Development of a Pharmacogenetic Lab-on-Chip Assay Based on the In-Check Technology to Screen for Genetic Variations Associated to Adverse Drug Reactions to Common Chemotherapeutic Agents




Abstract
1. Introduction
2. Materials and Methods
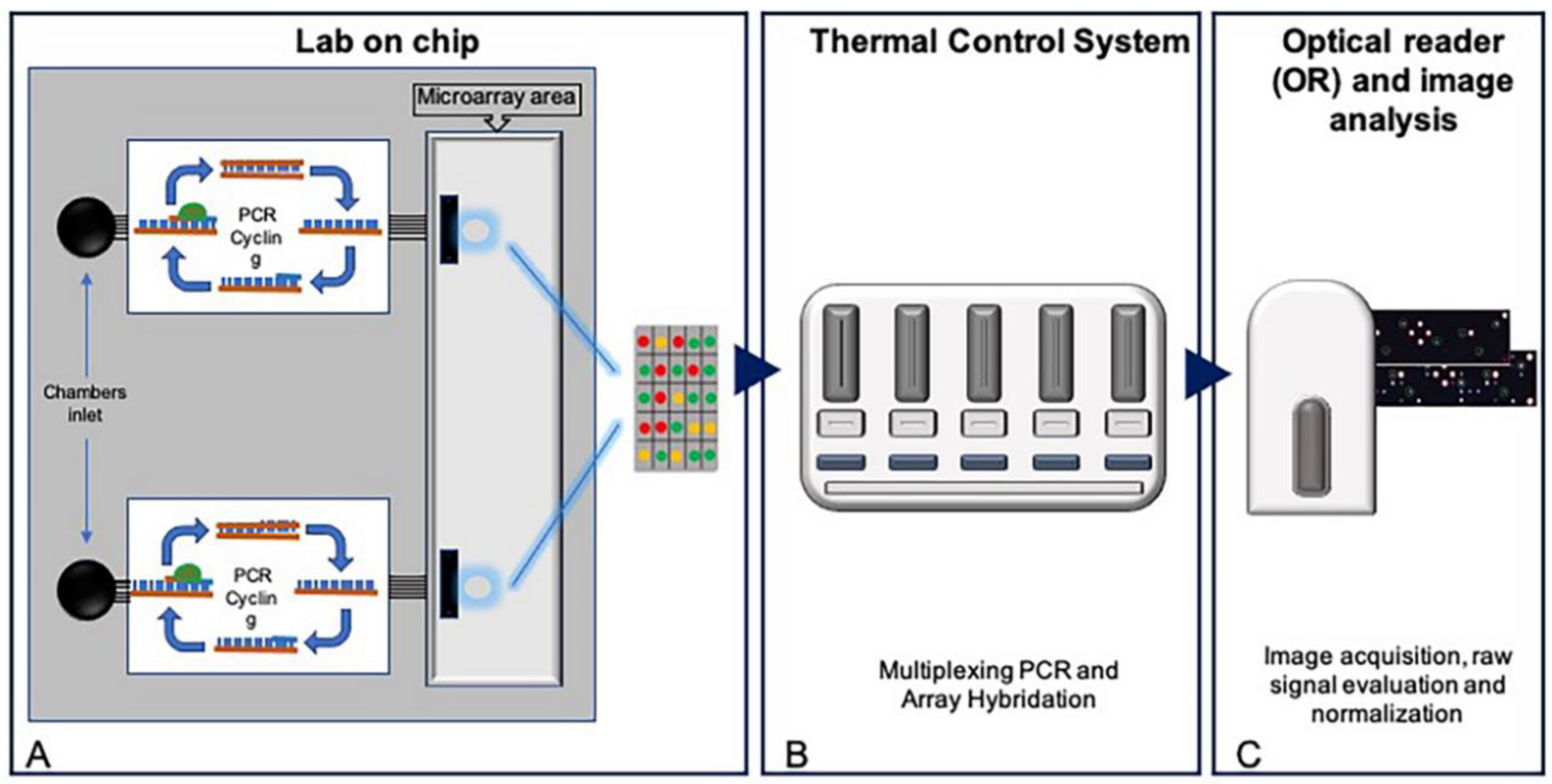
2.1. Development of a Pharmacogenetic Lab-On-Chip
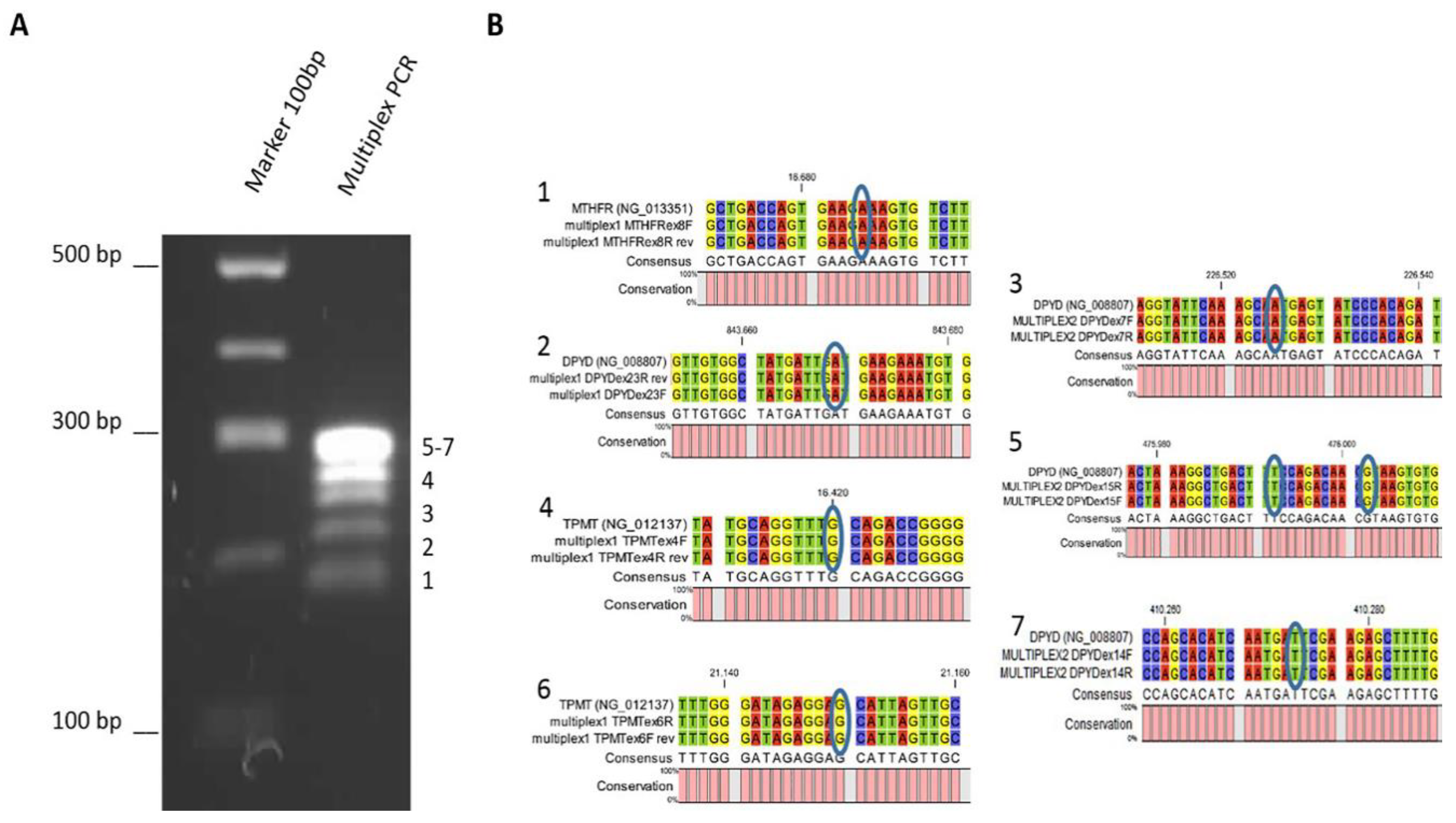
2.2. Asymmetric Multiplex PCR and Gene Sequencing
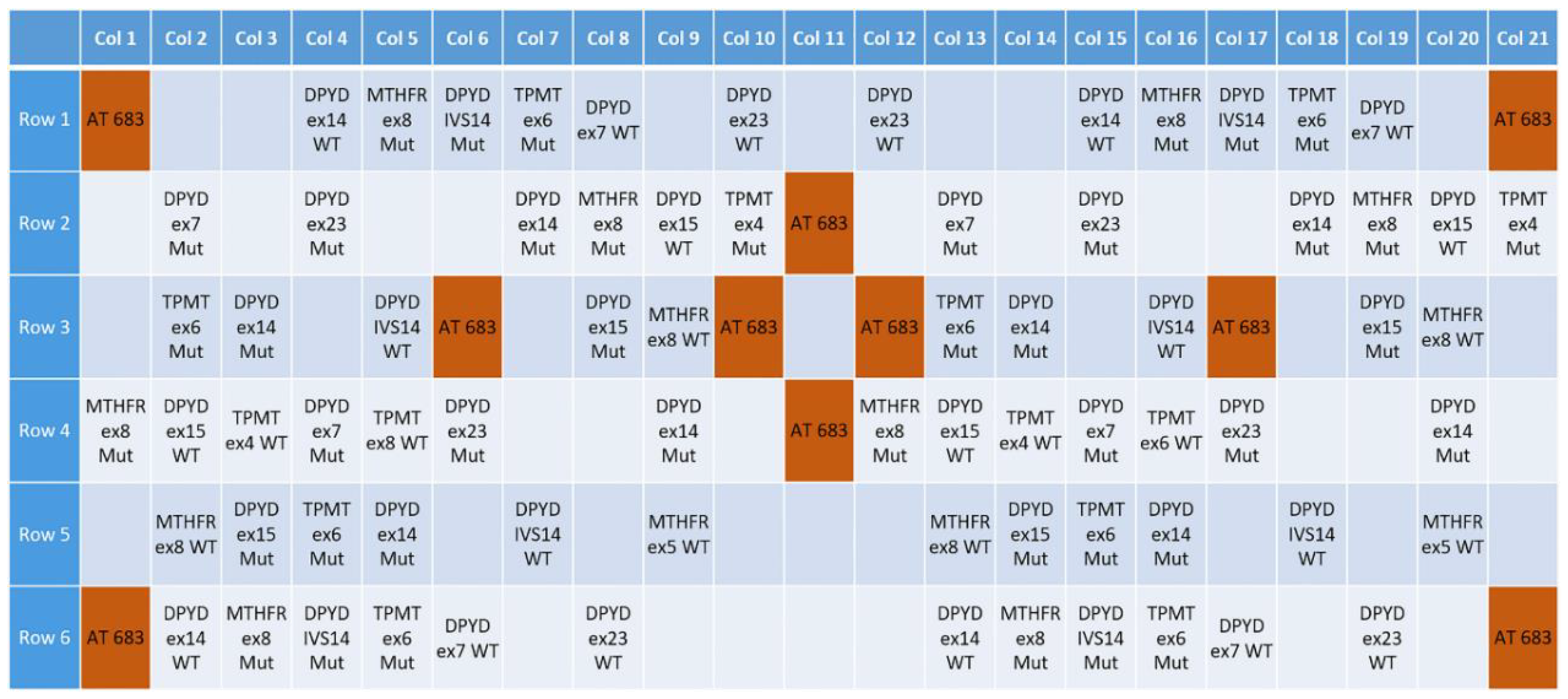
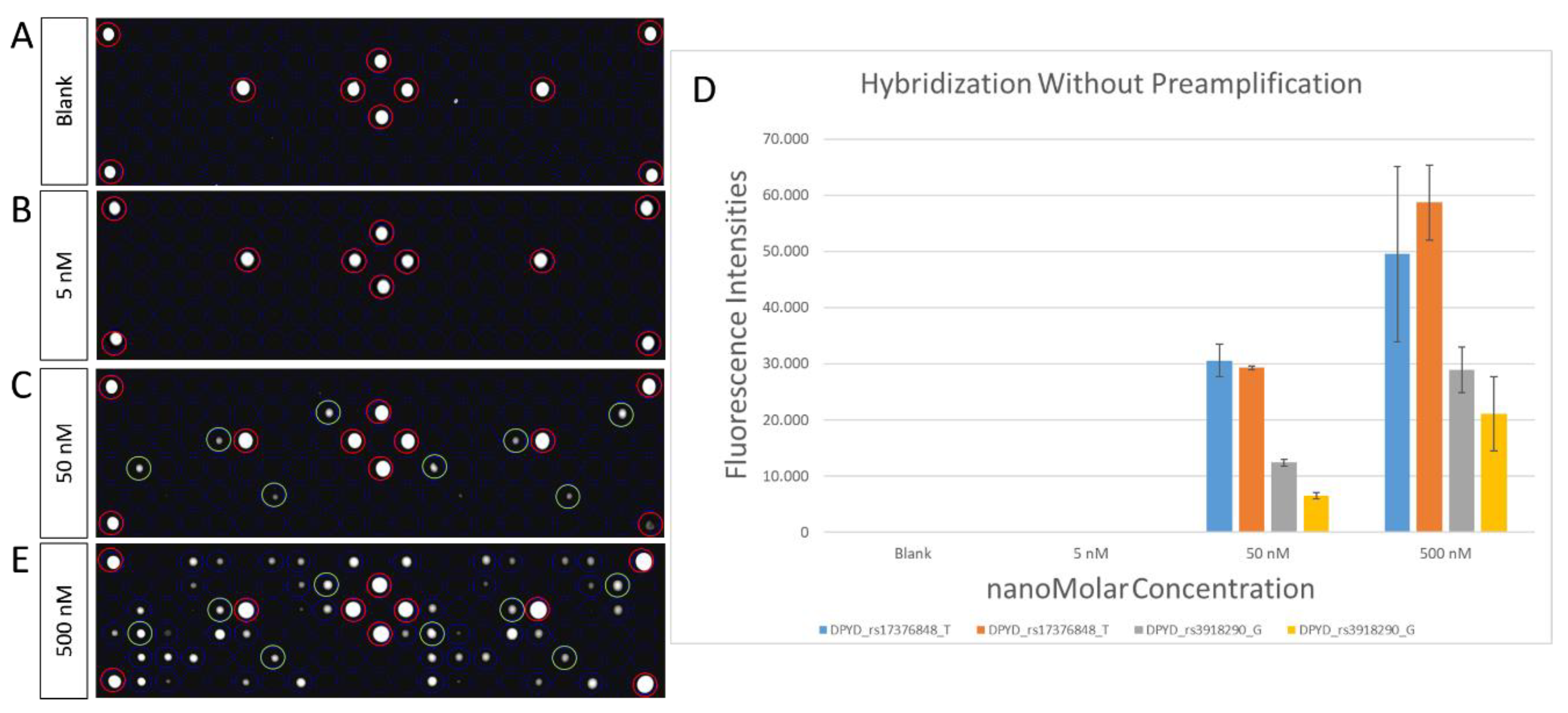
2.3. Microarray Hybridization and Scanning
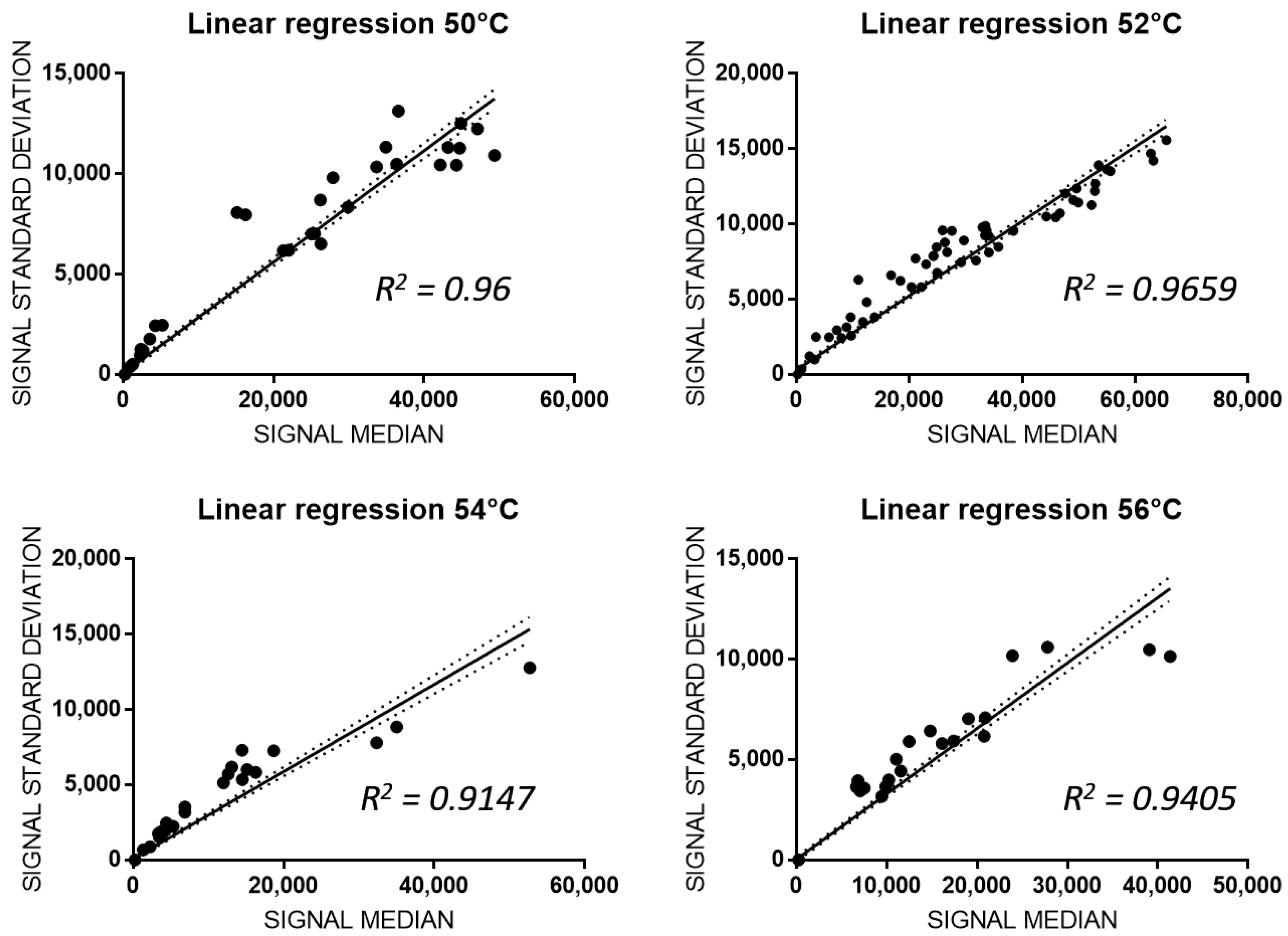
2.4. Statistical Analysis
3. Results
3.1. Design of a Pharmacogenetic LoC Assay for the In-Check Platform
3.2. Multiplex PCR Optimization
3.3. Analitical Variability of PGx-LoC
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forman, D.; Bray, F.; Brewster, D.; Gombe Mbalawa, C.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Ferlay, J. Cancer Incidence in Five Continents; Electronic Version; IARC: Lyon, France, 2013; Volume X. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA A Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Danesi, R.; Di Paolo, A.; Bocci, G.; Crea, F.; Del Tacca, M. Pharmacogenetics in oncology. Eur. J. Cancer Suppl. 2008, 6, 74–78. [Google Scholar] [CrossRef][Green Version]
- Meyer, U.A. Pharmacogenetics and adverse drug reactions. Lancet 2000, 356, 1667–1671. [Google Scholar] [CrossRef]
- Ozdemir, V.; Shear, N.H.; Kalow, W. What will be the role of pharmacogenetics in evaluating drug safety and minimising adverse effects? Drug Saf. 2001, 24, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Spear, B.B.; Heath-Chiozzi, M.; Huff, J. Clinical application of pharmacogenetics. Trends Mol. Med. 2001, 7, 201–204. [Google Scholar] [CrossRef]
- Stingl, J.C.; Brockmoller, J. Personalised pharmacogenetics. Evidence-based guidelines and clinical application of pharmacogenetic diagnostics. Bundesgesundheitsblatt Gesundh. Gesundh. 2013, 56, 1509–1521. [Google Scholar] [CrossRef]
