Multiplex Digital Quantification of β-Lactamase Genes in Antibiotic-Resistant Bacteria by Counting Gold Nanoparticle Labels on Silicon Microchips

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Silicon Microchips Fabrication
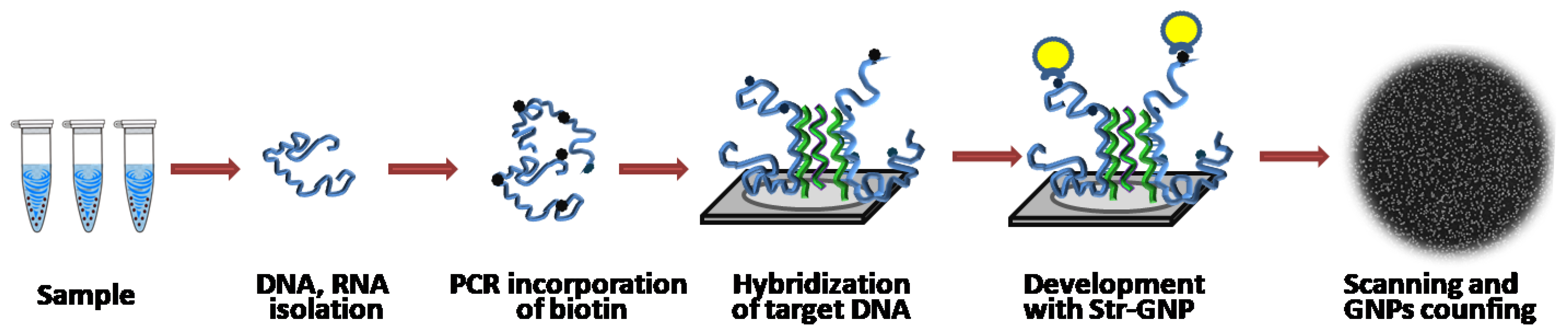
2.3. Amplification and Labeling of Target DNA
2.4. Hybridization of Target DNA on Silicon Microchips
2.5. Digital Detection and Data Processing
3. Results and Discussion
