Tween 20 | Denaturation: 99 °C 10 min | 1 fmol | Chamois (Rupicapra rupicapra) DNA |
| Hybridization: 55 °C 3 h | |||||||||
| Bead immobilization: 20 °C 30 min | |||||||||
| Elution: 95 °C 20 min | |||||||||
| [15] Goldman et al. 2013 | None (micelle drag tag-containg running buffer) | γ-carbon modified PNA (γPNA) amphiphile labelled with FITC | Cy5-labeled for DNA target or YOYO-1-stained duplex for RNA target | Capillary Electrophoresis equipped for laser-induced fluorescence detection | None | 1 × TBE (Tris/Borate/EDTA) | Hybridization: 95 °C 5 min, cooled to room temperature in 1 h | Single base mismatch discrimina-tion (10 nm or 2 × 1011 molecules) | Short 22-nucleotide RNA or DNA |
| YOYO-1 staining: 1 h at room temperature | |||||||||
| [17] Thieme et al. 2008 | Streptavidin-coated magnetic beads | 5′ Biotin labeled DNA + 2 unlabeled DNA helper probes | 3′ DIG-labeled DNA | Microplate fluorescence reader; Anti-DIG Fab fragment-AP conjugate + BBTP | 1 × SSC, 0.01% SDS in DEPC-treated water | 5 × SSC, 20% formamide, 3% dextran sulfate, 0.2% Tween 20, 0.02% Ficoll 400, 0.02% PVP, 1% blocking reagent in 100 mM maleic acid with 150 mM NaCl, all mixed in DEPC-treated water | Plate incubation: 50 °C 5 min | <1 fmol of RNA per well | Escherichia coli mRNA |
| Hybridization: 50 °C 30 min | |||||||||
| Immobilization: 50 °C 30 min | |||||||||
| Wash 1: 50 °C 2 min | |||||||||
| Wash 2: 30 °C 2 min (twice) | |||||||||
| Conjugation: 30 °C 30 min | |||||||||
| Substrate application: 37 °C 20 min | |||||||||
| [25] Zhang and Appella 2007 | DNA-BIND® 96-well plate | PNAα covalently attached to plate | Biotin-labeled PNAβ | Plate reader; avidin-HRP + TMB | Wash 1 and 3: PBS; Wash 2: 0.1% SDS in 0.1 × SSC | Hybridization: 0.15 M NaCl Blocking buffer: 3% BSA and 25 mM lysine in 50 mM Na2HPO4/NaH2PO4, 1 mM EDTA | Attach PNAα to plate: 37 °C 1 h | 10−5 fmol of DNA | Bacillus anthracis DNA (Anthrax) |
| Wash 1: 33°C 1 min (3 times) | |||||||||
| Blocking: 37°C 30 min | |||||||||
| Hybridization: 45 °C 3 h | |||||||||
| Wash 2: 33 °C 30 min (twice) | |||||||||
| Blocking: 37°C 30 min | |||||||||
| Conjugation: 37 °C 30 min | |||||||||
| Wash 3: 37 °C 1 min (3 times) | |||||||||
| Substrate application: 37 °C 20 min | |||||||||
| [14] Goffredi et al. 2006 | Biotin-coated polystyrene prongs | 5′-Biotinylated DNA, conjugated to streptavidin | Double DIG-labeled DNA at both 5′ and 3′ ends | Plate reader; anti-DIG-HRP + TMB-ELISA; robotic workstation | 50 mM Tris, 150 mM NaCl, 0.05% Tween 20 | 2M GuSCN, 50 mM Tris, 10 mM EDTA, 0.5% Tween 20, pH 8.6 | 25 to 30 °C for all steps | 5 larvae/mL of lysate | Barnacle 18S rRNA |
| Attach capture probe to prong: 8 min | |||||||||
| 1st Hybridization (capture): 8 min | |||||||||
| 2nd Hybridization (signal): 8 min | |||||||||
| 1st Wash: 2 min | |||||||||
| Antibody application: 5 min | |||||||||
| 2nd Wash: 2 min (twice) | |||||||||
| Substrate application: 5 min | |||||||||