- Bosch, T.M.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H.M. Genetic Polymorphisms of Drug-Metabolising Enzymes and Drug Transporters in the Chemotherapeutic Treatment of Cancer. Clin. Pharmacokinet. 2012, 45, 253–285. [Google Scholar] [CrossRef]
- Roco, A.; Quinones, L.; Agundez, J.A.; Garcia-Martin, E.; Squicciarini, V.; Miranda, C.; Garay, J.; Farfan, N.; Saavedra, I.; Caceres, D.; et al. Frequencies of 23 functionally significant variant alleles related with metabolism of antineoplastic drugs in the chilean population: Comparison with caucasian and asian populations. Front. Genet. 2012, 3, 229. [Google Scholar] [CrossRef]
- Chang, W.-C.; Tanoshima, R.; Ross, C.J.; Carleton, B.C. Challenges and Opportunities in Implementing Pharmacogenetic Testing in Clinical Settings. Annu. Rev. Pharmacol. Toxicol. 2020, 61. [Google Scholar] [CrossRef]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenomics and individualized drug therapy. Annu. Rev. Med. 2006, 57, 119–137. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Milani, L.; Ingelman-Sundberg, M. Pharmacogenomic Biomarkers for Improved Drug Therapy-Recent Progress and Future Developments. AAPS J. 2017, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Scionti, F.; Di Martino, M.T.; Caracciolo, D.; Pensabene, L.; Tassone, P.; Tagliaferri, P. Pharmacogenomics Biomarker Discovery and Validation for Translation in Clinical Practice. Clin. Transl. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mujica, M.L.; Zhang, Y.; Bédioui, F.; Gutiérrez, F.; Rivas, G. Label-free graphene oxide–based SPR genosensor for the quantification of microRNA21. Anal. Bioanal. Chem. 2020, 412, 3539–3546. [Google Scholar] [CrossRef] [PubMed]
- Grodzinski, P.; Liu, R.H.; Lee, A.P. Integrated DNA Biochips: Past, Present and Future. In Integrated Biochips for DNA Analysis; Liu, R.H., Lee, A.P., Eds.; Springer: New York, NY, USA, 2007. [Google Scholar] [CrossRef]
- Hardiman, G. Applications of microarrays and biochips in pharmacogenomics. In Pharmacogenomics in Drug Discovery and Development; Springer: Berlin/Heidelberg, Germany, 2008; pp. 21–30. [Google Scholar]