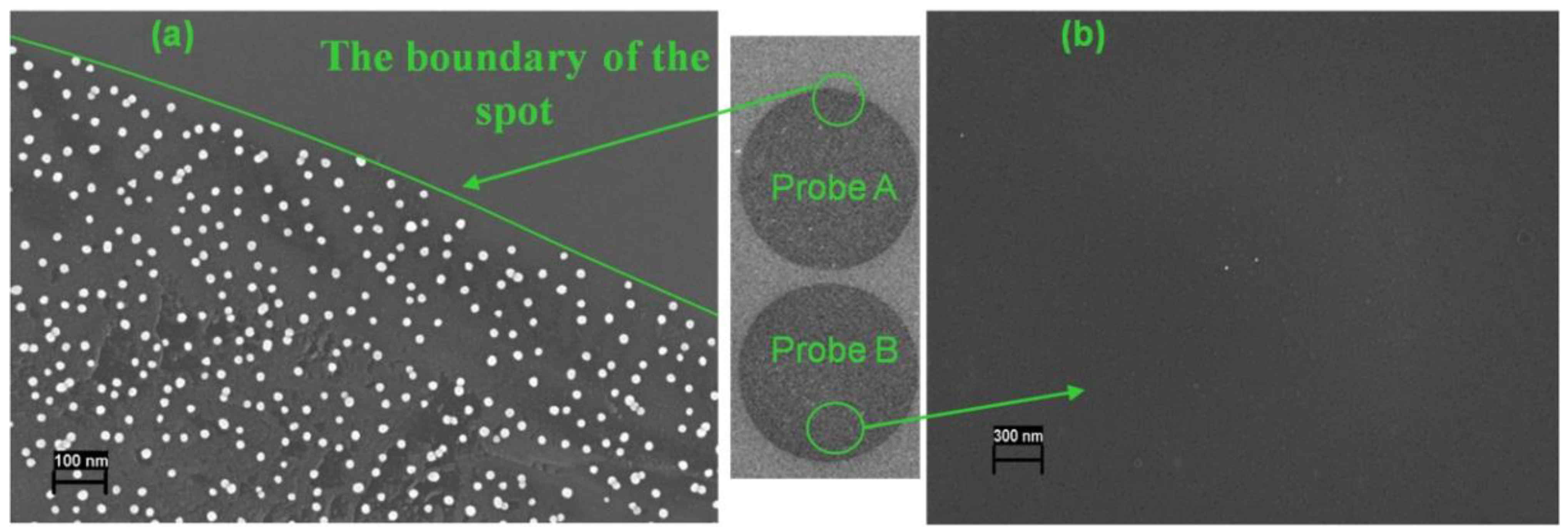
3.1. Assay Principle, Counting of Gold Nanoparticles on Silicon Microchips
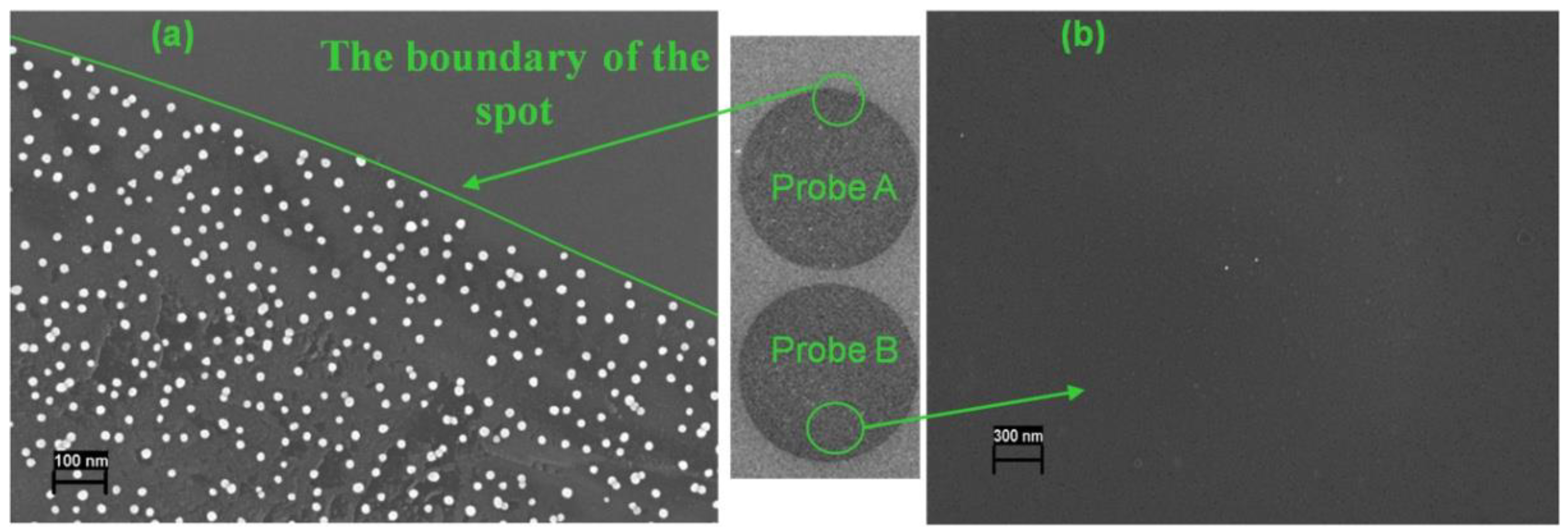
3.2. Analysis of Cleaved Silicon Microchips
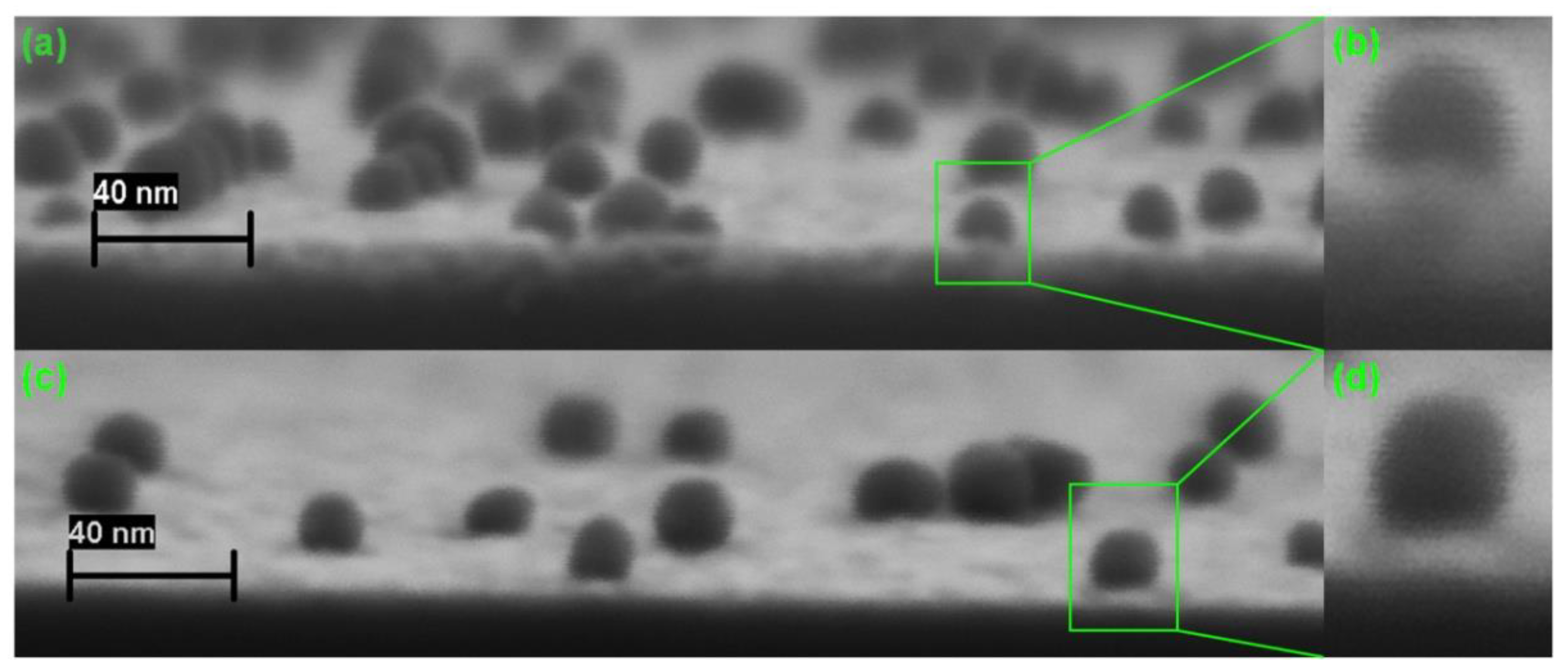
3.3. Optimization of Digital Analysis of DNA Duplexes by SEM
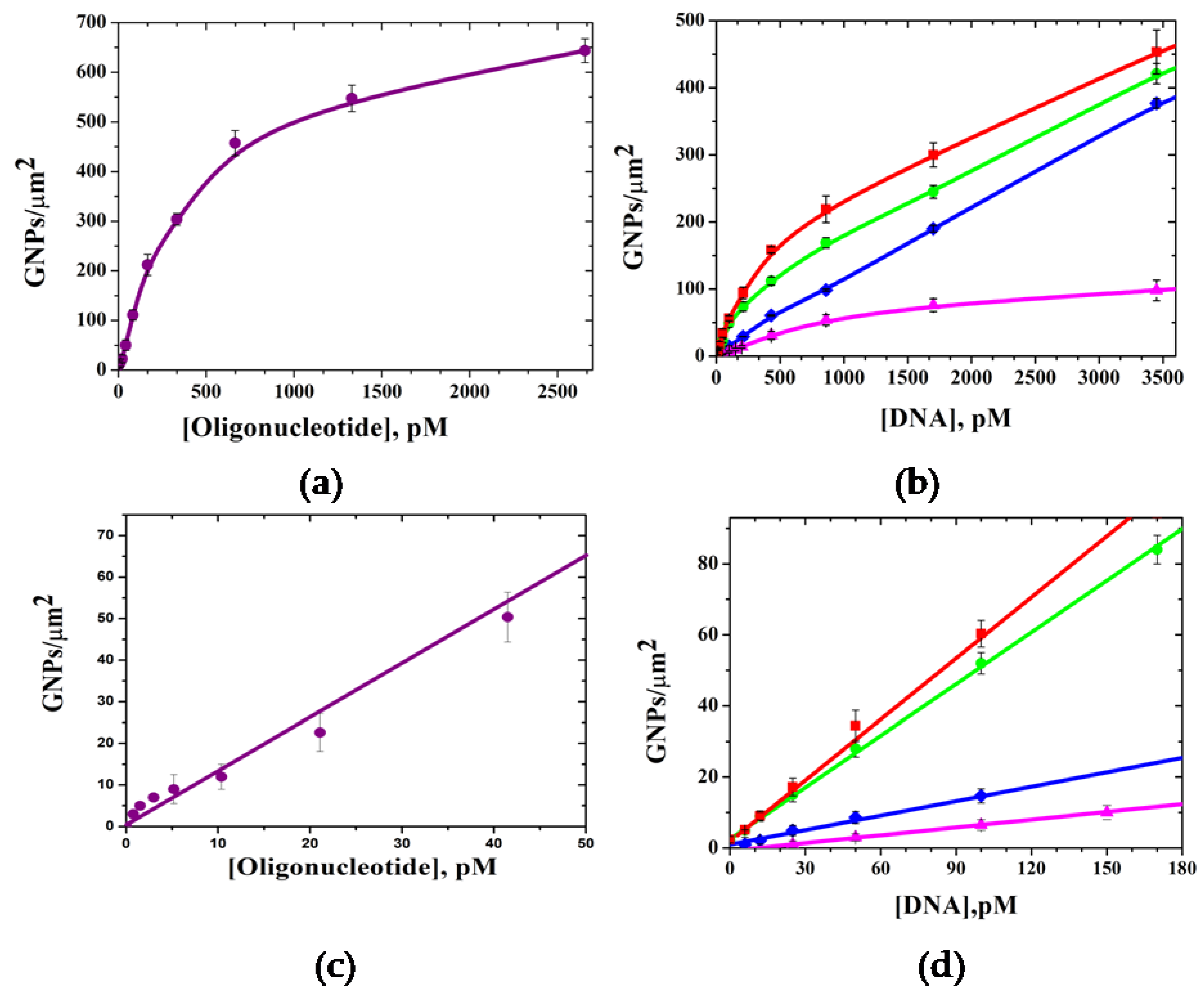
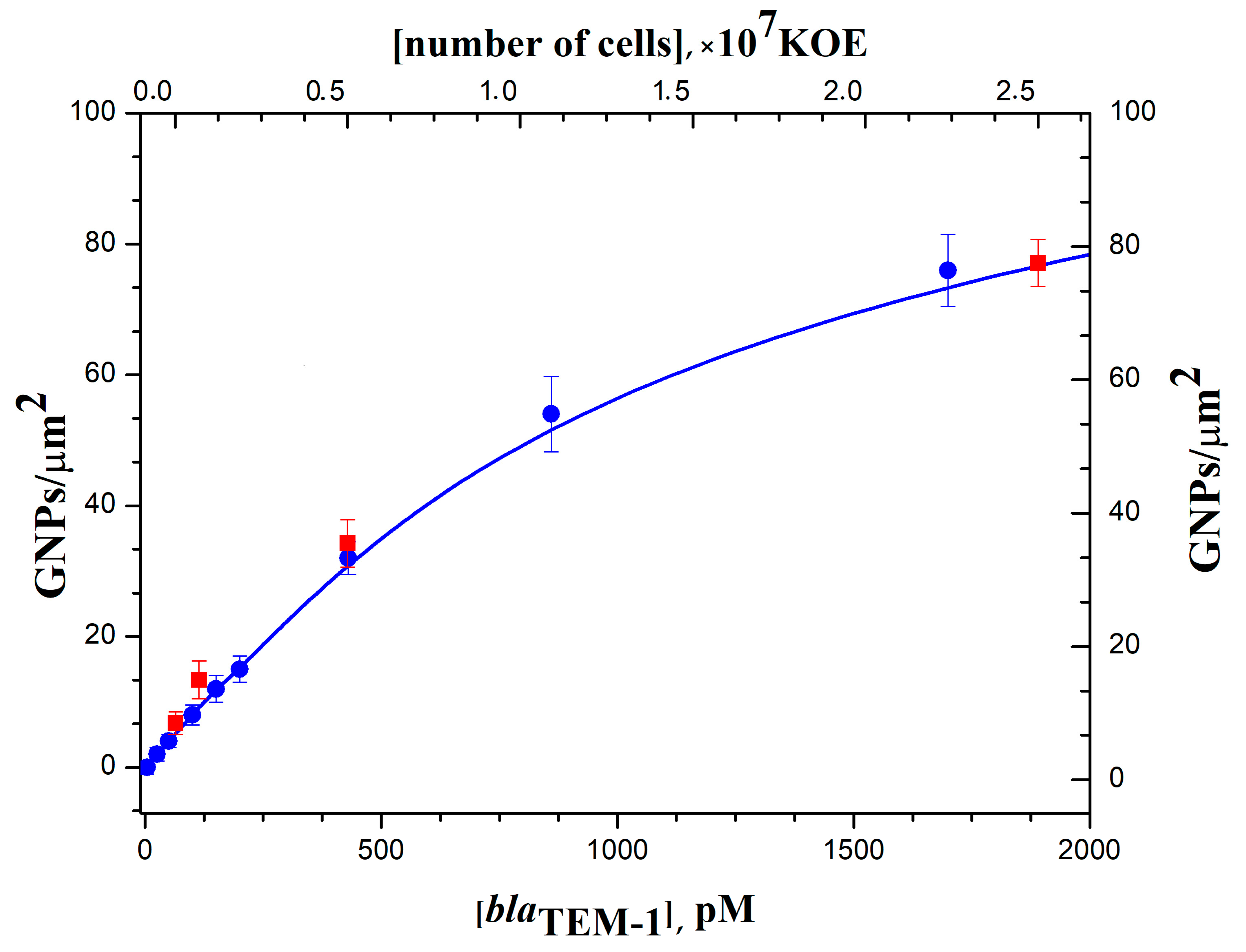
3.4. Digital Quantification of Nucleic Acids on Silicon Microchips
