A FRET-Based Biosensor for the Src N-Terminal Regulatory Element

 , ,
, , 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
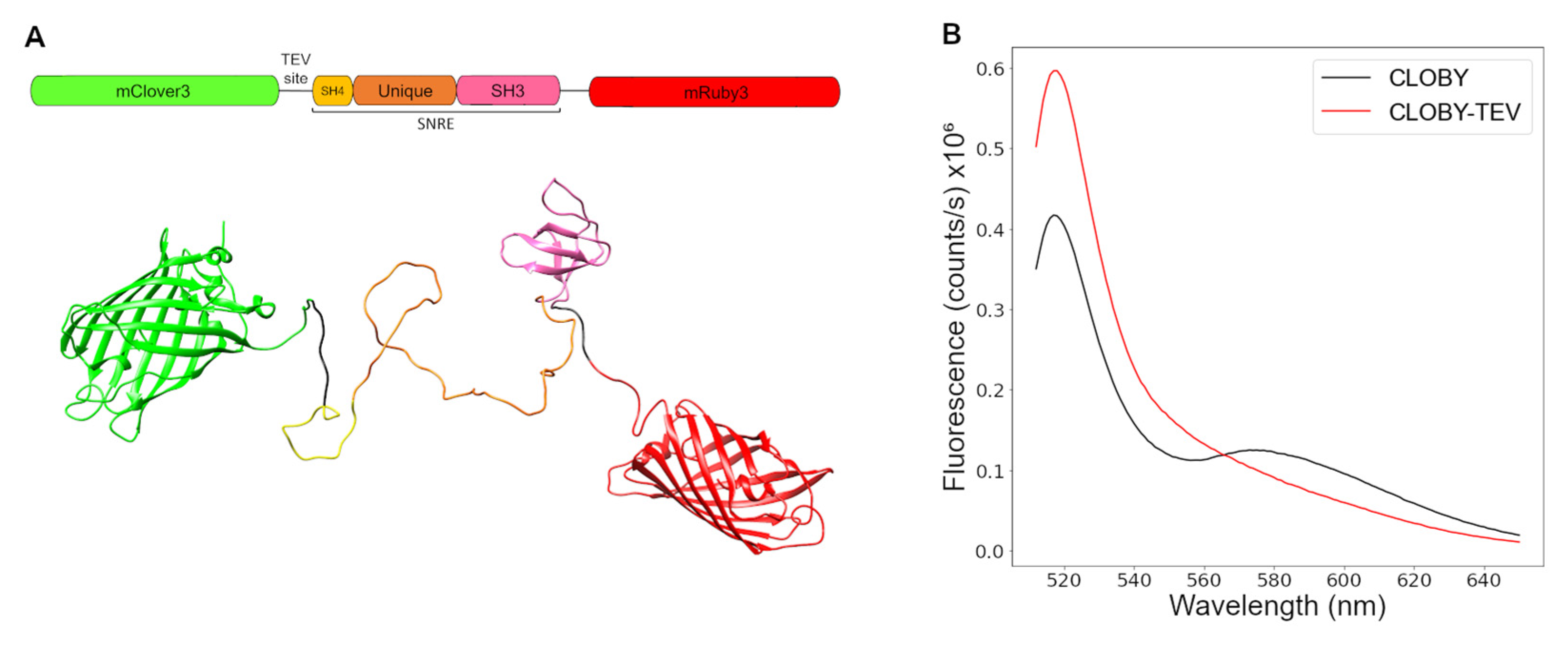
3.1. Biosensor Design
3.2. TEV Treatment Confirms Intramolecular FRET at Low Concentrations
3.3. Biosensor Characterization
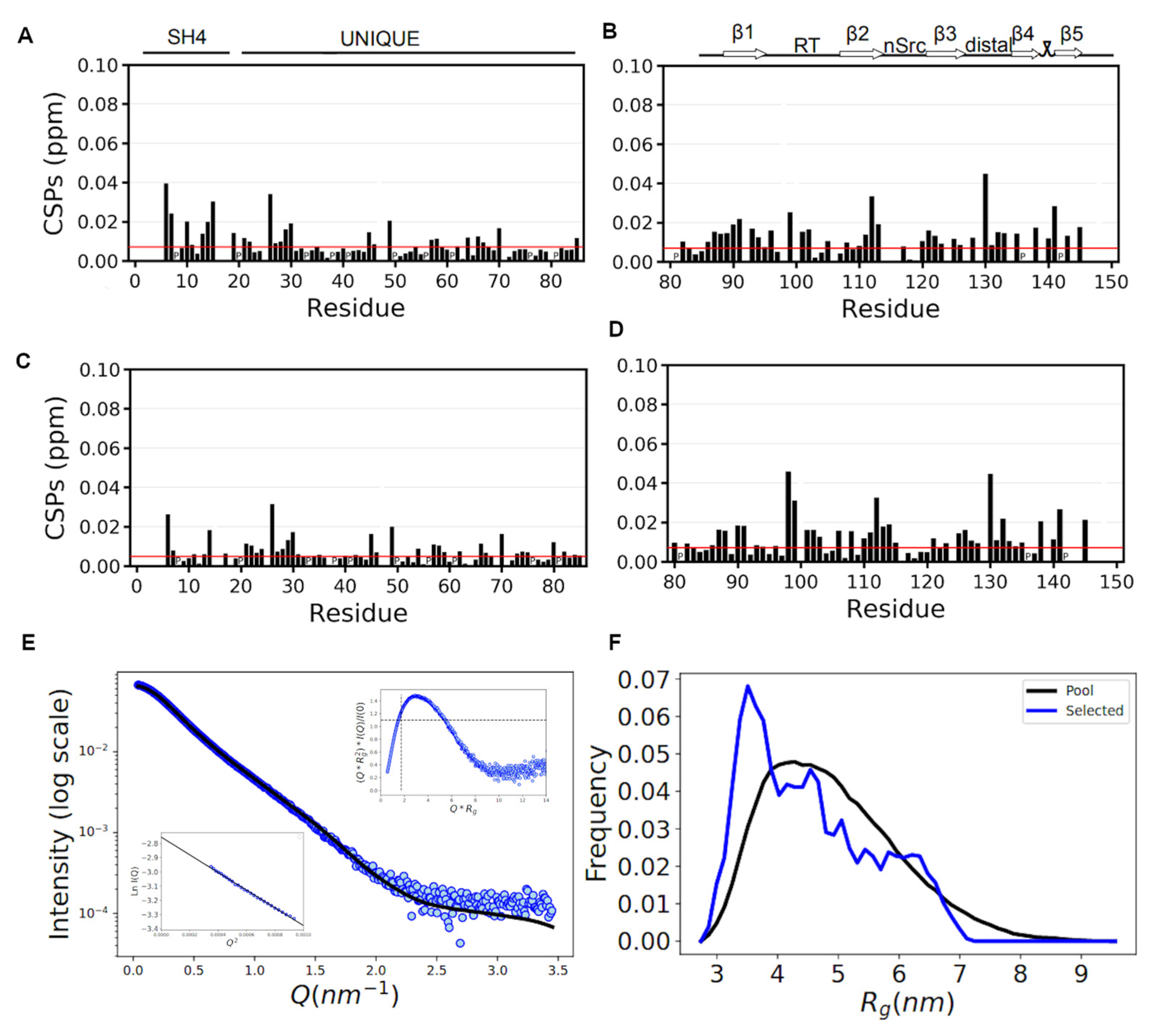
3.3.1. NMR Confirms That the SNRE Is Preserved in the CLOBY Biosensor
3.3.2. SAXS Data Confirm That the Core of the Biosensor Remains Disordered
3.3.3. Transient Weak Self-Association Does Not Affect Intramolecular FRET
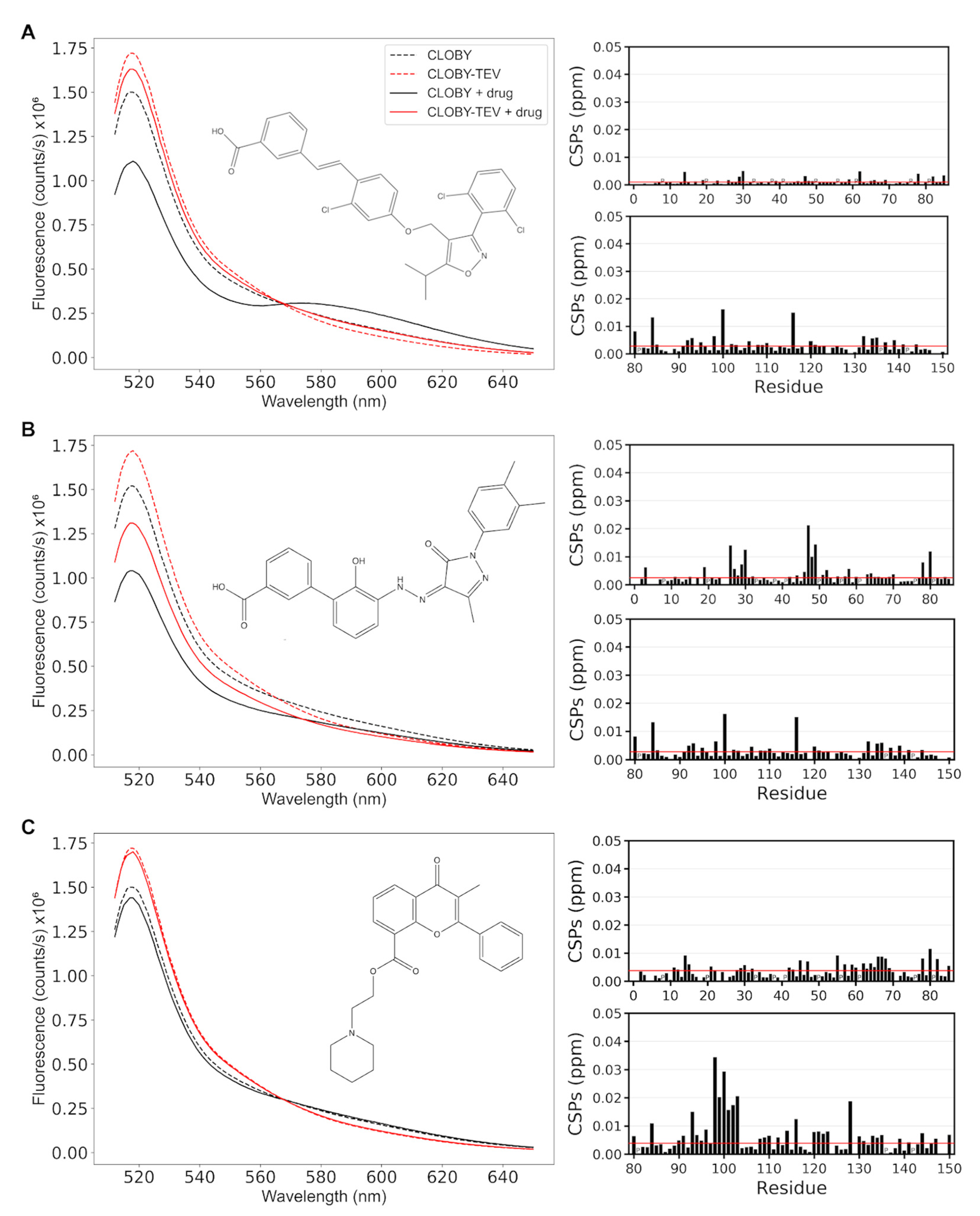
3.4. CLOBY Fluorescence Is Affected by Ligands Binding to the SNRE High-Throughput Screening of a Chemical Library Using the CLOBY Biosensor
3.5. NMR Validation of Screening Hits
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N. Intrinsically disordered proteins and their ‘Mysterious’ (meta)physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, D.; Tabaro, F.; Mičetić, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidović, R.; Dosztányi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D219–D227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhao, S.; Dunker, A.K. Intrinsically Disordered Proteins Link Alternative Splicing and Post-translational Modifications to Complex Cell Signaling and Regulation. J. Mol. Biol. 2018, 430, 2342–2359. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D 2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Iovanna, J.L.; Neira, J.L. Designing and repurposing drugs to target intrinsically disordered proteins for cancer treatment: Using NUPR1 as a paradigm. Mol. Cell. Oncol. 2019, 6, e1612678. [Google Scholar] [CrossRef]