- Glotov, A.; Nasedkina, T.; Ivaschenko, T.; Urasov, R.; Surzhikov, S.; Pan’kov, S.; Chudinov, A.; Baranov, V.; Zasedatelev, A. Development of a biochip for analyzing polymorphism of the biotransformation genes. Mol. Biol. 2005, 39, 357–365. [Google Scholar] [CrossRef]
- Bürger, J. Biochips-tools of 21st century medicine. Versicherungsmedizin 2006, 58, 9–13. [Google Scholar] [PubMed]
- Petralia, S.; Verardo, R.; Klaric, E.; Cavallaro, S.; Alessi, E.; Schneider, C. In-Check system: A highly integrated silicon Lab-on-Chip for sample preparation, PCR amplification and microarray detection of nucleic acids directly from biological samples. Sens. Actuators B Chem. 2013, 187, 99–105. [Google Scholar] [CrossRef]
- Palmieri, M.; Alessi, E.; Conoci, S.; Marchi, M.; Panvini, G. Developments of the in-check platform for diagnostic applications. In Microfluidics, BioMEMS, and Medical Microsystems VI; International Society for Optics and Photonics: Bellingham, WA, USA, 2008; p. 688602. [Google Scholar]
- Pernagallo, S.; Ventimiglia, G.; Cavalluzzo, C.; Alessi, E.; Ilyine, H.; Bradley, M.; Diaz-Mochon, J.J. Novel biochip platform for nucleic acid analysis. Sensors 2012, 12, 8100–8111. [Google Scholar] [CrossRef]
- Guarnaccia, M.; Iemmolo, R.; San Biagio, F.; Alessi, E.; Cavallaro, S. Genotyping of KRAS Mutational Status by the In-Check Lab-on-Chip Platform. Sensors 2018, 18, 131. [Google Scholar] [CrossRef]
- Patel, S.; Nanda, R.; Sahoo, S.; Mohapatra, E. Biosensors in Health Care: The Milestones Achieved in Their Development towards Lab-on-Chip-Analysis. Biochem. Res. Int. 2016, 2016, 3130469. [Google Scholar] [CrossRef]
- Guarnaccia, M.; Gentile, G.; Alessi, E.; Schneider, C.; Petralia, S.; Cavallaro, S. Is this the real time for genomics? Genomics 2014, 103, 177–182. [Google Scholar] [CrossRef]
- Wu, L.R.; Fang, J.Z.; Khodakov, D.; Zhang, D.Y. Nucleic Acid Quantitation with Log–Linear Response Hybridization Probe Sets. ACS Sens. 2020, 5, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Leimanis, S.; Hernández, M.; Fernández, S.; Boyer, F.; Burns, M.; Bruderer, S.; Glouden, T.; Harris, N.; Kaeppeli, O.; Philipp, P.; et al. A Microarray-based Detection System