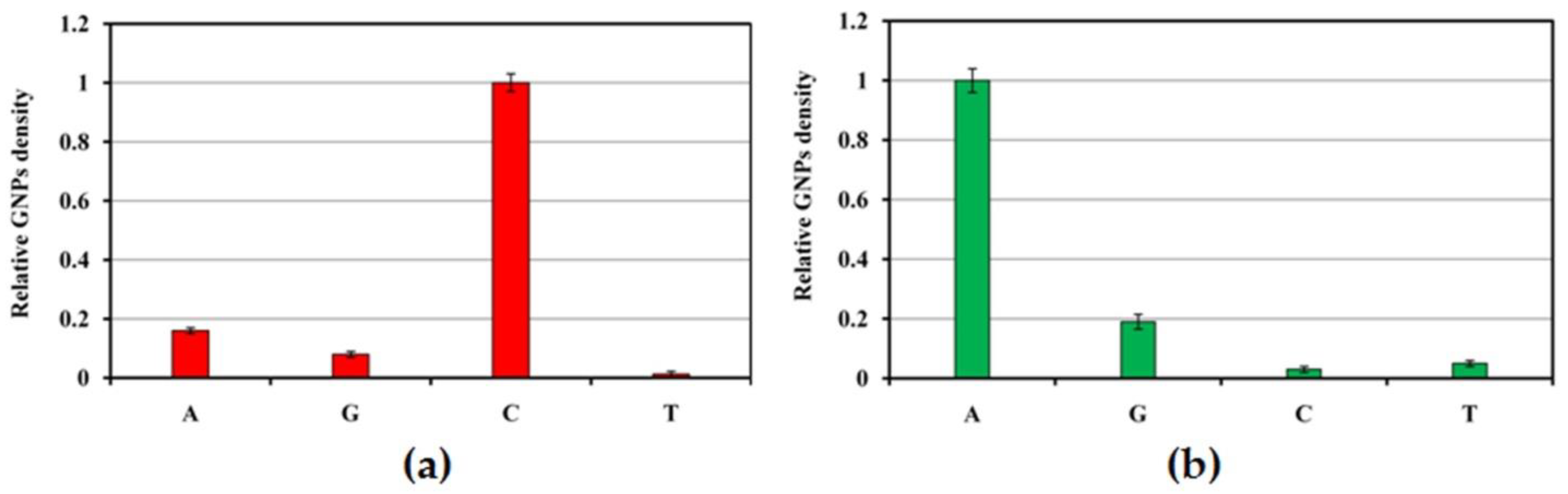
3.5. Digital Identification of Single Nucleotide Polymorphism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Du, Y.; Dong, S. Nucleic Acid Biosensors: Recent Advances and Perspectives. Anal. Chem. 2017, 89, 189–215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, J.; Xu, J.-J.; Zhang, S.; Chen, H.-Y. Advances in DNA/RNA detection using nanotechnology. Adv. Clin. Chem. 2019, 91, 31–98. [Google Scholar] [CrossRef] [PubMed]
- Scheler, O.; Glynn, B.; Kurg, A. Nucleic acid detection technologies and marker molecules in bacterial diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 489–500. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics 2020; World Health Organization: Geneva, Switzerland, 2020; Available online: https://apps.who.int/iris/bitstream/handle/10665/332070/9789240005105-eng.pdf (accessed on 2 April 2022).