| [16] Clancy et al. 2017 | Microarray glass slide | DNA covalently attached to the microarray glass slide | 5′ Cy5-labeled DNA | Microarray scanner | Wash 1: 2 × SSC, 0.5% SDS; | Solution-phase pre-hybridization: 2 × SSC, 0.5% SDS, 1 µM reporter probe | Pre-hybridization: 30 °C 20 min | 1 pM or 0.03 fmol of miRNA | Breast cancer related microRNA |
| Slide preheating: 30 °C 10 min | |||||||||
| Wash 2: 2 × SSC; | Hybridization: 30 °C 1 h | ||||||||
| Wash 3: 0.2 × SSC | Wash 1: 30 °C 10 min | ||||||||
| Wash 2&3: room temp 10 min | |||||||||
| [21] Zammatteo et al. 1997 | Amine-grafted magnetic beads or polystyrene plate | DNA covalently attached to beads or plates | Biotinylated or radiolabeled DNA | Liquid scintillation counter for radio-labeled probe or plate reader (+ HRP-streptavidine + TMB) for biotinylated probe | Radiolabeled: 0.1 × SSC | 4 × SSC, 10 × Denhart, 200 µg/mL DNA salmon sperm | Hybridization for both probe types: 60 °C for 2 h | 0.03 fmol of HCMV DNA | Human cytomegalovirus (HCMV) DNA |
| Biotinylated: buffer 1: 100mM maleic acid, pH 7.5, 150 mM NaCl, 0.3% Tween 20; blocking buffer: 100 mM maleic acid, pH 7.5, 150 mM NaCl, 0.1% Gloria milk powder; buffer 2: 100 mM maleic acid, pH 7.5, 150 mM NaCl | Biotinylated: streptavidin-peroxidase diluted in blocking buffer 23 °C for 45 min and TMB incubated 10 min in the dark | ||||||||
| [27] Scholin et al. 1996 | Nylon beads | DNA conjugated to beads | 5′ Biotin-labeled DNA | Visual inspection or photography | 50 mM Tris HCI, 10 mM EDTA, 100 mM NaCl, 1% (v/v) SDS, 1% (v/v) N-Iauryl sarcosine, pH 8.0 | Hybridization Buffer I: 100 mM Tris, 17 mM EDTA, 8.35% formamide, 5 M GuSCN, pH 7.5; Hybridization Buffer II: 100 mM Tris, 17 mM EDTA, 8.35% formamide, 3 M GuSCN, pH 7.5 | Primary hybridization (target to bead): 23–25 °C for 30 min | Not reported | Pseudo-nitzschia australis large subunit (LSU) rRNA |
| Secondary hybridization (signal probe to target): 23–25 °C 30 min | |||||||||
| Washing: 23–25 °C 2 min (2X) | |||||||||
| Conjugation: 23–25 °C 30 min | |||||||||
| Substrate application: 23–25 °C 30 min |
| References | Solid Support | Detection Instrument & Method | Target Genus/Group | Target Gene | Capture Probe | Signal Probe |
|---|---|---|---|---|---|---|
| [43] Matsunaga et al. 2001 | BMP (Bacterial Magnetic Particle) | Luminometer; Immunosorbent method: anti-DIG-AP used for signal detection after addition of the CDP-Star™ Substrate with Emerald-II™ Enhancer | Anabaena, Microcystis, Nostoc, Oscillatoria, Synechococcus | 16S rRNA amplicon | Anabaena1 562–579 nt ABACWWACAATGCCACCT; Anabaena2 647–666 nt CCAGGAATTCCCTCTGCCC; Microcystis 585–604 nt TTAAGCAACCTGATTTGA; Nostoc1 569–587 nt ACAGCAGACTTACATTG; Nostoc2 628–636 nt ACGTACTCTAGCTATG; Oscillatoria 802–823 nt ACAGGCHACACCTAGTCTCCATC; Synechococcus 575–593 nt RGGCTTTGACARCAGACT | CYA-781R: 781–800 nt GACTACTGGGGTATCTAATCCCATT |
| [44] Matsunaga et al. 2001 | BMP and MAG-microarray | Fluorescence stereomicroscope; Immunosorbent method: anti-DIG-AP used for signal detection after additon of the AttoPhos® AP substrate | ditto | ditto | ditto (R = A or G; Y = C or T; W= A or T; K = G or T; M= A or C; S = G or C; H = A, C or T; V = A, C, or G; D = A, G or T; B = T or G; N = A, C, G or T) | ditto |