for Genetically Modified (GM) Food Ingredients. Plant Mol. Biol. 2006, 61, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; McLeod, H.L.; Weinshilboum, R.M. Genomics and drug response. N. Engl. J. Med. 2011, 364, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Gutierrez, M.; Doroshow, J.H.; Murgo, A.J. Drug development in oncology: Classical cytotoxics and molecularly targeted agents. Br. J. Clin. Pharmacol. 2006, 62, 15–26. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Vreken, P.; Abeling, N.G.; Bakker, H.D.; Meinsma, R.; Van Lenthe, H.; De Abreu, R.A.; Smeitink, J.A.; Kayserili, H.; Apak, M.Y.; et al. Genotype and phenotype in patients with dihydropyrimidine dehydrogenase deficiency. Hum. Genet. 1999, 104, 1–9. [Google Scholar] [CrossRef]
- Caudle, K.E.; Thorn, C.F.; Klein, T.E.; Swen, J.J.; McLeod, H.L.; Diasio, R.B.; Schwab, M. Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin. Pharmacol. Ther. 2013, 94, 640–645. [Google Scholar] [CrossRef]
- Garziera, M.; Bidoli, E.; Cecchin, E.; Mini, E.; Nobili, S.; Lonardi, S.; Buonadonna, A.; Errante, D.; Pella, N.; D’Andrea, M.; et al. HLA-G 3’UTR Polymorphisms Impact the Prognosis of Stage II-III CRC Patients in Fluoropyrimidine-Based Treatment. PLoS ONE 2015, 10, e0144000. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Del Re, M.; Cinieri, S.; Michelucci, A.; Salvadori, S.; Loupakis, F.; Schirripa, M.; Cremolini, C.; Crucitta, S.; Barbara, C.; Di Leo, A. DPYD* 6 plays an important role in fluoropyrimidine toxicity in addition to DPYD* 2A and c. 2846A> T: A comprehensive analysis in 1254 patients. Pharm. J. 2019, 19, 556–563. [Google Scholar] [CrossRef]
- Iachetta, F.; Bonelli, C.; Romagnani, A.; Zamponi, R.; Tofani, L.; Farnetti, E.; Nicoli, D.; Damato, A.; Banzi, M.; Casali, B. The clinical relevance of multiple DPYD polymorphisms on patients candidate for fluoropyrimidine based-chemotherapy. An Italian case-control study. Br. J. Cancer 2019, 120, 834–839. [Google Scholar] [CrossRef]