- Eichenberger, E.M.; Thaden, J.T. Epidemiology and Mechanisms of Resistance of Extensively Drug Resistant Gram-Negative Bacteria. Antibiotics 2019, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 2019, 11, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Niazi, J.H. Biosensors for detecting viral and bacterial infections using host biomarkers: A review. Analyst 2021, 145, 7825–7848. [Google Scholar] [CrossRef] [PubMed]
- Therisod, R.; Tardif, M.; Marcoux, P.R.; Picard, E.; Jager, J.-B.; Hadji, E.; Peyrade, D.; Houdré, R. Gram-type differentiation of bacteria with 2D hollow photonic crystal cavities. Appl. Phys. Lett. 2018, 113, 111101. [Google Scholar] [CrossRef]
- Conteduca, D.; Brunetti, G.; Dell’Olio, F.; Armenise, M.N.; Krauss, T.F.; Cimenelli, C. Monitoring of individual bacteria using electro-photonic traps. Biomed. Opt. Express 2019, 10, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Baron, V.O.; Chen, M.; Clark, S.O.; Williams, A.; Dholakia, K.; Gillespie, S.H. Detecting Phenotypically Resistant Mycobacterium tuberculosis Using Wavelength Modulated Raman Spectroscopy. Methods Mol. Biol. 2018, 1736, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, J.C.; Ruseska, I.; Wright, J.B.; Costerton, J.W. Tobramycin resistance of Pseudomonas aeruginosa cells growing as a biofilm on urinary catheter material. Antimicrob. Agents Chemother. 1985, 27, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, G.; Conteduca, D.; Armenise, M.N.; Ciminelli, C. Novel Micro-Nano Optoelectronic Biosensor for Label-Free Real-Time Biofilm Monitoring. Biosensors 2021, 11, 361. [Google Scholar] [CrossRef]
- Kim, S.; Yu, G.; Kim, T.; Shin, K.; Yoon, J. Rapid bacterial detection with an interdigitated array electrode by electrochemical impedance spectroscopy. Electrochim. Acta 2012, 82, 126–131. [Google Scholar] [CrossRef]
- Wang, Y.; Reardon, C.P.; Read, N.; Thorpe, S.; Evans, A.; Todd, N.; Van Der Woude, M.; Krauss, T.F. Attachment and antibiotic response of early-stage biofilms studied using resonant hyperspectral imaging. NPJ Biofilms Microbiomes 2020, 6, 57. [Google Scholar] [CrossRef]
- Ferrier, D.C.; Shaver, M.P.; Hands, P.J.W. Micro- and nano-structure based oligonucleotide sensors. Biosens. Bioelectron. 2015, 68, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedin, C.; Crotti, S.; Tasciotti, E.; Agostini, M. Diagnostic devices for circulating biomarkers detection and quantification. Curr. Med. Chem. 2018, 25, 4304–4327. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Wu, Y.; Bennett, D.; Tilley, R.D.; Gooding, J.J. How nanoparticles transform single molecule measurements into quantitative sensors. Adv. Mater. 2019, 32, e1904339. [Google Scholar] [CrossRef]
- Cretich, M.; Daaboul, G.G.; Sola, L.; Unlu, M.S.; Chiari, M. Digital detection of biomarkers assisted by nanoparticles: Application to diagnostics. Trends Biotechnol. 2015, 33, 343–351. [Google Scholar] [CrossRef]
- Kataoka, M.; Ishida, K.; Ogasawara, K.; Nozaki, T.; Satoh, Y.I.; Sata, T.; Sato, Y.; Hasegawa, H.; Nakajima, N. Serial section array scanning electron microscopy analysis of cells from lung autopsy specimens following fatal A/H1N1 2009 pandemic influenza virus infection. J. Virol. 2019, 93, e00644-19. [Google Scholar] [CrossRef] [Green Version]
- Gopinath, S.C.B.; Awazu, K.; Fujimaki, M.; Shimizu, K.; Shima, T. Observations of immuno-gold conjugates on influenza viruses using waveguide-mode sensors. PLoS ONE 2013, 8, e69121. [Google Scholar] [CrossRef]
- Jiang, C.; Lu, H.; Zhang, H.; Shen, Y.; Lu, Y. Recent advances on in situ SEM mechanical and electrical characterization of low-dimensional nanomaterials. Scanning 2017, 2017, 1985149. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, G.; Costa, J.P.C.; Costa, P.I.; Zaghete, M.A.; Mazon, T. Electrochemical immunosensor based on ZnO nanorods-Au nanoparticles nanohybrids for ovarian cancer antigen CA-125 detection. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 76, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschou, D.; Trantidou, T.; Regoutz, A.; Carta, D.; Morgan, H.; Prodromakis, T. Surface and electrical characterization of Ag/AgCl pseudo-reference electrodes manufactured with commercially available PCB technologies. Sensors 2015, 15, 18102–18113. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Ding, X.; Yang, Y.Y.; Xu, Q.-H. Metal nanoparticles for diagnosis and therapy of bacterial infection. Adv. Healthc. Mater. 2018, 7, e1701392. [Google Scholar] [CrossRef]
