Integrating Biosensors in Organs-on-Chip Devices: A Perspective on Current Strategies to Monitor Microphysiological Systems





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
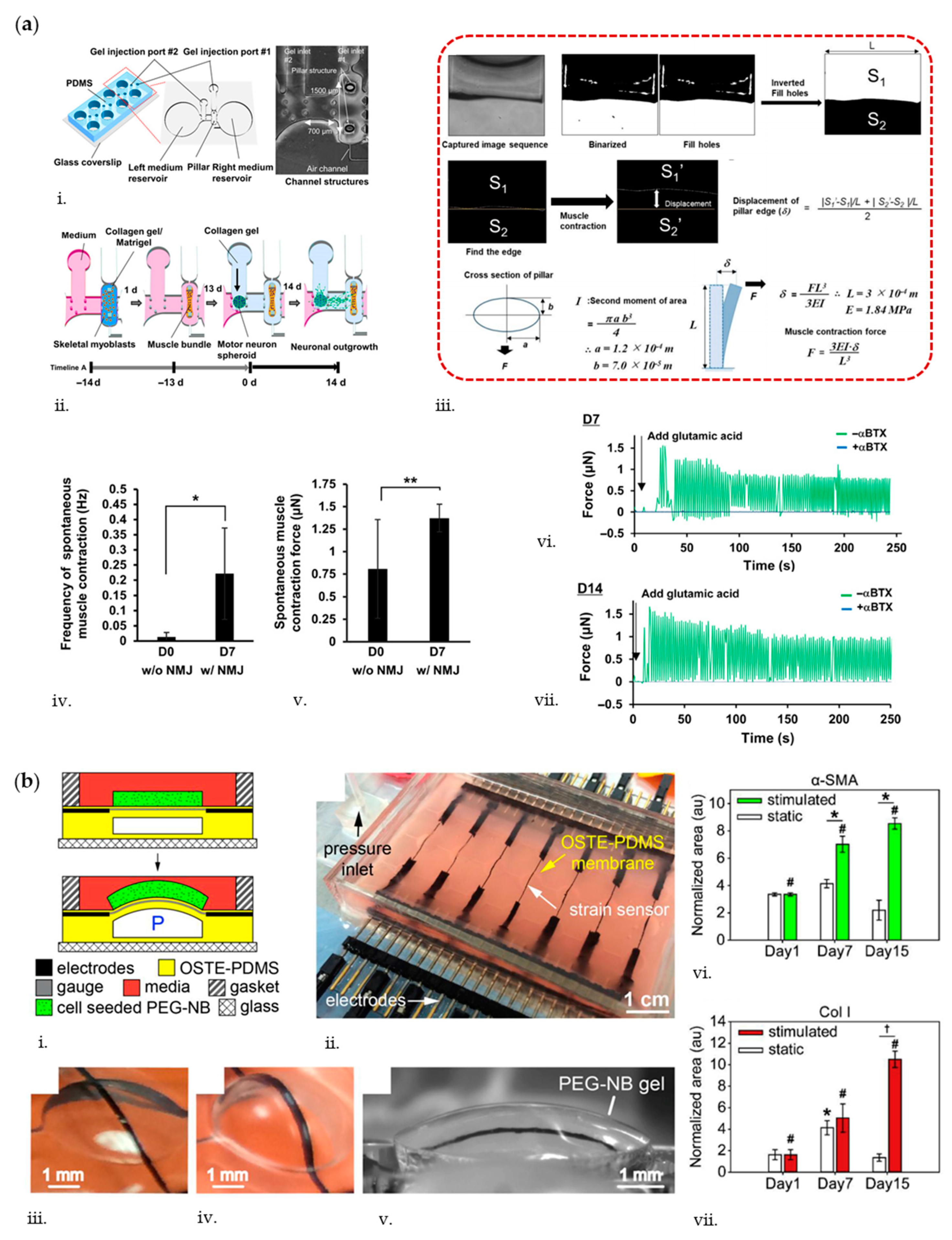{kind=link}
{kind=link}
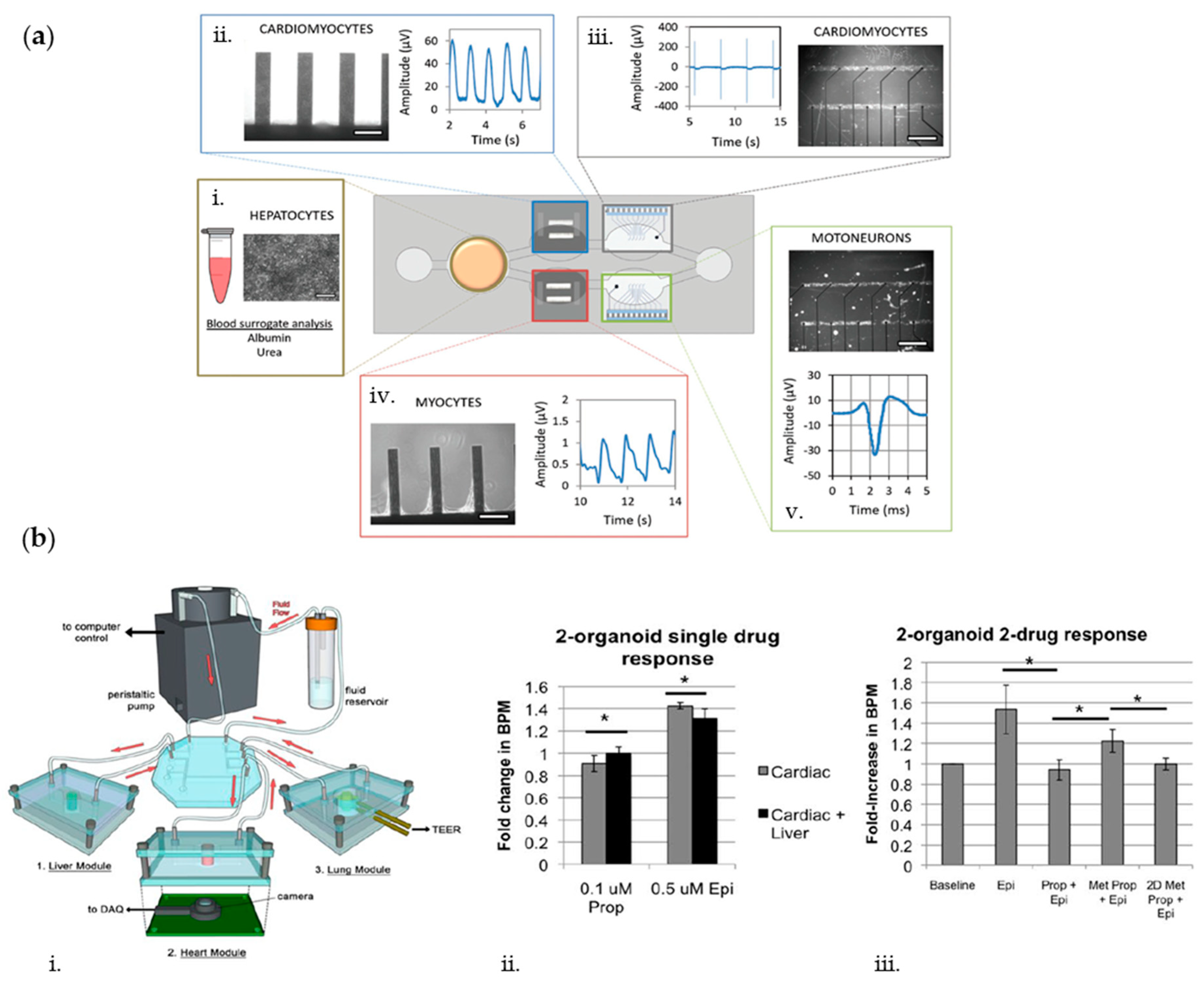{kind=link}
Abstract
1. Introduction
2. Biosensors for Measuring OoC’ Metabolic Activity
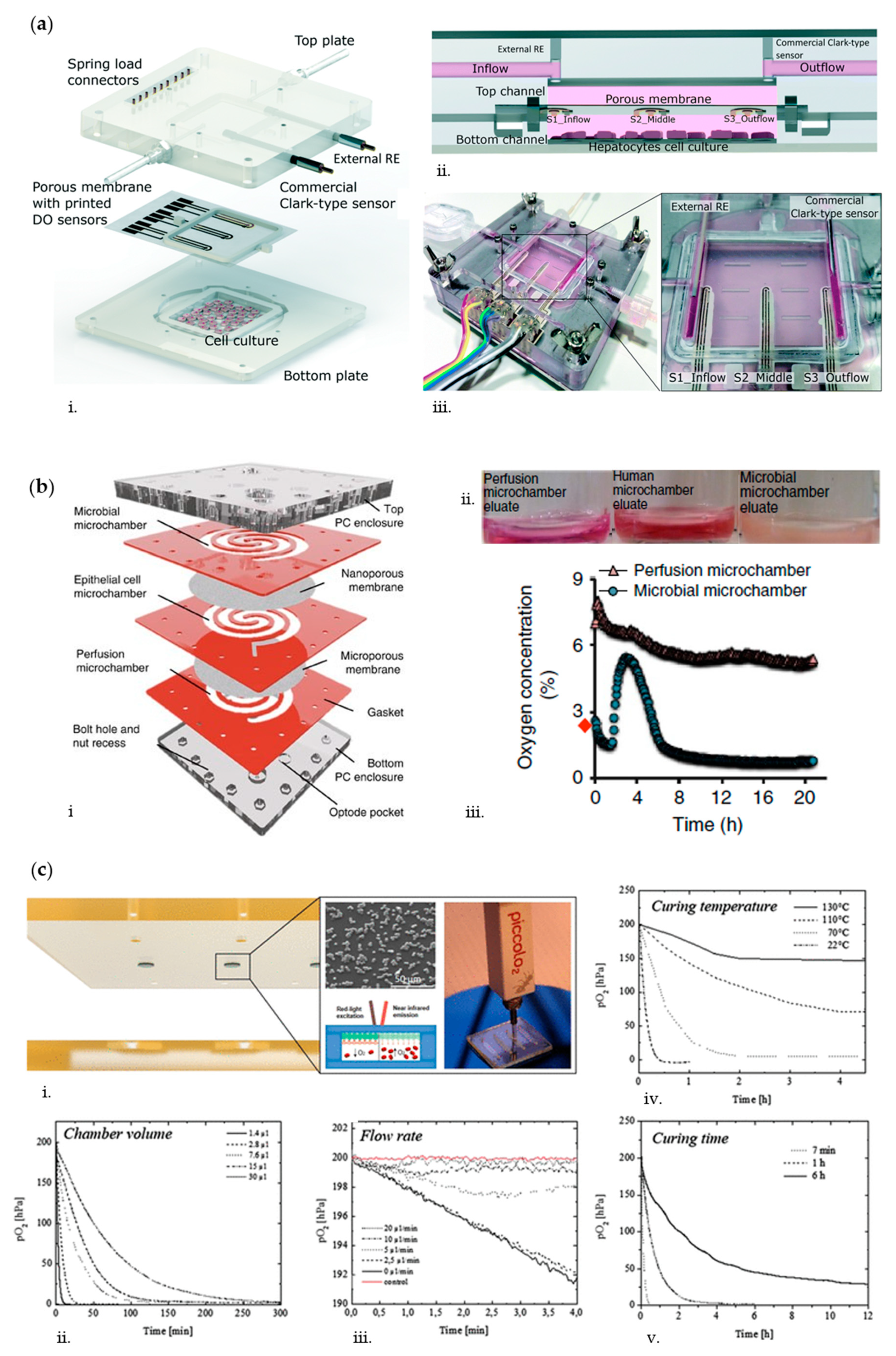
2.1. Oxygen
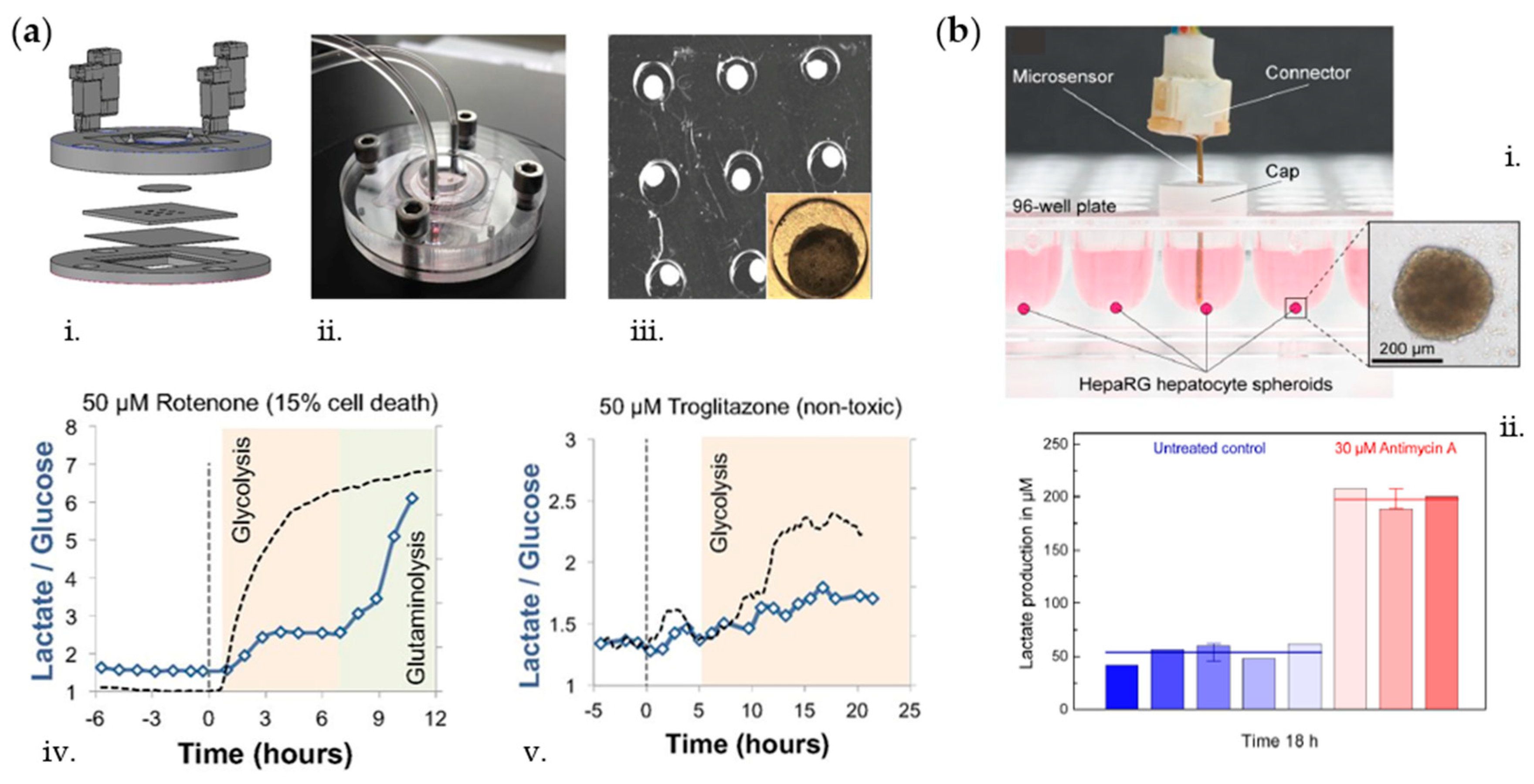
2.2. Glucose and Lactate
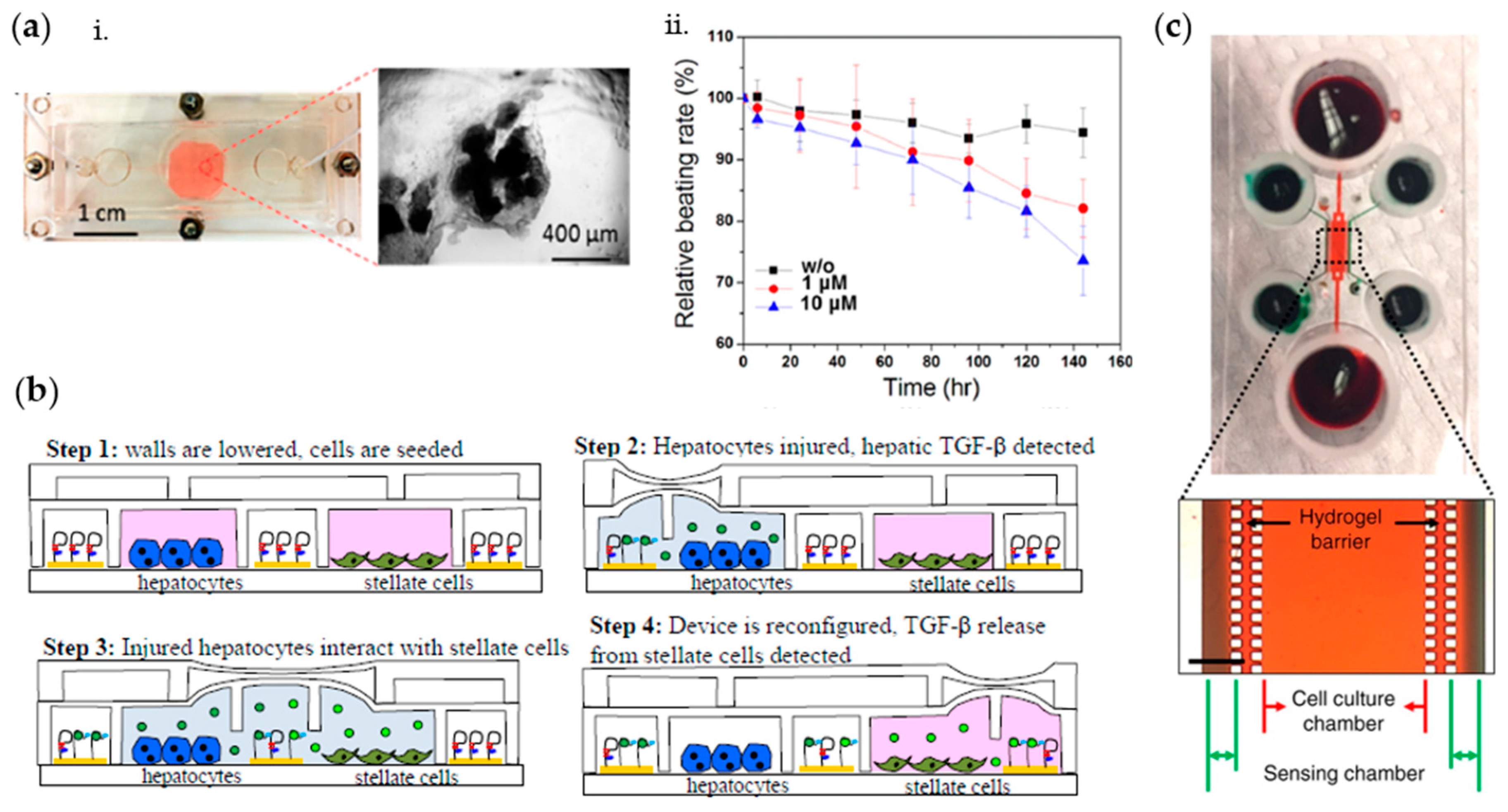
2.3. Cytokines and Other Metabolites
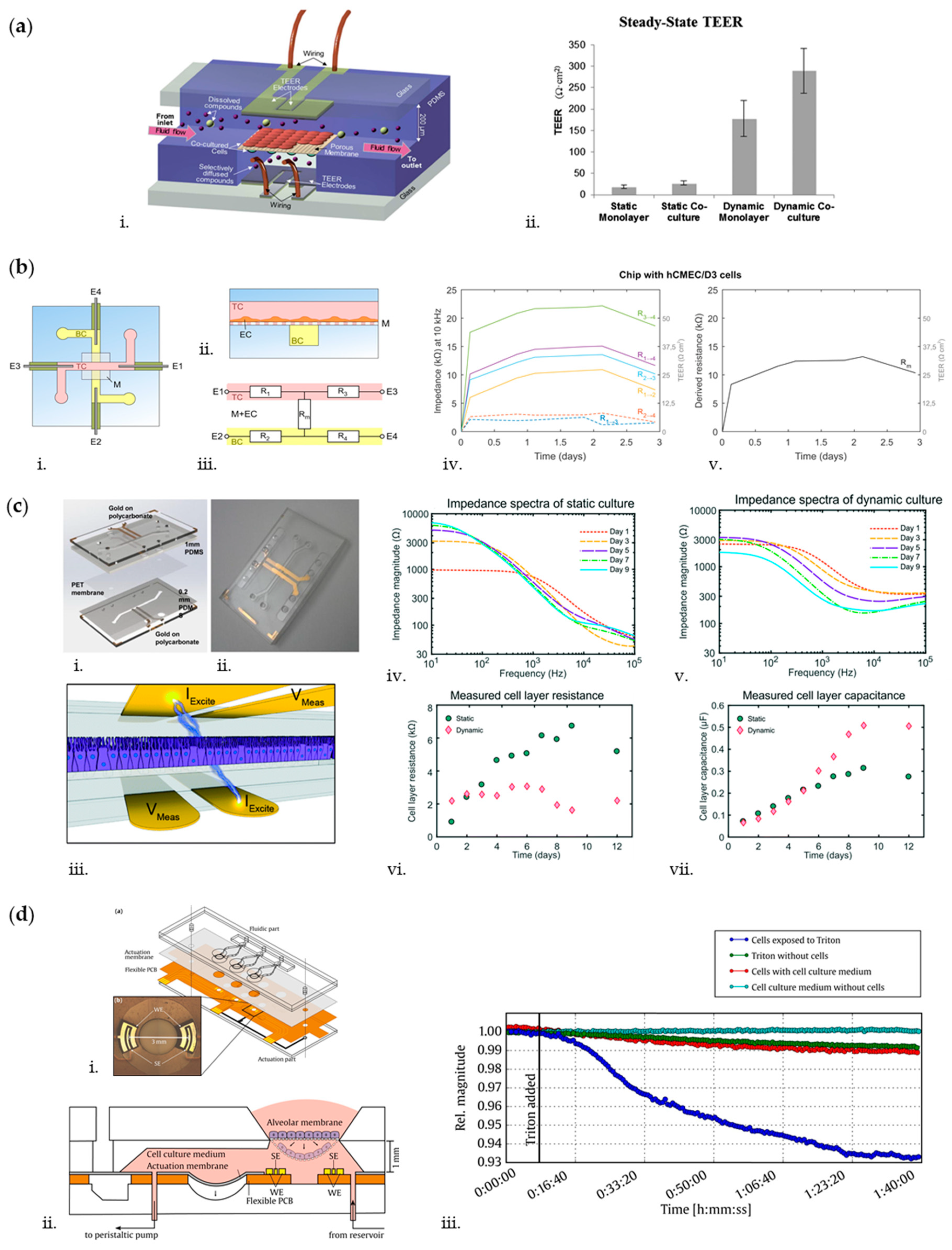
3. Biosensors for Measuring OoC’ Endothelial and Epithelial Barrier-Related Features
4. Biosensors for Measuring OoC’ Electrical Activity
5. Biosensors for Measuring OoC’ Mechanical Activity
6. Biosensors for Measuring OoC’ Electromechanical Activity
7. Multisensors for Analysis of Multiorgans-on-Chip
8. Outlook and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Caplin, J.D.; Granados, N.G.; James, M.R.; Montazami, R.; Hashemi, N. Microfluidic Organ-on-a-Chip Technology for Advancement of Drug Development and Toxicology. Adv. Healthc. Mater. 2015, 4, 1426–1450. [Google Scholar] [CrossRef] [PubMed]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef]
- Li, X.; Tian, T. Recent advances in an organ-on-a-chip: Biomarker analysis and applications. Anal. Methods 2018, 10, 3122–3130. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 6194. [Google Scholar] [CrossRef]
- Ribeiro, A.J.S.; Yang, X.; Patel, V.; Madabushi, R.; Strauss, D.G. Liver Microphysiological Systems for Predicting and Evaluating Drug Effects. Clin. Pharmacol. Ther. 2019, 106, 139–147. [Google Scholar] [CrossRef]
- Jorfi, M.; D’Avanzo, C.; Kim, D.Y.; Irimia, D. Three-Dimensional Models of the Human Brain Development and Diseases. Adv. Healthc. Mater. 2018, 7, 1–36. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 1–14. [Google Scholar] [CrossRef]
- Ugolini, G.S.; Cruz-Moreira, D.; Visone, R.; Redaelli, A.; Rasponi, M. Microfabricated physiological models for in vitro drug screening applications. Micromachines 2016, 7, 233. [Google Scholar] [CrossRef]
- Zheng, F.; Fu, F.; Cheng, Y.; Wang, C.; Zhao, Y.; Gu, Z. Organ-on-a-Chip Systems: Microengineering to Biomimic Living Systems. Small 2016, 12, 2253–2282. [Google Scholar] [CrossRef]
- Kodzius, R.; Schulze, F.; Gao, X.; Schneider, M.R. Organ-on-chip technology: Current state and future developments. Genes 2017, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From Three-Dimensional Cell Culture to Organs-on-Chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Hernández, J.E.; Villalba-Rodríguez, A.M.; Romero-Castillo, K.D.; Aguilar-Aguila-Isaías, M.A.; García-Reyes, I.E.; Hernández-Antonio, A.; Ahmed, I.; Sharma, A.; Parra-Saldívar, R.; Iqbal, H.M.N. Organs-on-a-Chip Module: A Review from the Development and Applications Perspective. Micromachines 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Radisic, M. Organ-on-A-chip devices advance to market. Lab Chip 2017, 17, 2395–2420. [Google Scholar] [CrossRef] [PubMed]
- Haddrick, M.; Simpson, P.B. Organ-on-a-chip technology: Turning its potential for clinical benefit into reality. Drug Discov. Today 2019, 24, 1217–1223. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Vunjak-Novakovic, G. Organs-on-a-Chip: A Fast Track for Engineered Human Tissues in Drug Development. Cell Stem Cell 2018, 22, 310–324. [Google Scholar] [CrossRef]
- Ishida, S. Organs-on-a-chip: Current applications and consideration points for in vitro ADME-Tox studies. Drug Metab. Pharmacokinet. 2018, 33, 49–54. [Google Scholar] [CrossRef]
- Rothbauer, M.; Rosser, J.M.; Zirath, H.; Ertl, P. Tomorrow today: Organ-on-a-chip advances towards clinically relevant pharmaceutical and medical in vitro models. Curr. Opin. Biotechnol. 2019, 55, 81–86. [Google Scholar] [CrossRef]
- Artzy-Schnirman, A.; Hobi, N.; Schneider-Daum, N.; Guenat, O.T.; Lehr, C.M.; Sznitman, J. Advanced in vitro lung-on-chip platforms for inhalation assays: From prospect to pipeline. Eur. J. Pharm. Biopharm. 2019, 144, 11–17. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Yuan Hsin, H.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 2016, 16, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Schneider, O.; Zeifang, L.; Fuchs, S.; Sailer, C.; Loskill, P. User-Friendly and Parallelized Generation of Human Induced Pluripotent Stem Cell-Derived Microtissues in a Centrifugal Heart-on-a-Chip. Tissue Eng. Part A 2019, 25, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Chen, Z.; Zhang, X.; Luo, Y.; Wu, Z.; Lu, Y.; Liu, T.; Zhao, W.; Lin, B. A liver-chip-based alcoholic liver disease model featuring multi-non-parenchymal cells. Biomed. Microdevices 2019, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-J.; Otieno, M.A.; Ronxhi, J.; Lim, H.-K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 2019, 11, eaax5516. [Google Scholar] [CrossRef] [PubMed]