- Varzari, A.; Deyneko, I.V.; Tudor, E.; Turcan, S. Polymorphisms of glutathione S-transferase and methylenetetrahydrofolate reductase genes in Moldavian patients with ulcerative colitis: Genotype-phenotype correlation. Meta Gene 2016, 7, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chiusolo, P.; Reddiconto, G.; Casorelli, I.; Laurenti, L.; Sora, F.; Mele, L.; Annino, L.; Leone, G.; Sica, S. Preponderance of methylenetetrahydrofolate reductase C677T homozygosity among leukemia patients intolerant to methotrexate. Ann. Oncol. 2002, 13, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, X.; Xu, L. Impact of methylenetetrahydrofolate reductase (MTHFR) polymorphisms on methotrexate-induced toxicities in acute lymphoblastic leukemia: A meta-analysis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2012, 33, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Etienne, M.C.; Formento, J.L.; Chazal, M.; Francoual, M.; Magne, N.; Formento, P.; Bourgeon, A.; Seitz, J.F.; Delpero, J.R.; Letoublon, C.; et al. Methylenetetrahydrofolate reductase gene polymorphisms and response to fluorouracil-based treatment in advanced colorectal cancer patients. Pharmacogenetics 2004, 14, 785–792. [Google Scholar] [CrossRef]
- Ramos-Esquivel, A.; Chinchilla, R.; Valle, M. Association of C677T and A1298C MTHFR Polymorphisms and Fluoropyrimidine-induced Toxicity in Mestizo Patients With Metastatic Colorectal Cancer. Anticancer Res. 2020, 40, 4263–4270. [Google Scholar] [CrossRef]
- Wang, L.; Pelleymounter, L.; Weinshilboum, R.; Johnson, J.A.; Hebert, J.M.; Altman, R.B.; Klein, T.E. Very important pharmacogene summary: Thiopurine S-methyltransferase. Pharm. Genom. 2010, 20, 401–405. [Google Scholar] [CrossRef]
- Relling, M.V.; Gardner, E.E.; Sandborn, W.J.; Schmiegelow, K.; Pui, C.H.; Yee, S.W.; Stein, C.M.; Carrillo, M.; Evans, W.E.; Hicks, J.K.; et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin. Pharmacol. Ther. 2013, 93, 324–325. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Allelic Variants (hg38) | MAF (1000 Genomes) | Antineoplastic Drug | Level(s) |
|---|---|---|---|---|
| DPYD (NG_008807.2) | rs2297595 g.226525A > G p.Met166Val | C = 0.0565/283 | Cetuximab, fluorouracil, capecitabine, oxaliplatin, bevacizumab | 2A |