- Kim, M.Y.; Na, I.; Kim, J.S.; Son, S.H.; Choi, S.; Lee, S.E.; Kim, J.H.; Jang, K.; Alterovitz, G.; Chen, Y.; et al. Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2. Sci. Adv. 2019, 5, eaav9810. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by SRC family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [Green Version]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [Green Version]
- Allgayer, H.; Boyd, D.D.; Heiss, M.M.; Abdalla, E.K.; Curley, S.A.; Gallick, G.E. Activation of src kinase in primary colorectal carcinoma: An indicator of poor clinical prognosis. Cancer 2002, 94, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Liu, Z.; Hu, H.-H.; Yang, Y.; Li, T.Y.; Lin, Z.-Z.; Ye, J.; Chen, J.; Huang, X.; Liu, D.-T.; et al. Proto-oncogene Src links lipogenesis via lipin-1 to breast cancer malignancy. Nat. Commun. 2020, 11, 5842. [Google Scholar] [CrossRef]
- Kuranami, S.; Yokobori, T.; Mogi, A.; Altan, B.; Yajima, T.; Onozato, R.; Azuma, Y.; Iijima, M.; Kosaka, T.; Kuwano, H. Src kinase-associated phosphoprotein2 expression is associated with poor prognosis in non-small cell lung cancer. Anticancer Res. 2015, 35, 2411–2415. [Google Scholar]