- Jalili-Firoozinezhad, S.; Gazzaniga, F.S.; Calamari, E.L.; Camacho, D.M.; Fadel, C.W.; Bein, A.; Swenor, B.; Nestor, B.; Cronce, M.J.; Tovaglieri, A.; et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat. Biomed. Eng. 2019, 3, 520–531. [Google Scholar] [CrossRef]
- Beaurivage, C.; Naumovska, E.; Chang, Y.; Elstak, E.; Nicolas, A.; Wouters, H.; van Moolenbroek, G.; Lanz, H.; Trietsch, S.; Joore, J.; et al. Development of a Gut-on-a-Chip Model for High Throughput Disease Modeling and Drug Discovery. Int. J. Mol. Sci. 2019, 20, 5661. [Google Scholar] [CrossRef]
- Schutgens, F.; Rookmaaker, M.B.; Margaritis, T.; Rios, A.; Ammerlaan, C.; Jansen, J.; Gijzen, L.; Vormann, M.; Vonk, A.; Viveen, M.; et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 2019, 37, 303–313. [Google Scholar] [CrossRef]
- Jang, K.J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.M.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, 1–16. [Google Scholar] [CrossRef]
- Agrawal, G.; Aung, A.; Varghese, S. Skeletal muscle-on-a-chip: An in vitro model to evaluate tissue formation and injury. Lab Chip 2017, 17, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-P.; Deng, X.; Chen, D.-Y.; Zhu, D.; Tong, J.-L.; Zhao, T.; Ma, J.-H.; Liu, Y.-Q. A microfluidic chip-based co-culture of fibroblast-like synoviocytes with osteoblasts and osteoclasts to test bone erosion and drug evaluation. R. Soc. Open Sci. 2018, 5, 180528. [Google Scholar] [CrossRef] [PubMed]
- Sheyn, D.; Cohn-Yakubovich, D.; Ben-David, S.; De Mel, S.; Chan, V.; Hinojosa, C.; Wen, N.; Hamilton, G.A.; Gazit, D.; Gazit, Z. Bone-chip system to monitor osteogenic differentiation using optical imaging. Microfluid. Nanofluidics 2019, 23, 99. [Google Scholar] [CrossRef] [PubMed]
- Occhetta, P.; Mainardi, A.; Votta, E.; Vallmajo-Martin, Q.; Ehrbar, M.; Martin, I.; Barbero, A.; Rasponi, M. Hyperphysiological compression of articular cartilage induces an osteoarthritic phenotype in a cartilage-on-a-chip model. Nat. Biomed. Eng. 2019, 3, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, Z.; Li, E.N.; Li, X.; Del Duke, C.J.; Shen, H.; Hao, T.; O’Donnell, B.; Bunnell, B.A.; Goodman, S.B.; et al. Osteochondral Tissue Chip Derived From iPSCs: Modeling OA Pathologies and Testing Drugs. Front. Bioeng. Biotechnol. 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L.K. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab Chip 2017, 17, 448–459. [Google Scholar] [CrossRef]
- Ugolini, G.S.; Occhetta, P.; Saccani, A.; Re, F.; Krol, S.; Rasponi, M.; Redaelli, A. Design and validation of a microfluidic device for blood–brain barrier monitoring and transport studies. J. Micromech. Microeng. 2018, 28, 044001. [Google Scholar] [CrossRef]
- Iannielli, A.; Ugolini, G.S.; Cordiglieri, C.; Bido, S.; Rubio, A.; Colasante, G.; Valtorta, M.; Cabassi, T.; Rasponi, M.; Broccoli, V. Reconstitution of the Human Nigro-striatal Pathway on-a-Chip Reveals OPA1-Dependent Mitochondrial Defects and Loss of Dopaminergic Synapses. Cell Rep. 2019, 29, 4646–4656. [Google Scholar] [CrossRef]
- Taylor, A.M.; Dieterich, D.C.; Ito, H.T.; Kim, S.A.; Schuman, E.M. Microfluidic Local Perfusion Chambers for the Visualization and Manipulation of Synapses. Neuron 2010, 66, 57–68. [Google Scholar] [CrossRef]
- Visone, R.; Talò, G.; Occhetta, P.; Cruz-Moreira, D.; Lopa, S.; Pappalardo, O.A.; Redaelli, A.; Moretti, M.; Rasponi, M. A microscale biomimetic platform for generation and electro-mechanical stimulation of 3D cardiac microtissues. APL Bioeng. 2018, 2, 046102. [Google Scholar] [CrossRef]
- Occhetta, P.; Isu, G.; Lemme, M.; Conficconi, C.; Oertle, P.; Räz, C.; Visone, R.; Cerino, G.; Plodinec, M.; Rasponi, M.; et al. A three-dimensional: In vitro dynamic micro-tissue model of cardiac scar formation. Integr. Biol. 2018, 10, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Navaee, F.; Stradolini, F.; Renaud, P.; Carrara, S. Organs-on-chip monitoring: Sensors and other strategies. Microphysiol. Syst. 2018, 1, 1. [Google Scholar] [CrossRef]
- Modena, M.M.; Chawla, K.; Misun, P.M.; Hierlemann, A. Smart Cell Culture Systems: Integration of Sensors and Actuators into Microphysiological Systems. ACS Chem. Biol. 2018, 13, 1767–1784. [Google Scholar] [CrossRef] [PubMed]
- Kratz; Höll; Schuller; Ertl; Rothbauer Latest Trends in Biosensing for Microphysiological Organs-on-a-Chip and Body-on-a-Chip Systems. Biosensors 2019, 9, 110. [CrossRef]
- Kieninger, J.; Weltin, A.; Flamm, H.; Urban, G.A. Microsensor systems for cell metabolism-from 2D culture to organ-on-chip. Lab Chip 2018, 18, 1274–1291. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Águas, A.C.P.; Rainer, A.; Forte, G. Microfluidic organ/body-on-a-chip devices at the convergence of biology and microengineering. Sensors 2015, 15, 31142–31170. [Google Scholar] [CrossRef]