- Mocan, T.; Matea, C.T.; Pop, T.; Mosteanu, O.; Buzoianu, A.D.; Puia, C.; Iancu, C.; Mocan, L. Development of nanoparticle based optical sensors for pathogenic bacteria detection. J. Nanobiotechnol. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.M.; Fan, S.F.; Wang, Q.L.; Lv, Q.Y.; Song, X.; Cui, H.F. An enzyme-free electrochemical sandwich DNA assay based on the use of hybridization chain reaction and gold nanoparticles: Application to the determination of the DNA of Helicobacter pylori. Microchim. Acta 2019, 187, 73. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tilley, R.D.; Gooding, J.J. The challenges and solutions in developing ultrasensitive biosensors. J. Am. Chem. Soc. 2019, 141, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Takei, H.; Yasuda, K. Quantitative evaluation of a gold-nanoparticle labeling method for detecting target DNAs on DNA microarrays. Sens. Actuators B Chem. 2010, 144, 6–10. [Google Scholar] [CrossRef]
- Hughes, D.; Andersson, D.I. Evolutionary trajectories to antibiotic resistance. Annu. Rev. Microbiol. 2017, 71, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K. Past and present perspectives of β-lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippon, A.; Slama, P.; Dény, P.; Labia, R. A structure-based classification of class A β-lactamases, a broadly diverse family of enzymes. Clin. Microbiol. Rev. 2016, 29, 29–57. [Google Scholar] [CrossRef] [Green Version]
- Avershina, E.; Shapovalova, V.; Shipulin, G. Fighting Antibiotic Resistance in Hospital-Acquired Infections: Current State and Emerging Technologies in Disease Prevention, Diagnostics and Therapy. Front. Microbiol. 2021, 12, 707330. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Presnova, G.V.; Rubtsova, M.Y.; Presnov, D.E.; Grigorenko, V.G.; Yaminsky, I.V.; Egorov, A.M. Streptavidin conjugates with gold nanoparticles for visualization of single DNA interactions on the silicon surface. Biochem. Suppl. Ser. B Biomed. Chem. 2014, 8, 164–167. [Google Scholar] [CrossRef]
- Lamture, J.B.; Beattie, K.L.; Burke, B.E.; Eggers, M.D.; Ehrlich, D.J.; Fowler, R.; Hollis, M.A.; Kosicki, B.B.; Reich, R.K.; Smith, S.R. Direct detection of nucleic acid hybridization on the surface of a charge coupled device. Nucleic Acids Res. 1994, 22, 2121–2125. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, M.Y.; Ulyashova, M.M.; Edelstein, M.V.; Egorov, A.M. Oligonucleotide microarrays with horseradish peroxidase-based detection for the identification of extended-spectrum β-lactamases. Biosens. Bioelectron. 2010, 26, 1252–1260. [Google Scholar] [CrossRef]
- Dubrovin, E.V.; Presnova, G.V.; Rubtsova, M.Y.; Egorov, A.M.; Grigorenko, V.G.; Yaminsky, I.V. The use of atomic force microscopy for 3D analysis of nucleic acid hybridization on microarrays. Acta Nat. 2015, 7, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Taton, T.A.; Mirkin, C.A. Array-Based Electrical Detection of DNA with Nanoparticle Probes. Science 2002, 295, 1503–1506. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, H.; Jeong, J.; Kwon, J.N.; Jung, Y.; Chung, B.H. Label-free and naked eye detection of PNA/DNA hybridization using enhancement of gold nanoparticles. Chem. Commun. 2010, 46, 3315–3317. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Jung, J.; Chung, B.H. Scanometric analysis of DNA microarrays using DNA intercalator-conjugated gold nanoparticles. Chem. Commun. 2012, 48, 7601–7603. [Google Scholar] [CrossRef] [PubMed]
- Ulyashova, M.M.; Rubtsova, M.Y.; Egorov, A.M. Colorimetric detection of immobilized horseradish peroxidase based on the co-oxidation of benzidine derivatives and 4-chloro-1-naphthol. Russ. Chem. Bull. 2011, 60, 656–661. [Google Scholar] [CrossRef]
- Rizzi, G.; Lee, J.-R.; Guldberg, P.; Dufva, M.; Wang, S.X.; Hansen, M.F. Denaturation strategies for detection of double stranded PCR products on GMR magnetic biosensor array. Biosens. Bioelectron. 2017, 93, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, N.; Rizzi, G.; Chang, S.E.; Cheung, P.; Utz, P.J.; Wang, S.H. Quantification of cDNA on GMR biosensor array towards point-of-care gene expression analysis. Biosens. Bioelectron. 2019, 130, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Cao, X.; Yang, Y.; Liu, M.G.; Wang, Y.F. Array-based nano-amplification technique was applied in detection of hepatitis E virus. J. Biochem. Mol. Biol. 2006, 39, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Misra, A. SNP genotyping: Technologies and biomedical applications. Annu. Rev. Biomed. Eng. 2007, 9, 289–320. [Google Scholar] [CrossRef] [PubMed]

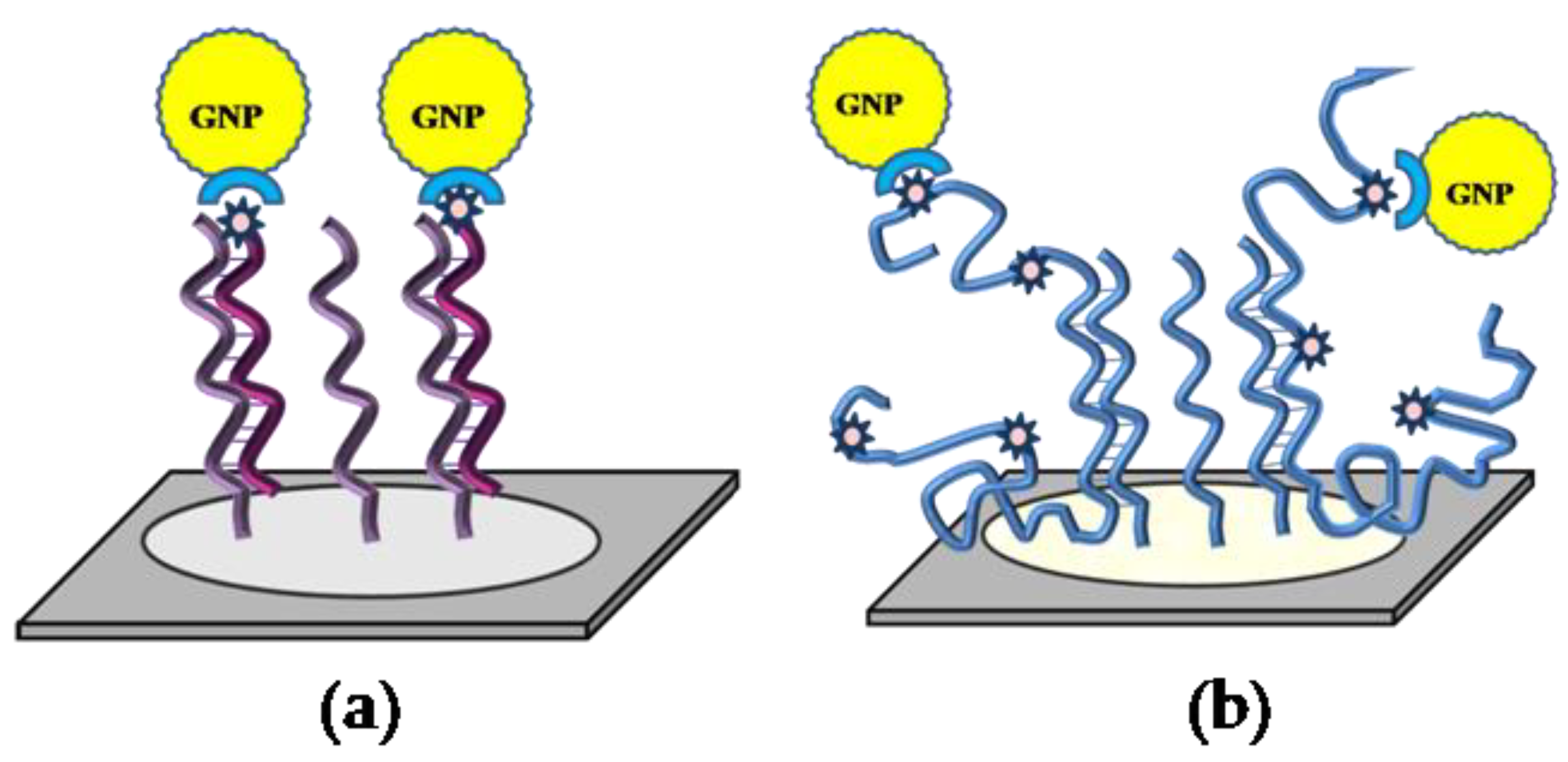
 —capture oligonucleotide probes immobilized on the microchip surface,
—capture oligonucleotide probes immobilized on the microchip surface,  —a conjugate of streptavidin with gold nanoparticles,
—a conjugate of streptavidin with gold nanoparticles,  —target oligonucleotides labeled with biotin,
—target oligonucleotides labeled with biotin,  —target DNA labeled with biotin.