- Donahue, T.R.; Tran, L.M.; Hill, R.; Li, Y.; Kovochich, A.; Calvopina, J.H.; Patel, S.G.; Wu, N.; Hindoyan, A.; Farrell, J.J.; et al. Integrative survival-based molecular profiling of human pancreatic cancer. Clin. Cancer Res. 2012, 18, 1352–1363. [Google Scholar] [CrossRef] [Green Version]
- Arbesú, M.; Maffei, M.; Cordeiro, T.N.; Teixeira, J.M.C.; Pérez, Y.; Bernado, P.; Roche, S.; Pons, M. The Unique Domain Forms a Fuzzy Intramolecular Complex in Src Family Kinases. Structure 2017, 25, 630–640.e4. [Google Scholar] [CrossRef] [Green Version]
- Maffei, M.; Arbesú, M.; Le Roux, A.-L.; Amata, I.; Roche, S.; Pons, M. The SH3 domain acts as a scaffold for the N-terminal intrinsically disordered regions of c-Src. Structure 2015, 23, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016, 6, 20889. [Google Scholar] [CrossRef]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A guide to fluorescent protein FRET pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef]
- Rickles, R.J.; Botfield, M.C.; Zhou, X.M.; Henry, P.A.; Brugge, J.S.; Zoller, M.J. Phage display selection of ligand residues important for Src homology 3 domain binding specificity. Proc. Natl. Acad. Sci. USA 1995, 92, 10909–10913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roux, A.-L.; Mohammad, I.-L.; Mateos, B.; Arbesú, M.; Gairí, M.; Khan, F.A.; Teixeira, J.M.; Pons, M. A Myristoyl-Binding Site in the SH3 Domain Modulates c-Src Membrane Anchoring. iScience 2019, 12, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, J.M.C.; Skinner, S.P.; Arbesú, M.; Breeze, A.L.; Pons, M. Farseer-NMR: Automatic