- Rothbauer, M.; Wartmann, D.; Charwat, V.; Ertl, P. Recent advances and future applications of microfluidic live-cell microarrays. Biotechnol. Adv. 2015, 33, 948–961. [Google Scholar] [CrossRef]
- Pires, N.M.M.; Dong, T.; Hanke, U.; Hoivik, N. Recent developments in optical detection technologies in lab-on-a-chip devices for biosensing applications. Sensors 2014, 14, 15458–15479. [Google Scholar] [CrossRef]
- Gruber, P.; Marques, M.P.C.; Szita, N.; Mayr, T. Integration and application of optical chemical sensors in microbioreactors. Lab Chip 2017, 17, 2693–2712. [Google Scholar] [CrossRef]
- Wang, T.; Green, R.; Nair, R.R.; Howell, M.; Mohapatra, S.; Guldiken, R.; Mohapatra, S.S. Surface acoustic waves (SAW)-based biosensing for quantification of cell growth in 2D and 3D Cultures. Sensors 2015, 15, 32045–32055. [Google Scholar] [CrossRef]
- Maoz, B.M.; Herland, A.; Henry, O.Y.F.; Leineweber, W.D.; Yadid, M.; Doyle, J.; Mannix, R.; Kujala, V.J.; Fitzgerald, E.A.; Parker, K.K.; et al. Organs-on-Chips with combined multi-electrode array and transepithelial electrical resistance measurement capabilities. Lab Chip 2017, 17, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Stauffer, F.; Simona, B.R.; Zhang, F.; Zhang, Z.M.; Huang, N.P.; Vörös, J. Multifunctional 3D electrode platform for real-time in situ monitoring and stimulation of cardiac tissues. Biosens. Bioelectron. 2018, 112, 149–155. [Google Scholar] [CrossRef] [PubMed]
- van de Wijdeven, R.; Ramstad, O.H.; Valderhaug, V.D.; Köllensperger, P.; Sandvig, A.; Sandvig, I.; Halaas, Ø. A novel lab-on-chip platform enabling axotomy and neuromodulation in a multi-nodal network. Biosens. Bioelectron. 2019, 140, 111329. [Google Scholar] [CrossRef]
- Caluori, G.; Pribyl, J.; Pesl, M.; Jelinkova, S.; Rotrekl, V.; Skladal, P.; Raiteri, R. Non-invasive electromechanical cell-based biosensors for improved investigation of 3D cardiac models. Biosens. Bioelectron. 2019, 124–125, 129–135. [Google Scholar] [CrossRef]
- Liu, H.; MacQueen, L.A.; Usprech, J.F.; Maleki, H.; Sider, K.L.; Doyle, M.G.; Sun, Y.; Simmons, C.A. Microdevice arrays with strain sensors for 3D mechanical stimulation and monitoring of engineered tissues. Biomaterials 2018, 172, 30–40. [Google Scholar] [CrossRef]
- Zirath, H.; Rothbauer, M.; Spitz, S.; Bachmann, B.; Jordan, C.; Müller, B.; Ehgartner, J.; Priglinger, E.; Mühleder, S.; Redl, H.; et al. Every breath you take: Non-invasive real-time oxygen biosensing in two-and three-dimensional microfluidic cell models. Front. Physiol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Rennert, K.; Steinborn, S.; Gröger, M.; Ungerböck, B.; Jank, A.-M.; Ehgartner, J.; Nietzsche, S.; Dinger, J.; Kiehntopf, M.; Funke, H.; et al. A microfluidically perfused three dimensional human liver model. Biomaterials 2015, 71, 119–131. [Google Scholar] [CrossRef]
- Moya, A.; Ortega-Ribera, M.; Guimerà, X.; Sowade, E.; Zea, M.; Illa, X.; Ramon, E.; Villa, R.; Gracia-Sancho, J.; Gabriel, G. Online oxygen monitoring using integrated inkjet-printed sensors in a liver-on-a-chip system. Lab Chip 2018, 18, 2023–2035. [Google Scholar] [CrossRef]
- Weltin, A.; Slotwinski, K.; Kieninger, J.; Moser, I.; Jobst, G.; Wego, M.; Ehret, R.; Urban, G.A. Cell culture monitoring for drug screening and cancer research: A transparent, microfluidic, multi-sensor microsystem. Lab Chip 2014, 14, 138–146. [Google Scholar] [CrossRef]
- Misun, P.M.; Rothe, J.; Schmid, Y.R.F.; Hierlemann, A.; Frey, O. Multi-analyte biosensor interface for real-time monitoring of 3D microtissue spheroids in hanging-drop networks. Microsyst. Nanoeng. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fernández-Garibay, X.; Castaño, A.G.; De Chiara, F.; Hernández-Albors, A.; Balaguer-Trias, J.; Ramón-Azcón, J. Muscle-on-a-chip with an on-site multiplexed biosensing system for: In situ monitoring of secreted IL-6 and TNF-α. Lab Chip 2019, 19, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Young, A.T.; Rivera, K.R.; Erb, P.D.; Daniele, M.A. Monitoring of Microphysiological Systems: Integrating Sensors and Real-Time Data Analysis toward Autonomous Decision-Making. ACS Sens. 2019, 4, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; Hafny-Rahbi, B.E.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Klimant, I.; Wolfbeis, O.S. Oxygen-Sensitive Luminescent Materials Based on Silicone-Soluble Ruthenium Diimine Complexes. Anal. Chem. 1995, 67, 3160–3166. [Google Scholar] [CrossRef]
- Oomen, P.E.; Skolimowski, M.D.; Verpoorte, E. Implementing oxygen control in chip-based cell and tissue culture systems. Lab Chip 2016, 16, 3394–3414. [Google Scholar] [CrossRef]
- Ronkainen, N.J.; Halsall, H.B.; Heineman, W.R. Electrochemical biosensors. Chem. Soc. Rev. 2010, 39, 1747–1763. [Google Scholar] [CrossRef]