| rs55886062 g.410273T > A g.410273T > G p.Ile560Asn; p.Ile560Ser | C = 0.0002/1 | Capecitabine, fluorouracil, Pyrimidine analogues, tegafur | 1A | |
| rs17376848 g.475992T > C p.Phe632Phe | G = 0.0521/261 | Leucovorin, fluorouracil, capecitabine, oxaliplatin | 3 | |
| rs3918290 g.476002G > A IVS14 | T = 0.0030/15 | Capecitabine, fluorouracil, Pyrimidine analogues, tegafur | 1A | |
| rs67376798 g.843669A > T p.Asp949Val | A = 0.0022/11 | Cetuximab, oxaliplatin, bevacizumab, leucovorin, tegafur, fluorouracil, capecitabine, Pyrimidine analogues | 1A | |
| MTHFR (NG_013351) | rs1801131 g.16685A > C p.Glu429Ala | G = 0.2494/1249 | Leucovorin, capecitabine, fluorouracil, oxaliplatin, methotrexate, bevacizumab, carboplatin, cisplatin, cyanocobalamin, folic acid, or pemetrexed | 3 |
| TPMT (NG_012137.1) | rs1800462 g.16420G > C p.Ala80Pro | G = 0.0022/11 | s-adenosylmethionine, purine analogues, mercaptopurine, azathioprine, thioguanine | 1A |
| rs1800460 g.21147G > A p.Ala154Thr | T = 0.0128/64 | s-adenosylmethionine, mercaptopurine, purine analogues, azathioprine, thioguanine, cisplatin | 1A/3 |
| mPCR | Region of Interest | Primer F | GC % | TM | Primer R | GC % | TM | Product Length |
|---|---|---|---|---|---|---|---|---|
| #1 | MTHFR exon8 | TTTGGGGAGCTGAAGGACTAC | 52 | 61.2 | CACTCCAGCATCACTCACTTT | 48 | 59.5 | 177 |
| DPYD exon23 | TGCAGTACCTTGGAACATTTGG | 45 | 60.1 | TGCAGAAGAGCAATATTTGGCA | 41 | 58.4 | 245 | |
| TPMT exon4 | GATCTGCTTTCCTGCATGTTC | 48 | 59.5 | TCCAGGAATTTCGGTGATTGG | 48 | 59.5 | 269 | |
| TPMT exon6 | GGACGCTGCTCATCTTCTTA | 50 | 58.4 | GACAAAGCTAGTATTGGATTTAGGT | 36 | 60.9 | 295 | |
| #2 | DPYD exon7 | ACTGAAAATGTACTGCTCATTGCT | 38 | 60.3 | CCCCAATCGAGCCAAAAAGG | 55 | 60.5 | 265 |
| DPYD exon15 | TGTTTCCCCCAGAATCATCCG | 52 | 61.2 | TGCATCAGCAAAGCAACTG | 47 | 55 | 287 | |
| DPYD exon14 | AGAAATGGCCGGATTGAAGT | 45 | 56.4 | GACAGAAAGGAAGGAAAGAAACTAA | 36 | 60.9 | 300 |
| Probe Name | Probe Sequences (5′-3′) | 5′-End Modification | Probe Type |
|---|---|---|---|
