Design of Multiplex Lateral Flow Tests: A Case Study for Simultaneous Detection of Three Antibiotics


 and
and 
Abstract
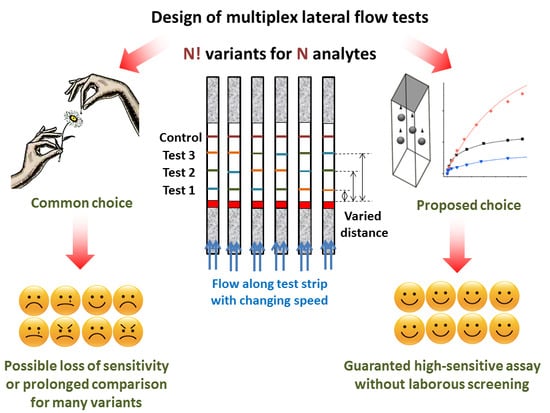
1. Introduction
2. Materials and Methods
2.1. Reactants
2.2. Synthesis of Gold Nanoparticles and Their Conjugation with Antibodies
2.3. Preparation of the Test Strips
2.4. Performing Lateral Flow Immunoassay
2.5. Data Processing
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- O’Farrell, B. Evolution in Lateral Flow–Based Immunoassay Systems. In Lateral Flow Immunoassay; Wong, R., Tse, H., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 1–33. ISBN 978-1-59745-240-3. [Google Scholar]
- Wang, K.; Qin, W.; Hou, Y.; Xiao, K.; Yan, W. The application of lateral flow immunoassay in point of care testing: A review. Nano Biomed. Eng. 2016, 8, 172–183. [Google Scholar] [CrossRef]
- Huang, X.; Aguilar, Z.P.; Xu, H.; Lai, W.; Xiong, Y. Membrane-based lateral flow immunochromatographic strip with nanoparticles as reporters for detection: A review. Biosens. Bioelectron. 2015, 75, 166–180. [Google Scholar] [CrossRef]
- Anfossi, L.; Baggiani, C.; Giovannoli, C.; D’Arco, G.; Giraudi, G. Lateral-flow immunoassays for mycotoxins and phycotoxins: A review. Anal. Bioanal. Chem. 2013, 405, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Upadhyay, N.; Nara, S. Recent advancements in lateral flow immunoassays: A journey for toxin detection in food. Crit. Rev. Food Sci. Nutr. 2018, 58, 1715–1734. [Google Scholar] [CrossRef] [PubMed]
- Dincer, C.; Bruch, R.; Kling, A.; Dittrich, P.S.; Urban, G.A. Multiplexed point-of-care testing – xPOCT. Trends Biotechnol. 2017, 35, 728–742. [Google Scholar] [CrossRef]
- Sanchis, A.; Salvador, J.P.; Marco, M.P. Multiplexed immunochemical techniques for the detection of pollutants in aquatic environments. TRAC Trends Anal. Chem. 2018, 106, 1–10. [Google Scholar] [CrossRef]
- Li, Y.-F.; Sun, Y.-M.; Beier, R.C.; Lei, H.T.; Gee, S.; Hammock, B.D.; Wang, H.; Wang, Z.; Sun, X.; Shen, Y.-D.; et al. Immunochemical techniques for multianalyte analysis of chemical residues in food and the environment: A review. TRAC Trends Anal. Chem. 2017, 88, 25–40. [Google Scholar] [CrossRef]
- Zherdev, A.V.; Dzantiev, B.B. Ways to Reach Lower Detection Limits of Lateral Flow Immunoassays. In Rapid Test—Advances in Design, Format and Diagnostic Applications; IntechOpen: London, UK, 2018; pp. 9–43. ISBN 978-1-78923-901-0. [Google Scholar]
- Anfossi, L.; Di Nardo, F.; Cavalera, S.; Giovannoli, C.; Baggiani, C. Multiplex lateral flow immunoassay: An overview of strategies towards high-throughput point-of-need testing. Biosensors 2019, 9, 2. [Google Scholar] [CrossRef]
- Bishop, J.D.; Hsieh, H.V.; Gasperino, D.J.; Weigl, B.H. Sensitivity enhancement in lateral flow assays: A systems perspective. Lab. Chip. 2019, 19, 2486–2499. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Q.; Han, M.; Zhou, J.; Gong, L.; Niu, Y.; Zhang, Y.; He, L.; Zhang, L. Development and optimization of a multiplex lateral flow immunoassay for the simultaneous determination of three mycotoxins in corn, rice and peanut. Food Chem. 2016, 213, 478–484. [Google Scholar] [CrossRef]