- Alborzinia, H.; Can, S.; Holenya, P.; Scholl, C.; Lederer, E.; Kitanovic, I.; Wölfl, S. Real-Time Monitoring of Cisplatin-Induced Cell Death. PLoS ONE 2011, 6, e19714. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta-Bioenerg. 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Bavli, D.; Prill, S.; Ezra, E.; Levy, G.; Cohen, M.; Vinken, M.; Vanfleteren, J.; Jaeger, M.; Nahmias, Y. Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E2231–E2240. [Google Scholar] [CrossRef]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Ahmad, J.; Farshori, N.N.; Saquib, Q.; Jahan, S.; Kashyap, M.P.; Ahamed, M.; Musarrat, J.; Al-Khedhairy, A.A. Rotenone-induced oxidative stress and apoptosis in human liver HepG2 cells. Mol. Cell. Biochem. 2013, 384, 59–69. [Google Scholar] [CrossRef]
- Julie, N.L.; Julie, I.M.; Kende, A.I.; Wilson, G.L. Mitochondrial dysfunction and delayed hepatotoxicity: Another lesson from troglitazone. Diabetologia 2008, 51, 2108–2116. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic. Biol. Med. 2013, 55, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Fritz, J.V.; Glaab, E.; Desai, M.S.; Greenhalgh, K.; Frachet, A.; Niegowska, M.; Estes, M.; Jäger, C.; Seguin-Devaux, C.; et al. A microfluidics-based in vitro model of the gastrointestinal human–microbe interface. Nat. Commun. 2016, 7, 11535. [Google Scholar] [CrossRef] [PubMed]
- Mauleon, G.; Fall, C.P.; Eddington, D.T. Precise Spatial and Temporal Control of Oxygen within In Vitro Brain Slices via Microfluidic Gas Channels. PLoS ONE 2012, 7, e43309. [Google Scholar] [CrossRef]
- Bonnekoh, P.; Kuroiwa, T.; Oschlies, U.; Hossmann, K.A. Barbiturate promotes post-ischemic reaggregation of polyribosomes in gerbil hippocampus. Neurosci. Lett. 1992, 146, 75–78. [Google Scholar] [CrossRef]
- Sticker, D.; Rothbauer, M.; Ehgartner, J.; Steininger, C.; Liske, O.; Liska, R.; Neuhaus, W.; Mayr, T.; Haraldsson, T.; Kutter, J.P.; et al. Oxygen Management at the Microscale: A Functional Biochip Material with Long-Lasting and Tunable Oxygen Scavenging Properties for Cell Culture Applications. ACS Appl. Mater. Interfaces 2019, 11, 9730–9739. [Google Scholar] [CrossRef]
- Carioscia, J.A.; Stansbury, J.W.; Bowman, C.N. Evaluation and Control of Thiol-ene/Thiol-epoxy Hybrid Networks. Polymer 2007, 23, 1–7. [Google Scholar]
- Brennan, M.D.; Rexius-Hall, M.L.; Elgass, L.J.; Eddington, D.T. Oxygen control with microfluidics. Lab Chip 2014, 14, 4305–4318. [Google Scholar] [CrossRef]
- Weltin, A.; Hammer, S.; Noor, F.; Kaminski, Y.; Kieninger, J.; Urban, G.A. Accessing 3D microtissue metabolism: Lactate and oxygen monitoring in hepatocyte spheroids. Biosens. Bioelectron. 2017, 87, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yu, P.; Hao, J.; Wang, Y.; Ohsaka, T.; Mao, L. Continuous and simultaneous electrochemical measurements of glucose, lactate, and ascorbate in rat brain following brain ischemia. Anal. Chem. 2014, 86, 3895–3901. [Google Scholar] [CrossRef] [PubMed]
- Dervisevic, E.; Tuck, K.L.; Voelcker, N.H.; Cadarso, V.J. Recent Progress in Lab-On-a-Chip Systems for the Monitoring of Metabolites for Mammalian and Microbial Cell Research. Sensors 2019, 19, 5027. [Google Scholar] [CrossRef] [PubMed]
- Bange, A.; Halsall, H.B.; Heineman, W.R. Microfluidic immunosensor systems. Biosens. Bioelectron. 2005, 20, 2488–2503. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.A.; Ahmed, M.U. Electrochemical immunosensors and their recent nanomaterial-based signal amplification strategies: A review. RSC Adv. 2016, 6, 24995–25014. [Google Scholar] [CrossRef]
- Stenken, J.A.; Poschenrieder, A.J. Bioanalytical Chemistry of Cytokines-A Review. Anal. Chim. Acta 2015, 853, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Qi, M.; Hutchinson, M.R.; Yang, G.; Goldys, E.M. Recent advances in cytokine detection by immunosensing. Biosens. Bioelectron. 2016, 79, 810–821. [Google Scholar] [CrossRef]
- Li, X.; Soler, M.; Özdemir, C.I.; Belushkin, A.; Yesilköy, F.; Altug, H. Plasmonic nanohole array biosensor for label-free and real-time analysis of live cell secretion. Lab Chip 2017, 17, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.R.; Kilic, T.; Zhang, Y.S.; Avci, H.; Hu, N.; Kim, D.; Branco, C.; Aleman, J.; Massa, S.; Silvestri, A.; et al. Label-Free and Regenerative Electrochemical Microfluidic Biosensors for Continual Monitoring of Cell Secretomes. Adv. Sci. 2017, 4, 1600522. [Google Scholar] [CrossRef] [PubMed]