| DPYD_ g.226525A | TTTTTTTGGTATTCAAAGCAATGAGTA | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.226525A | TTTTTTTAGGTATTCAAAGCAATGAGT | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.226525G | TTTTTTTGGTATTCAAAGCAGTGAGTA | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.226525G | TTTTTTTAGGTATTCAAAGCAGTGAGT | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.410273T | TTTTTTTCATCAATGATTCGAAGAGCT | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.410273T | TTTTTTTCACATCAATGATTCGAAGAG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.410273G | TTTTTTTTGAGTCGAAGAGCTTTTGAA | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.410273G | TTTTTTTAATGAGTCGAAGAGCTTTTG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.410273A | TTTTTTTTGAATCGAAGAGCTTTTGAA | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.410273A | TTTTTTTAATGAATCGAAGAGCTTTTG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.475992T | TTTTTTTTAAAGGCTGACTTTCCAGAC | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.475992T | TTTTTTTGAACTAAAGGCTGACTTTCC | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.475992C | TTTTTTTTAAAGGCTGACTTCCCAGAC | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.475992C | TTTTTTTGAACTAAAGGCTGACTTCCC | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.476002G | TTTTTTTTTCCAGACAACGTAAGTGTG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.476002G | TTTTTTTCTTTCCAGACAACGTAAGTG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.476002A | TTTTTTTTTCCAGACAACATAAGTGTG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.476002A | TTTTTTTCTTTCCAGACAACATAAGTG | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.843669A | TTTTTTTGGCTATGATTGATGAAGAAAT | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.843669A | TTTTTTTGTGGCTATGATTGATGAAGAA | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.843669T | TTTTTTTGGCTATGATTGTTGAAGAAAT | 5′-C6-NH2 | CAPTURE PROBE |
| DPYD_ g.843669T | TTTTTTTGTGGCTATGATTGTTGAAGAA | 5′-C6-NH2 | CAPTURE PROBE |
| MTHFR_ g.16685A | TTTTTTTCAGTGAAGAAAGTGTCTTTG | 5′-C6-NH2 | CAPTURE PROBE |
| MTHFR_ g.16685A | TTTTTTTCCAGTGAAGAAAGTGTCTTT | 5′-C6-NH2 | CAPTURE PROBE |