- Peng, J.; Wang, Y.; Liu, L.; Kuang, H.; Li, A.; Xu, C. Multiplex lateral flow immunoassay for five antibiotics detection based on gold nanoparticle aggregations. RSC Adv. 2016, 6, 7798–7805. [Google Scholar] [CrossRef]
- Foubert, A.; Beloglazova, N.V.; Gordienko, A.; Tessier, M.D.; Drijvers, E.; Hens, Z.; De Saeger, S. Development of a rainbow lateral flow immunoassay for the simultaneous detection of four mycotoxins. J. Agric. Food Chem. 2017, 65, 7121–7130. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Jin, M.; Du, P.; Chen, G.; Zhang, C.; Zhang, Y.; Shao, Y.; Wang, J. Development of immunoassays for multi-residue detection of small molecule compounds. Food Agric. Immunol. 2018, 29, 638–652. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, X.; Wang, Z.; Luo, P.; Xu, L.; Zheng, Q.; Kuang, H. A sensitive lateral flow immunoassay for the multiple residues of five adamantanes. Food Agric. Immunol. 2019, 30, 647–661. [Google Scholar] [CrossRef]
- Millipore, E.M.D. Rapid Lateral Flow Test Strips: Considerations for Product Development; EMD Millipore Corporation: Billerica, MA, USA, 2013; 36p. [Google Scholar]
- Mansfield, M.A. Nitrocellulose Membranes for Lateral Flow Immunoassays: A Technical Treatise. In Lateral Flow Immunoassay; Wong, R., Tse, H., Eds.; Humana Press Springer: Totowa, NJ, USA, 2009; pp. 95–114. ISBN 978-1-59745-240-3. [Google Scholar]
- Mendes, R.E.; Farrell, D.J.; Sader, H.S.; Streit, J.M.; Jones, R.N. Update of the telavancin activity in vitro tested against a worldwide collection of Gram-positive clinical isolates (2013), when applying the revised susceptibility testing method. Diagn. Micr. Infec. Dis. 2015, 81, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Martínez-Puchol, S.; Ruiz-Roldán, L.; Pons, M.J.; Del Valle Mendoza, J.; Ruiz, J. Development and characterisation of highly antibiotic resistant Bartonella bacilliformis mutants. Sci. Rep. 2016, 6, 33584. [Google Scholar] [CrossRef]
- Joshi, M.; Kaur, S.; Shergill, B.; Sood, S.; Tulika, M. Isolation of multidrug-resistant bacteria from the hospital environment. Int. J. Res. Pharm. Sci. 2019, 10, 990–996. [Google Scholar] [CrossRef]
- Wang, M.; Cai, C.; Zhang, B.; Liu, H. Characterization and mechanism analysis of lincomycin biodegradation with Clostridium sp. strain LCM-B isolated from lincomycin mycelial residue (LMR). Chemosphere 2018, 193, 611–617. [Google Scholar] [CrossRef]
- Ahmad, A.; Græsboll, K.; Christiansen, L.E.; Toft, N.; Matthews, L.; Nielsen, S.S. Pharmacokinetic-pharmacodynamic model to evaluate intramuscular tetracycline treatment protocols to prevent antimicrobial resistance in pigs. Antimicrob. Agents Chemother. 2015, 59, 1634–1642. [Google Scholar] [CrossRef]
- Berlina, A.N.; Bartosh, A.V.; Zherdev, A.V.; Xu, C.; Dzantiev, B.B. Development of immunochromatographic assay for determination of tetracycline in human serum. Antibiotics 2018, 7, 99. [Google Scholar] [CrossRef]
- Taranova, N.A.; Berlina, A.N.; Zherdev, A.V.; Dzantiev, B.B. ‘Traffic light’ immunochromatographic test based on multicolor quantum dots for the simultaneous detection of several antibiotics in milk. Biosens. Bioelectr. 2015, 63, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, O.D.; Zvereva, E.A.; Popravko, D.S.; Zherdev, A.V.; Xu, C.; Dzantiev, B.B. An immunochromatographic test system for the determination of lincomycin in foodstuffs of animal origin. J. Chromatogr. B 2020, 1141, 122014. [Google Scholar] [CrossRef]