- Baganizi, D.R.; Leroy, L.; Laplatine, L.; Fairley, S.J.; Heidmann, S.; Menad, S.; Livache, T.; Marche, P.N.; Roupioz, Y. A Simple microfluidic platform for long-term analysis and continuous dual-imaging detection of T-Cell secreted IFN-γ and IL-2 on antibody-based biochip. Biosensors 2015, 5, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.R.; Zhang, Y.S.; Kim, D.J.; Manbohi, A.; Avci, H.; Silvestri, A.; Aleman, J.; Hu, N.; Kilic, T.; Keung, W.; et al. Aptamer-Based Microfluidic Electrochemical Biosensor for Monitoring Cell-Secreted Trace Cardiac Biomarkers. Anal. Chem. 2016, 88, 10019–10027. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Patel, D.; Kwa, T.; Haque, A.; Matharu, Z.; Stybayeva, G.; Gao, Y.; Diehl, A.M.; Revzin, A. Liver injury-on-a-chip: Microfluidic co-cultures with integrated biosensors for monitoring liver cell signaling during injury. Lab Chip 2015, 15, 4467–4478. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Matharu, Z.; Rahimian, A.; Revzin, A. Detecting multiple cell-secreted cytokines from the same aptamer-functionalized electrode. Biosens. Bioelectron. 2015, 64, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Son, K.J.; Gheibi, P.; Stybayeva, G.; Rahimian, A.; Revzin, A. Detecting cell-secreted growth factors in microfluidic devices using bead-based biosensors. Microsyst. Nanoeng. 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Riahi, R.; Shaegh, S.A.M.; Ghaderi, M.; Zhang, Y.S.; Shin, S.R.; Aleman, J.; Massa, S.; Kim, D.; Dokmeci, M.R.; Khademhosseini, A. Automated microfluidic platform of bead-based electrochemical immunosensor integrated with bioreactor for continual monitoring of cell secreted biomarkers. Sci. Rep. 2016, 6, 24598. [Google Scholar] [CrossRef]
- Wong, J.F.; Simmons, C.A. Microfluidic assay for the on-chip electrochemical measurement of cell monolayer permeability. Lab Chip 2019, 19, 1060–1070. [Google Scholar] [CrossRef]
- Matter, K.; Balda, M.S. Functional analysis of tight junctions. Methods 2003, 30, 228–234. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Vargas-Osorio, Z.; Da Silva-Candal, A.; Piñeiro, Y.; Iglesias-Rey, R.; Sobrino, T.; Campos, F.; Castillo, J.; Rivas, J. Multifunctional Superparamagnetic Stiff Nanoreservoirs for Blood Brain Barrier Applications. Nanomaterials 2019, 9, 449. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol.-Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Nishihara, H.; Kanda, T. Blood–brain barrier dysfunction in immuno-mediated neurological diseases. Immunol. Med. 2018, 41, 120–128. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Merali, Z.; Huang, K.; Mikulis, D.; Silver, F.; Kassner, A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS ONE 2017, 12, e0171558. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T.; Takahashi, T.; Nakashima, I. Oligoclonal bands and periventricular lesions in multiple sclerosis will not increase blood-brain barrier permeability. J. Neurol. Sci. 2018, 387, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, S.; Zheng, J.; Li, Y.; Huang, H. Recent progress in microfluidic models of the blood-brain barrier. Micromachines 2019, 10, 375. [Google Scholar] [CrossRef]
- Heidari, H.; Taylor, H. Review Article: Capturing the physiological complexity of the brain’s neuro-vascular unit in vitro. Biomicrofluidics 2018, 12, 051502. [Google Scholar] [CrossRef]
- Booth, R.; Kim, H. Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB). Lab Chip 2012, 12, 1784–1792. [Google Scholar] [CrossRef]
- Cucullo, L.; Hossain, M.; Puvenna, V.; Marchi, N.; Janigro, D. The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci. 2011, 12, 40. [Google Scholar] [CrossRef]
- Vastag, M.; Keseru, G. Current in vitro and in silico models of blood-brain barrier penetration: A practical view. Curr. Opin. Drug Discov. Dev. 2009, 12, 115–124. [Google Scholar]
- Kim, S.; Buonocore, J.; Park, J.; Welsh, C.J.; Li, J.; Han, A. A Three-Dimensional Arrayed Microfluidic Blood-Brain Barrier Model With Integrated Electrical Sensor Array. IEEE Trans. Biomed. Eng. 2018, 65, 431–439. [Google Scholar]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic blood–brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Griep, L.M.; Wolbers, F.; De Wagenaar, B.; Ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; Van Der Meer, A.D.; Van Den Berg, A. BBB on CHIP: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Khafagy, E.S.; Khanafer, K.; Takayama, S.; Elsayed, M.E.H. Organization of Endothelial Cells, Pericytes, and Astrocytes into a 3D Microfluidic in Vitro Model of the Blood-Brain Barrier. Mol. Pharm. 2016, 13, 895–906. [Google Scholar] [CrossRef]
- van der Helm, M.W.; Odijk, M.; Frimat, J.-P.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L.I. Direct quantification of transendothelial electrical resistance in organs-on-chips. Biosens. Bioelectron. 2016, 85, 924–929. [Google Scholar] [CrossRef]
- van der Helm, M.W.; Odijk, M.; Frimat, J.P.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L.I. Fabrication and validation of an organ-on-chip system with integrated electrodes to directly quantify transendothelial electrical resistance. J. Vis. Exp. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef]
- Henry, O.Y.F.; Villenave, R.; Cronce, M.J.; Leineweber, W.D.; Benz, M.A.; Ingber, D.E. Organs-on-chips with integrated electrodes for trans-epithelial electrical resistance (TEER) measurements of human epithelial barrier function. Lab Chip 2017, 17, 2264–2271. [Google Scholar] [CrossRef]