—capture oligonucleotide probes immobilized on the microchip surface, —a conjugate of streptavidin with gold nanoparticles, —target oligonucleotides labeled with biotin, —target DNA labeled with biotin.
—target DNA labeled with biotin.
—capture oligonucleotide probes immobilized on the microchip surface, —a conjugate of streptavidin with gold nanoparticles, —target oligonucleotides labeled with biotin, —target DNA labeled with biotin.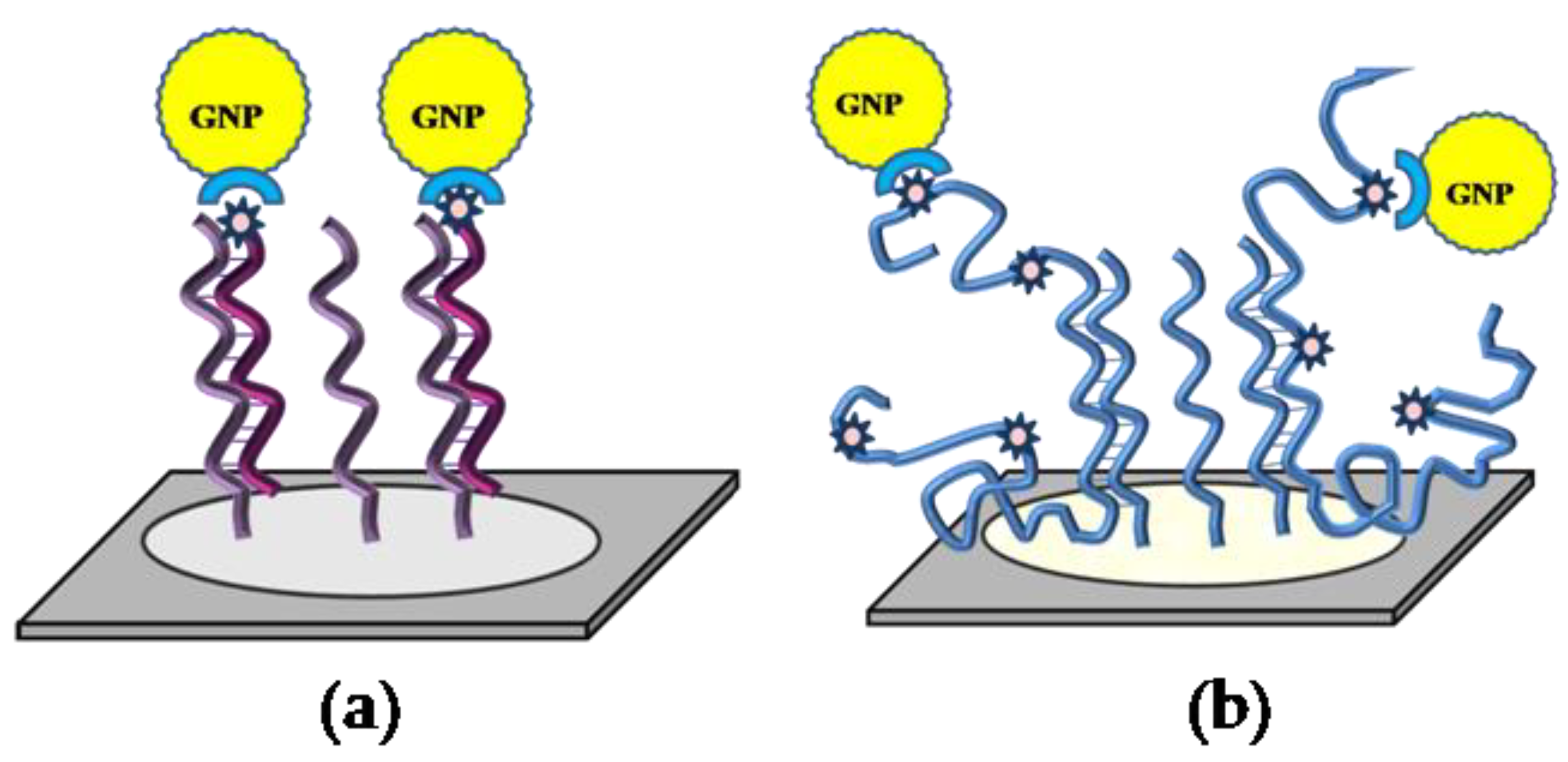





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe Name | Sequence, 5′-3′ | Length, Bases |
|---|---|---|
| Probe A | GATTGGACGAGTCAGGAGC | 19 |
| Probe B | TTCTAGACAGCCACTCATA | 19 |
| Probe C | Biotin-GCTCCTGACTCGTCCAATC | 19 |
| Probe CTX-M | ATATCGCGGTGATCTGGCC | 19 |
| Probe TEM | CCAGAAACGCTGGTGAAAGT | 20 |
| Probe VIM | GTGGTTGTGCCGTTCAT | 17 |
| CTX-M-3_167_A | GACCGTACCGAGACGACGTTAAAC | 24 |
| CTX-M-3_167_G | GACCGTACCGAGGCGACGTTAAAC | 24 |
| CTX-M-3_167_C | GACCGTACCGAGCCGACGTTAAAC | 24 |
| CTX-M-3_167_T | GACCGTACCGAGTCGACGTTAAAC | 24 |
| CTX-M-3_240_A | GGCAGCGGTGACTATGGCAC | 20 |
| CTX-M-3_240_G | GGCAGCGGTGGCTATGGCAC | 20 |
| CTX-M-3_240_C | GGCAGCGGTGCCTATGGCAC | 20 |
| CTX-M-3_240_T | GGCAGCGGTGTCTATGGCAC | 20 |
| MF * | 150 KX | 75 KX | 35 KX | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NF ** | ||||||||||
| Xav | CV% | D | Xav | CV% | D | Xav | CV% | D | ||
| Concentration of Probe C, 5 pM | ||||||||||
| 3 | 24 | 29 | 8 | 77 | 8.9 | 7 | 302 | 4.6 | 6 | |
| 6 | 23 | 27 | 8 | 76 | 5,9 | 7 | 296 | 3.1 | 6 | |
| 9 | 23 | 22 | 8 | 77 | 4.9 | 6 | 302 | 2.4 | 6 | |
| Concentration of Probe C, 500 pM | ||||||||||
| 3 | 1193 | 3.5 | 426 | 4599 | 3.7 | 407 | 20, 956 | 3.1 | 403 | |
| 6 | 1190 | 2.7 | 425 | 4567 | 2.9 | 412 | 21,060 | 2.3 | 405 | |
| Method/Detection Principle | DNA Target Size | Label | Limit of Detection | Reference |
|---|---|---|---|---|
| Determination of short oligonucleotides | ||||
| Hybridization on silicon microchips/counting of GNP labels | Oligonucleotide (19 b) | Indirect labeling of DNA duplexes with GNPs via streptavidin-biotin interaction | 0.04 pM | This work |
| Sandwich hybridization on plastic microchips/counting of GNP labels | Oligonucleotide (46 b) | Direct labeling of detection oligonucleotide probe with GNPs | 1 pM | [18] |
| Sandwich hybridization on the microelectrodes/detection of conductivity | Oligonucleotide (27 b) | Direct labeling of detection oligonucleotide probe with GNPs, silver enhancement | 0.5 pM (500 fM) | [39] |
| DNA hybridization with PNA probes/colorimetric detection of GNPs | Oligonucleotide (18 b) | Electrostatic interaction of DNA duplexes with GNPs, gold or silver enhancement | 10 pM | [40] |