| [46] Castiglioni et al. 2004 | Microarray spotted with universal “zip code” probes | ScanArray 4000 laser-scanning system (Cy3 with λex = 543 nm;λ em = 570 nm) | 19 cyanobacterial groups | 16S rRNA amplicons | Group-specific discriminating probes labeled with Cy3 dye at the 5’ end (see [46] for sequences) | Genus-specific common probes phosphorylated at the 5’ end and carrying a czip code at the 3’ end (see [46] for sequences) |
| [41] Preston et al. 2009 | 96-well plate or membrane array in Environmental sample processor (ESP) | Robotic processor/plate reader (A450) or CCD camera with digital image analysis system; colorimetric method: anti-DIG-HRP + HRP substrate | Synechococcus CCMP 1334 | 16S rRNA (in vitro transcripts or extracted RNA) | Picophyto496: 5’-Biotin-[C9 x 3]-GGCACGGAATTAGCCGWGGCTTA-3’ | EUB338: 5’-DIG-[C9]-GCWGCCWCCCGTAGGWGT-[C9]-DIG-3’; Univ519ab: 5’-DIG-[C9]-TTACCGCGGCKGCTGGCAC-[C9]-DIG-3’ |
| [47] Zhu et al. 2012 | Magnetic beads modified with isothiocyanate groups | Cary 50 UV-Vis Spectrophotometer (A405) | Microcystis | PC-IGS amplicon | TF: 5’-GCAATAAGTTTCCTACGG-NH2 | TR: 5’biotin-GGTATCTCCCAATAATCT-3’ |
| [48] Zhu et al. 2012 | Microcystis | PC-IGS amplicon | TF: 5’-GCAATAAGTTTCCTACGG-NH2 | TR: 5’biotin-GGTATCTCCCAATAATCT-3’ | ||
| Immunosorbent method: Alkaline phosphatase-streptavidin + enzymatic substrate p-nitrophenyl phosphate sodium | Microcystis | mcyJ amplicon | TJF: 5’-CCAACCTTCCACCGGGCTGCA-NH2 | TJR: 5’biotin-CGACCCACTCTAGGCAAACAATC-3’ | ||
| [50] Dearth 2017; [51] Dearth et al. 2022 | Streptavidin-coated 96-well plate | BioTek Synergy HT Plate reader (A450) & Affirm robotic processor; colorimetric method | Microcystis | 16S rRNA (extracted RNA) | MIC593: 5’ biotin-[C9 x 3]-AACCTGATTTGACGGCAGACTTGGCTGA-3’ | EUB338: 5’-DIG-[C9]-GCWGCCWCCCGTAGGWGT-[C9]-DIG-3’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, P.; Antrim, A.K.; Bickman, S.R.; Cooley, E.G.; Chung, S.H. Sandwich Hybridization Assay for In Situ Real-Time Cyanobacterial Detection and Monitoring: A Review. Biosensors 2022, 12, 640. https://doi.org/10.3390/bios12080640
Gong P, Antrim AK, Bickman SR, Cooley EG, Chung SH. Sandwich Hybridization Assay for In Situ Real-Time Cyanobacterial Detection and Monitoring: A Review. Biosensors. 2022; 12(8):640. https://doi.org/10.3390/bios12080640
Chicago/Turabian StyleGong, Ping, Anna K. Antrim, Sarah R. Bickman, Emily G. Cooley, and Seung Ho Chung. 2022. "Sandwich Hybridization Assay for In Situ Real-Time Cyanobacterial Detection and Monitoring: A Review" Biosensors 12, no. 8: 640. https://doi.org/10.3390/bios12080640
APA StyleGong, P., Antrim, A. K., Bickman, S. R., Cooley, E. G., & Chung, S. H. (2022). Sandwich Hybridization Assay for In Situ Real-Time Cyanobacterial Detection and Monitoring: A Review. Biosensors, 12(8), 640. https://doi.org/10.3390/bios12080640