treatment, analysis and plotting of large, multi-variable NMR data. J. Biomol. NMR 2018, 71, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardot, R.; Viguier, G.; Pérez, J.; Ounsy, M. FOXTROT: A Java-Based Application to Reduce and Analyse SAXS and WAXS Piles of 2D data at Synchrotron SOLEIL. In Proceedings of the 8th canSAS Meeting, Tokai, Japan, 14–16 April 2005. [Google Scholar]
- Panjkovich, A.; Svergun, D.I. CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle X-ray scattering data. Bioinformatics 2018, 34, 1944–1946. [Google Scholar] [CrossRef]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Remington, S.J. Fluorescent proteins: Maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 2006, 16, 714–721. [Google Scholar] [CrossRef]
- Bernadó, P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 2010, 39, 769–780. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar] [CrossRef] [Green Version]
- Zimnicka, A.M.; Chen, Z.; Toth, P.T.; Minshall, R.D. Live-cell fret imaging of phosphorylation-dependent caveolin-1 switch. Methods Mol. Biol. 2020, 2169, 71–80. [Google Scholar]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Botvinick, E.L.; Zhao, Y.; Berns, M.; Usami, S.; Tsien, R.Y.; Chien, S. Visualizing the mechanical activation of Src. Nature 2005, 434, 1040–1045. [Google Scholar] [CrossRef]
- Koudelkova, L.; Pataki, A.C.; Tolde, O.; Pavlik, V.; Nobis, M.; Gemperle, J.; Anderson, K.; Brábek, J.; Rosel, D. Novel FRET-Based Src Biosensor Reveals Mechanisms of Src Activation and Its Dynamics in Focal Adhesions. Cell Chem. Biol. 2019, 26, 255–268.e4. [Google Scholar] [CrossRef]