- Byzova, N.A.; Zvereva, E.A.; Zherdev, A.V.; Eremin, S.A.; Sveshnikov, P.G.; Dzantiev, B.B. Pretreatment-free immunochromatographic assay for the detection of streptomycin and its application to the control of milk and dairy products. Anal. Chim. Acta 2011, 701, 209–217. [Google Scholar] [CrossRef]
- Hermanson, G. Bioconjugate Techniques, 3rd ed.; Academic Press: London, UK, 2013; ISBN 9780123822390. [Google Scholar]
- Urusov, A.E.; Zherdev, A.V.; Dzantiev, B.B. Use of gold nanoparticle-labeled secondary antibodies to improve the sensitivity of an immunochromatographic assay for aflatoxin B1. Microchim. Acta 2014, 181, 1939–1946. [Google Scholar] [CrossRef]
- Urusov, A.E.; Petrakova, A.V.; Zherdev, A.V.; Dzantiev, B.B. «Multistage in one touch» design with a universal labelling conjugate for high-sensitive lateral flow immunoassays. Biosens. Bioelectr. 2016, 86, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Petrakova, A.V.; Urusov, A.E.; Gubaydullina, M.K.; Zherdev, A.V.; Dzantiev, B.B. “External” antibodies as the simplest tool for sensitive immunochromatographic tests. Talanta 2017, 175, 77–81. [Google Scholar] [CrossRef]
- Berlina, A.N.; Bartosh, A.V.; Sotnikov, D.V.; Zherdev, A.V.; Xu, C.; Dzantiev, B.B. Complexes of gold nanoparticles with antibodies in immunochromatography: Comparison of direct and indirect immobilization of antibodies for the detection of antibiotics. Nanotechnol. Russia 2018, 13, 430–438. [Google Scholar] [CrossRef]
- Urusov, A.E.; Petrakova, A.V.; Zvereva, E.A.; Zherdev, A.V. Indirect labeling of antibodies as a universal approach to increase sensitivity of lateral flow tests: A case study for mycotoxins detection. Open Biotechnol. J. 2019, 13, 113–121. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LOD Instrumental, ng/mL | IC50, ng/mL | LOD Visual, ng/mL | |
|---|---|---|---|
| Lincomycin | 0.5 ± 0.4 | 0.8 ± 0.5 | 1.3 ± 0.3 |
| Chloramphenicol | 0.4 ± 0.2 | 0.7 ± 0.3 | 1.2 ± 0.2 |
| Tetracycline | 1.1 ± 0.4 | 1.7 ± 0.4 | 2.7 ± 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartosh, A.V.; Sotnikov, D.V.; Hendrickson, O.D.; Zherdev, A.V.; Dzantiev, B.B. Design of Multiplex Lateral Flow Tests: A Case Study for Simultaneous Detection of Three Antibiotics. Biosensors 2020, 10, 17. https://doi.org/10.3390/bios10030017
Bartosh AV, Sotnikov DV, Hendrickson OD, Zherdev AV, Dzantiev BB. Design of Multiplex Lateral Flow Tests: A Case Study for Simultaneous Detection of Three Antibiotics. Biosensors. 2020; 10(3):17. https://doi.org/10.3390/bios10030017
Chicago/Turabian StyleBartosh, Anastasiya V., Dmitriy V. Sotnikov, Olga D. Hendrickson, Anatoly V. Zherdev, and Boris B. Dzantiev. 2020. "Design of Multiplex Lateral Flow Tests: A Case Study for Simultaneous Detection of Three Antibiotics" Biosensors 10, no. 3: 17. https://doi.org/10.3390/bios10030017
APA StyleBartosh, A. V., Sotnikov, D. V., Hendrickson, O. D., Zherdev, A. V., & Dzantiev, B. B. (2020). Design of Multiplex Lateral Flow Tests: A Case Study for Simultaneous Detection of Three Antibiotics. Biosensors, 10(3), 17. https://doi.org/10.3390/bios10030017