- van der Helm, M.W.; Henry, O.Y.F.; Bein, A.; Hamkins-Indik, T.; Cronce, M.J.; Leineweber, W.D.; Odijk, M.; van der Meer, A.D.; Segerink, L.I.; Ingber, D.E. Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy. Lab Chip 2019, 19, 452–463. [Google Scholar] [CrossRef]
- Odijk, M.; Van Der Meer, A.D.; Levner, D.; Kim, H.J.; Van Der Helm, M.W.; Segerink, L.I.; Frimat, J.P.; Hamilton, G.A.; Ingber, D.E.; Van Den Berg, A. Measuring direct current trans-epithelial electrical resistance in organ-on-a-chip microsystems. Lab Chip 2015, 15, 745–752. [Google Scholar] [CrossRef]
- Mermoud, Y.; Felder, M.; Stucki, J.D.; Stucki, A.O.; Guenat, O.T. Microimpedance tomography system to monitor cell activity and membrane movements in a breathing lung-on-chip. Sens. Actuators B Chem. 2018, 255, 3647–3653. [Google Scholar] [CrossRef]
- Wegener, J.; Seebach, J. Experimental tools to monitor the dynamics of endothelial barrier function: A survey of in vitro approaches. Cell Tissue Res. 2014, 355, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.J.; Gillette, M.U. New perspectives on neuronal development via microfluidic environments. Trends Neurosci. 2012, 35, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Vahidi, B.; Taylor, A.M.; Rhee, S.W.; Jeon, N.L. Microfluidic culture platform for neuroscience research. Nat. Protoc. 2006, 1, 2128–2136. [Google Scholar] [CrossRef]
- Jadhav, D.A.; Wei, L.; Shi, P. Compartmentalized Platforms for Neuro-Pharmacological Research. Curr. Neuropharmacol. 2015, 14, 72–86. [Google Scholar] [CrossRef]
- Taylor, A.M.; Rhee, S.W.; Tu, C.H.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. Microfluidic multicompartment device for neuroscience research. Langmuir 2003, 19, 1551–1556. [Google Scholar] [CrossRef]
- Taylor, A.M.; Cotman, C.W.; Cribbs, D.H.; Blurton-Jones, M.; Jeon, N.L.; Rhee, S.W. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.; Park, S.I.; Choe, Y.; Li, J.; Han, A. A microchip for quantitative analysis of CNS axon growth under localized biomolecular treatments. J. Neurosci. Methods 2014, 221, 166–174. [Google Scholar] [CrossRef]
- Kilinc, D.; Peyrin, J.M.; Soubeyre, V.; Magnifico, S.; Saias, L.; Viovy, J.L.; Brugg, B. Wallerian-like degeneration of central neurons after synchronized and geometrically registered mass axotomy in a three-compartmental microfluidic chip. Neurotox. Res. 2011, 19, 149–161. [Google Scholar] [CrossRef]
- Hosmane, S.; Fournier, A.; Wright, R.; Rajbhandari, L.; Siddique, R.; Yang, I.H.; Ramesh, K.T.; Venkatesan, A.; Thakor, N. Valve-based microfluidic compression platform: Single axon injury and regrowth. Lab Chip 2011, 11, 3888–3895. [Google Scholar] [CrossRef]
- Lu, X.; Kim-Han, J.S.; O’Malley, K.L.; Sakiyama-Elbert, S.E. A microdevice platform for visualizing mitochondrial transport in aligned dopaminergic axons. J. Neurosci. Methods 2012, 209, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Virlogeux, A.; Moutaux, E.; Christaller, W.; Genoux, A.; Bruyère, J.; Fino, E.; Charlot, B.; Cazorla, M.; Saudou, F. Reconstituting Corticostriatal Network on-a-Chip Reveals the Contribution of the Presynaptic Compartment to Huntington’s Disease. Cell Rep. 2018, 22, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Biffi, E.; Piraino, F.; Pedrocchi, A.; Fiore, G.B.; Ferrigno, G.; Redaelli, A.; Menegon, A.; Rasponi, M. A microfluidic platform for controlled biochemical stimulation of twin neuronal networks. Biomicrofluidics 2012, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Habibey, R.; Latifi, S.; Mousavi, H.; Pesce, M.; Arab-Tehrany, E.; Blau, A. A multielectrode array microchannel platform reveals both transient and slow changes in axonal conduction velocity. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gladkov, A.; Pigareva, Y.; Kutyina, D.; Kolpakov, V.; Bukatin, A.; Mukhina, I.; Kazantsev, V.; Pimashkin, A. Design of Cultured Neuron Networks in vitro with Predefined Connectivity Using Asymmetric Microfluidic Channels. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Moutaux, E.; Charlot, B.; Genoux, A.; Saudou, F.; Cazorla, M. An integrated microfluidic/microelectrode array for the study of activity-dependent intracellular dynamics in neuronal networks. Lab Chip 2018, 18, 3425–3435. [Google Scholar] [CrossRef]
- Shen, X.; Wu, J.; Wang, Z.; Chen, T. Characterization of in vitro neural functional connectivity on a neurofluidic device. Electrophoresis 2019, 40, 2996–3004. [Google Scholar] [CrossRef]
- Morin, F.; Nishimura, N.; Griscom, L.; LePioufle, B.; Fujita, H.; Takamura, Y.; Tamiya, E. Constraining the connectivity of neuronal networks cultured on microelectrode arrays with microfluidic techniques: A step towards neuron-based functional chips. Biosens. Bioelectron. 2006, 21, 1093–1100. [Google Scholar] [CrossRef]