- Liu, B.; Stone, O.J.; Pablo, M.; Herron, J.C.; Nogueira, A.T.; Dagliyan, O.; Grimm, J.B.; Lavis, L.D.; Elston, T.C.; Hahn, K.M. Biosensors based on peptide exposure show single molecule conformations in live cells. Cell 2021, 184, 5670–5685.e23. [Google Scholar] [CrossRef]
- Elia, N. Using unnatural amino acids to selectively label proteins for cellular imaging: A cell biologist viewpoint. FEBS J. 2021, 288, 1107–1117. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iruela, G.; Fernández, A.; Sagar, A.; Carvajal, F.J.; Bernadó, P.; Pons, M. A FRET-Based Biosensor for the Src N-Terminal Regulatory Element. Biosensors 2022, 12, 96. https://doi.org/10.3390/bios12020096
Iruela G, Fernández A, Sagar A, Carvajal FJ, Bernadó P, Pons M. A FRET-Based Biosensor for the Src N-Terminal Regulatory Element. Biosensors. 2022; 12(2):96. https://doi.org/10.3390/bios12020096
Chicago/Turabian StyleIruela, Guillermo, Alejandro Fernández, Amin Sagar, Francisco Javier Carvajal, Pau Bernadó, and Miquel Pons. 2022. "A FRET-Based Biosensor for the Src N-Terminal Regulatory Element" Biosensors 12, no. 2: 96. https://doi.org/10.3390/bios12020096
APA StyleIruela, G., Fernández, A., Sagar, A., Carvajal, F. J., Bernadó, P., & Pons, M. (2022). A FRET-Based Biosensor for the Src N-Terminal Regulatory Element. Biosensors, 12(2), 96. https://doi.org/10.3390/bios12020096