| MTHFR_ g.16685C | TTTTTTTGTGAAGGAAGTGTCTTTGAA | 5′-C6-NH2 | CAPTURE PROBE |
| MTHFR_ g.16685C | TTTTTTTCAGTGAAGGAAGTGTCTTTG | 5′-C6-NH2 | CAPTURE PROBE |
| TPMT_ g.16420G | TTTTTTTGTTTGCAGACCGGGGACA | 5′-C6-NH2 | CAPTURE PROBE |
| TPMT_ g.16420C | TTTTTTTGTTTCCAGACCGGGGACA | 5′-C6-NH2 | CAPTURE PROBE |
| TPMT_ g.21147G | TTTTTTTGGATAGAGGAGCATTAGTTG | 5′-C6-NH2 | CAPTURE PROBE |
| TPMT_ g.21147A | TTTTTTTATAGAGGAACATTAGTTGCC | 5′-C6-NH2 | CAPTURE PROBE |
| TPMT_ g.21147A | TTTTTTTGGGATAGAGGAACATTAGTT | 5′-C6-NH2 | CAPTURE PROBE |
| AT683 | AGTGAGGGAGGAGATGGAACCATCT | 5′-C6-NH2 | hybridization control |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iemmolo, R.; La Cognata, V.; Morello, G.; Guarnaccia, M.; Arbitrio, M.; Alessi, E.; Cavallaro, S. Development of a Pharmacogenetic Lab-on-Chip Assay Based on the In-Check Technology to Screen for Genetic Variations Associated to Adverse Drug Reactions to Common Chemotherapeutic Agents. Biosensors 2020, 10, 202. https://doi.org/10.3390/bios10120202
Iemmolo R, La Cognata V, Morello G, Guarnaccia M, Arbitrio M, Alessi E, Cavallaro S. Development of a Pharmacogenetic Lab-on-Chip Assay Based on the In-Check Technology to Screen for Genetic Variations Associated to Adverse Drug Reactions to Common Chemotherapeutic Agents. Biosensors. 2020; 10(12):202. https://doi.org/10.3390/bios10120202
Chicago/Turabian StyleIemmolo, Rosario, Valentina La Cognata, Giovanna Morello, Maria Guarnaccia, Mariamena Arbitrio, Enrico Alessi, and Sebastiano Cavallaro. 2020. "Development of a Pharmacogenetic Lab-on-Chip Assay Based on the In-Check Technology to Screen for Genetic Variations Associated to Adverse Drug Reactions to Common Chemotherapeutic Agents" Biosensors 10, no. 12: 202. https://doi.org/10.3390/bios10120202
APA StyleIemmolo, R., La Cognata, V., Morello, G., Guarnaccia, M., Arbitrio, M., Alessi, E., & Cavallaro, S. (2020). Development of a Pharmacogenetic Lab-on-Chip Assay Based on the In-Check Technology to Screen for Genetic Variations Associated to Adverse Drug Reactions to Common Chemotherapeutic Agents. Biosensors, 10(12), 202. https://doi.org/10.3390/bios10120202