- Pine, J. Recording action potentials from cultured neurons with extracellular microcircuit electrodes. J. Neurosci. Methods 1980, 2, 19–31. [Google Scholar] [CrossRef]
- Gross, G.W. Simultaneous Single Unit Recording in vitro with a Photoetched Laser Deinsulated Gold Multimicroelectrode Surface. IEEE Trans. Biomed. Eng. 1979, BME-26, 273–279. [Google Scholar] [CrossRef]
- Perelman, Y.; Ginosar, R. An integrated system for multichannel neuronal recording with spike/LFP separation, integrated A/D conversion and threshold detection. IEEE Trans. Biomed. Eng. 2007, 54, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Shimba, K.; Sakai, K.; Isomura, T.; Kotani, K.; Jimbo, Y. Axonal conduction slowing induced by spontaneous bursting activity in cortical neurons cultured in a microtunnel device. Integr. Biol. 2015, 7, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Riss, M.; Buitrago, J.O.; Claverol-Tinturé, E. Biophysics of microchannel-enabled neuron–electrode interfaces. J. Neural Eng. 2012, 9, 026010. [Google Scholar] [CrossRef] [PubMed]
- Hong, N.; Joo, S.; Nam, Y. Characterization of axonal spikes in cultured neuronal networks using microelectrode arrays and microchannel devices. IEEE Trans. Biomed. Eng. 2017, 64, 492–498. [Google Scholar] [CrossRef]
- Pedraza, E.; Karajić, A.; Raoux, M.; Perrier, R.; Pirog, A.; Lebreton, F.; Arbault, S.; Gaitan, J.; Renaud, S.; Kuhn, A.; et al. Guiding pancreatic beta cells to target electrodes in a whole-cell biosensor for diabetes. Lab Chip 2015, 15, 3880–3890. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Braun, M. Regulation of Insulin Secretion in Human Pancreatic Islets. Annu. Rev. Physiol. 2013, 75, 155–179. [Google Scholar] [CrossRef]
- Koutsouras, D.A.; Perrier, R.; Villarroel Marquez, A.; Pirog, A.; Pedraza, E.; Cloutet, E.; Renaud, S.; Raoux, M.; Malliaras, G.G.; Lang, J. Simultaneous monitoring of single cell and of micro-organ activity by PEDOT:PSS covered multi-electrode arrays. Mater. Sci. Eng. C 2017, 81, 84–89. [Google Scholar] [CrossRef]
- Lebreton, F.; Pirog, A.; Belouah, I.; Bosco, D.; Berney, T.; Meda, P.; Bornat, Y.; Catargi, B.; Renaud, S.; Raoux, M.; et al. Slow potentials encode intercellular coupling and insulin demand in pancreatic beta cells. Diabetologia 2015, 58, 1291–1299. [Google Scholar] [CrossRef]
- Raoux, M.; Bontorin, G.; Bornat, Y.; Lang, J.; Renaud, S. Bioelectronic Sensing of Insulin Demand. In Biohybrid Systems; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 191–202. [Google Scholar]
- Raoux, M.; Bornat, Y.; Quotb, A.; Catargi, B.; Renaud, S.; Lang, J. Non-invasive long-term and real-time analysis of endocrine cells on micro-electrode arrays. J. Physiol. 2012, 590, 1085–1091. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Trauner, D.; Rutter, G.A.; Hodson, D.J.; Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose Article Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef]
- Renaud, S.; Catargi, B.; Lang, J. Biosensors in Diabetes: How to get the most out of evolution and transpose it into a signal. IEEE Pulse 2014, 5, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Perrier, R.; Pirog, A.; Jaffredo, M.; Gaitan, J.; Catargi, B.; Renaud, S.; Raoux, M.; Lang, J. Bioelectronic organ-based sensor for microfluidic real-time analysis of the demand in insulin. Biosens. Bioelectron. 2018, 117, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, R.; Sherratt, M.J.; Cruickshank, J.K.; Derby, B. Characterizing the elastic properties of tissues. Mater Today 2012, 14, 96–105. [Google Scholar] [CrossRef]
- Palchesko, R.N.; Zhang, L.; Sun, Y.; Feinberg, A.W. Development of Polydimethylsiloxane Substrates with Tunable Elastic Modulus to Study Cell Mechanobiology in Muscle and Nerve. PLoS ONE 2012, 7, e51499. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Singh, S.P.; Schwartz, M.P.; Tokuda, E.Y.; Luo, Y.; Rogers, R.E.; Fujita, M.; Ahn, N.G.; Anseth, K.S. A synthetic modular approach for modeling the role of the 3D microenvironment in tumor progression. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Lin, C.H.; Wang, C.K.; Chen, Y.A.; Peng, C.C.; Liao, W.H.; Tung, Y.C. Measurement of in-plane elasticity of live cell layers using a pressure sensor embedded microfluidic device. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Tandon, N.; Cannizzaro, C.; Chao, P.P.-H.G.; Maidhof, R.; Marsano, A.; Au, H.T.H.; Radisic, M.; Vunjak-Novakovic, G. Electrical stimulation systems for cardiac tissue engineering. Nat. Protoc. 2009, 4, 155–173. [Google Scholar] [CrossRef]
- Feinberg, A.W.; Alford, P.W.; Jin, H.; Ripplinger, C.M.; Werdich, A.A.; Sheehy, S.P.; Grosberg, A.; Parker, K.K. Controlling the contractile strength of engineered cardiac muscle by hierarchal tissue architecture. Biomaterials 2012, 23, 5732–5741. [Google Scholar] [CrossRef]