| Hybridization on DNA microarrays/scanometric detection with optical scanner) | Oligonucleotide (21 b) | Labeling of the ds-DNA with DNA intercalator (daunorubicin) conjugated to GNPs, enhancement of the GNPs | 10 pM | [41] |
| Determination of long DNA | ||||
| Hybridization on silicon microchips/counting of GNP labels | Full-size gene of β-lactamase blaCTX-M-5 (870 bp) | Indirect labeling of DNA duplexes with GNPs via streptavidin-biotin interaction | 0.3 pM | This work |
| Hybridization on membrane microchips/colorimetric detection | Full-size gene of β-lactamase blaCTX-M-5 (870 bp) | Indirect labeling of DNA duplexes with horseradish peroxidase via streptavidin-biotin interaction | 0.71 nM (0.40 ng μL−1) | [42] |
| Hybridization on biosensor array/detection of magnetoresistive ratio | Synthetic ssDNA (167 p) GAPDH gene (the fragment size is not specified) | Indirect labeling of DNA duplexes with magnetic NPs via streptavidin-biotin interaction | 39 pM 0.1–1 pM depending on amount of amplification cycles | [43,44] |
| Sandwich hybridization on glass microchips/optical detection | Fragment of Hepatitis E virus RNA (DNA target of 500 bp) | Direct labeling of detection oligonucleotide probe with nano-gold, silver enhancement | 0.1 pM (100 fM) | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Presnova, G.V.; Presnov, D.E.; Filippova, A.A.; Tsiniaikin, I.I.; Ulyashova, M.M.; Rubtsova, M.Y. Multiplex Digital Quantification of β-Lactamase Genes in Antibiotic-Resistant Bacteria by Counting Gold Nanoparticle Labels on Silicon Microchips. Biosensors 2022, 12, 226. https://doi.org/10.3390/bios12040226
Presnova GV, Presnov DE, Filippova AA, Tsiniaikin II, Ulyashova MM, Rubtsova MY. Multiplex Digital Quantification of β-Lactamase Genes in Antibiotic-Resistant Bacteria by Counting Gold Nanoparticle Labels on Silicon Microchips. Biosensors. 2022; 12(4):226. https://doi.org/10.3390/bios12040226
Chicago/Turabian StylePresnova, Galina V., Denis E. Presnov, Anna A. Filippova, Ilia I. Tsiniaikin, Mariya M. Ulyashova, and Maya Yu. Rubtsova. 2022. "Multiplex Digital Quantification of β-Lactamase Genes in Antibiotic-Resistant Bacteria by Counting Gold Nanoparticle Labels on Silicon Microchips" Biosensors 12, no. 4: 226. https://doi.org/10.3390/bios12040226
APA StylePresnova, G. V., Presnov, D. E., Filippova, A. A., Tsiniaikin, I. I., Ulyashova, M. M., & Rubtsova, M. Y. (2022). Multiplex Digital Quantification of β-Lactamase Genes in Antibiotic-Resistant Bacteria by Counting Gold Nanoparticle Labels on Silicon Microchips. Biosensors, 12(4), 226. https://doi.org/10.3390/bios12040226