- Wang, Y.I.; Oleaga, C.; Long, C.J.; Esch, M.B.; McAleer, C.W.; Miller, P.G.; Hickman, J.J.; Shuler, M.L. Self-contained, low-cost Body-on-a-Chip systems for drug development. Exp. Biol. Med. 2017, 242, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Schmid, Y.R.F.; Bürgel, S.C.; Misun, P.M.; Hierlemann, A.; Frey, O. Electrical Impedance Spectroscopy for Microtissue Spheroid Analysis in Hanging-Drop Networks. ACS Sens. 2016, 1, 1028–1035. [Google Scholar] [CrossRef]
- Bürgel, S.C.; Diener, L.; Frey, O.; Kim, J.Y.; Hierlemann, A. Automated, multiplexed electrical impedance spectroscopy platform for continuous monitoring of microtissue spheroids. Anal. Chem. 2016, 88, 10876–10883. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, P.; Moritz, W.; Kelm, J.M.; Ullrich, N.D.; Agarkova, I.; Anson, B.D.; Suter, T.M.; Zuppinger, C. Development and Characterization of a Scaffold-Free 3D Spheroid Model of Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes. Tissue Eng.-Part C Methods 2015, 21, 852–861. [Google Scholar] [CrossRef]
- Gómez, A.M.; Valdivia, H.H.; Cheng, H.; Lederer, M.R.; Santana, L.F.; Mccune, S.A.; Altschuld, R.A.; Lederer, W.J.; Gomez, A.M.; Valdivia, H.H.; et al. Defective Excitation-Contraction Coupling in Experimental Cardiac Hypertrophy and Heart Failure. Science 1997, 276, 800–806. [Google Scholar] [CrossRef]
- Farman, G.P.; Tachampa, K.; Mateja, R.; Cazorla, O.; Lacampagne, A.; De Tombe, P.P. Blebbistatin: Use as inhibitor of muscle contraction. Pflug. Arch. Eur. J. Physiol. 2008, 455, 995–1005. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Sun, H.; Trivieri, M.G.; Koch, S.E.; Dawood, F.; Ackerley, C.; Yazdanpanah, M.; Wilson, G.J.; Schwartz, A.; Liu, P.P.; et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med. 2003, 9, 1187–1194. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, T.; Wang, P.; Hu, N. High-Throughput Assessment of Drug Cardiac Safety Using a High-Speed Impedance Detection Technology-Based Heart-on-a-Chip. Micromachines 2016, 7, 122. [Google Scholar] [CrossRef]
- Radisic, M.; Park, H.; Gerecht, S.; Cannizzaro, C.; Langer, R.; Vunjak-Novakovic, G. Biomimetic approach to cardiac tissue engineering. Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 1357–1368. [Google Scholar] [CrossRef]
- Qian, F.; Huang, C.; Lin, Y.D.; Ivanovskaya, A.N.; O’Hara, T.J.; Booth, R.H.; Creek, C.J.; Enright, H.A.; Soscia, D.A.; Belle, A.M.; et al. Simultaneous electrical recording of cardiac electrophysiology and contraction on chip. Lab Chip 2017, 17, 1732–1739. [Google Scholar] [CrossRef]
- Wang, T.; Hu, N.; Cao, J.; Wu, J.; Su, K.; Wang, P. A cardiomyocyte-based biosensor for antiarrhythmic drug evaluation by simultaneously monitoring cell growth and beating. Biosens. Bioelectron. 2013, 49, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Nguemo, F.; Šarić, T.; Pfannkuche, K.; Watzele, M.; Reppel, M.; Hescheler, J. In vitro model for assessing arrhythmogenic properties of drugs based on high-resolution impedance measurements. Cell. Physiol. Biochem. 2012, 29, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Munch, P.A.; Thoren, P.N.; Brown, A.M. Dual effects of norepinephrine and mechanisms of baroreceptor stimulation. Circ. Res. 1987, 61, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.F.L.; Huyer, L.D.; Lu, R.X.Z.; Drecun, S.; Radisic, M.; Zhang, B. InVADE: Integrated Vasculature for Assessing Dynamic Events. Adv. Funct. Mater. 2017, 27, 1–11. [Google Scholar] [CrossRef]
- Oleaga, C.; Lavado, A.; Riu, A.; Rothemund, S.; Carmona-Moran, C.A.; Persaud, K.; Yurko, A.; Lear, J.; Narasimhan, N.S.; Long, C.J.; et al. Long-Term Electrical and Mechanical Function Monitoring of a Human-on-a-Chip System. Adv. Funct. Mater. 2019, 29, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Shaegh, S.A.M.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Zhang, Y.S.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, E.; Palma, C.; Vesentini, S.; Occhetta, P.; Rasponi, M. Integrating Biosensors in Organs-on-Chip Devices: A Perspective on Current Strategies to Monitor Microphysiological Systems. Biosensors 2020, 10, 110. https://doi.org/10.3390/bios10090110
Ferrari E, Palma C, Vesentini S, Occhetta P, Rasponi M. Integrating Biosensors in Organs-on-Chip Devices: A Perspective on Current Strategies to Monitor Microphysiological Systems. Biosensors. 2020; 10(9):110. https://doi.org/10.3390/bios10090110
Chicago/Turabian StyleFerrari, Erika, Cecilia Palma, Simone Vesentini, Paola Occhetta, and Marco Rasponi. 2020. "Integrating Biosensors in Organs-on-Chip Devices: A Perspective on Current Strategies to Monitor Microphysiological Systems" Biosensors 10, no. 9: 110. https://doi.org/10.3390/bios10090110
APA StyleFerrari, E., Palma, C., Vesentini, S., Occhetta, P., & Rasponi, M. (2020). Integrating Biosensors in Organs-on-Chip Devices: A Perspective on Current Strategies to Monitor Microphysiological Systems. Biosensors, 10(9), 110. https://doi.org/10.3390/bios10090110