Design of Engineered Cyclodextrin Derivatives for Spontaneous Coating of Highly Porous Metal-Organic Framework Nanoparticles in Aqueous Media

 , , ,
, , , 

Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of 1-O-propargyl-13-O-acetyl-1,4,7,10,13-pentaoxatridecane (3)
2.3. Synthesis of Heptakis{6-deoxy-6-[4′-(2″,3″,4″,6″-tetra-O-acetyl-α-d-mannopyranosyloxymethyl)-1H- 1,2,3-triazol-1′-yl]}cyclomaltoheptaose (5)
2.4. Synthesis of Heptakis(6-deoxy-6-{4′-[14″-O-acetyl-(2″,5″,8″,11″,14″-pentaoxatetradecyl)]-1H-1,2,3-tri- azol-1′-yl})cyclomaltoheptose (6)
2.5. Synthesis of Heptakis(6-deoxy-6-{4′-[14″-O-(2‴,3‴,4‴,6‴-tetra-O-acetyl-α-d-mannopyranosyl)-2″, 5″,8″,11″,14″-pentaoxatetradecyl]-1H-1,2,3-triazol-1′-yl})cyclomaltoheptose (7)
2.6. Synthesis of Heptakis{6-deoxy-6-[4′-(α-d-mannopyranosyloxymethyl)-1H-1,2,3-triazol-1′-yl]}cyclomal- toheptaose Phosphate Sodium Salt (8)
2.7. Synthesis of Heptakis{6-deoxy-6-[4′-(13″-hydroxy-2″,5″,8″,11″-tetraoxatridecyl)-1H-1,2,3-triazol-1′- yl]}cyclomaltoheptose Phosphate Sodium Salt (9)
2.8. Synthesis of Heptakis{6-desoxi-6-{4′-[14″-O-(α-d-mannopyranosyl)-2″,5″,8″,11″,14″-pentaoxatetrade- cyl]-1H-1,2,3-triazol-1′-yl}}ciclomaltoheptose Phosphate Sodium Salt (10)
2.9. Synthesis of Heptakis(6-O-tert-butyldimethylsilyl)cyclomaltoheptaose (11)
2.10. Synthesis of 1-azido-1-deoxy-ω-O-methoxypentatetraconta(ethylene glycol) (14)
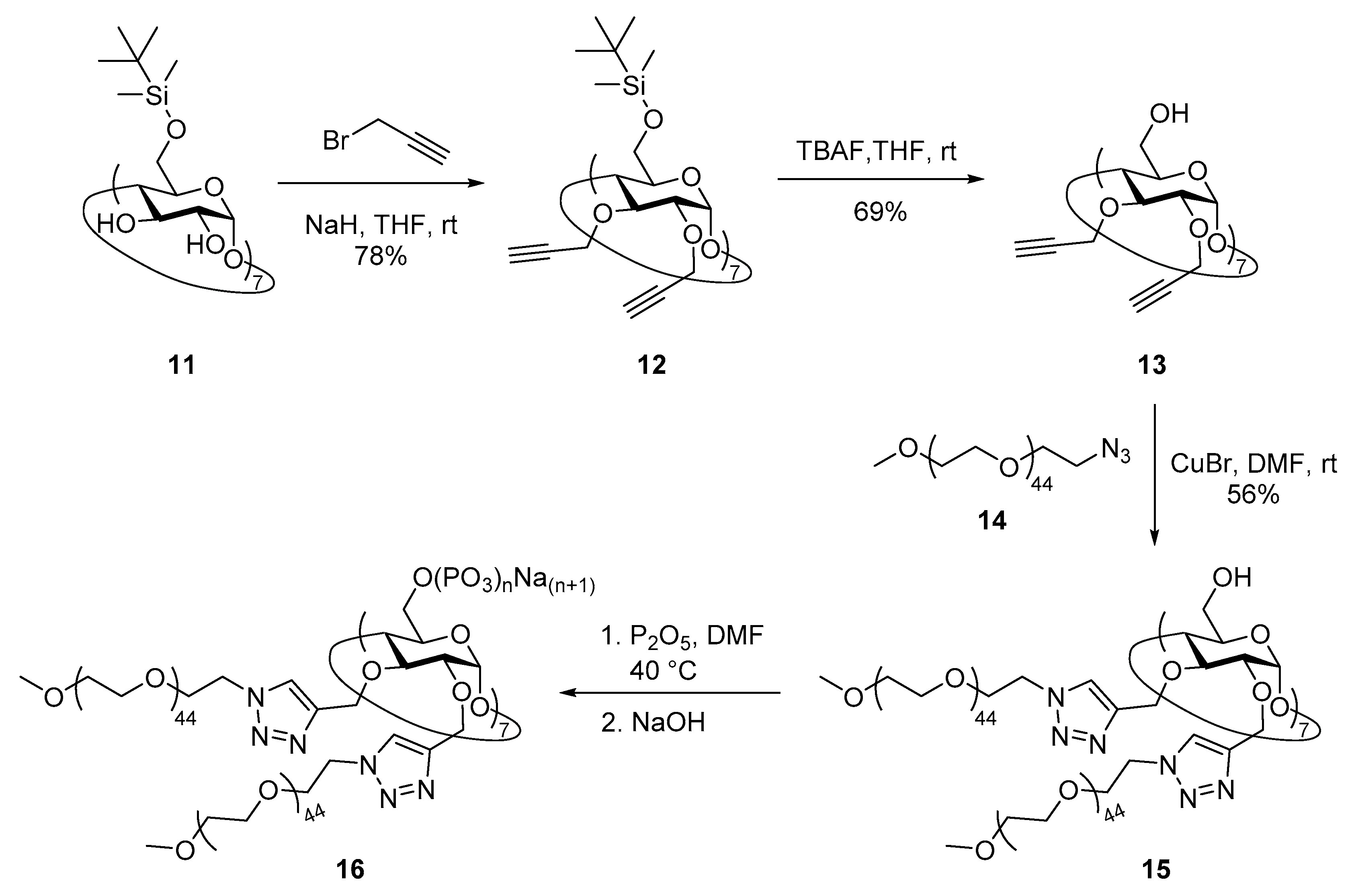
2.11. Synthesis of Heptakis{2,3-di-O-{1′-[methoxypentatetraconta(ethylene glycol)yl-1H-1,2,3-triazol-4′-yl]- methyl}}cyclomaltoheptaose (15)
2.12. Synthesis of Heptakis{2,3-di-O-{1′-[methoxypentatetraconta(ethylene glycol)yl-1H-1,2,3-triazol-4′-yl]- methyl}}cyclomaltoheptaose Phosphate Sodium Salt (16)
2.13. Synthesis and Characterization of MIL-100(Fe) nanoMOFs
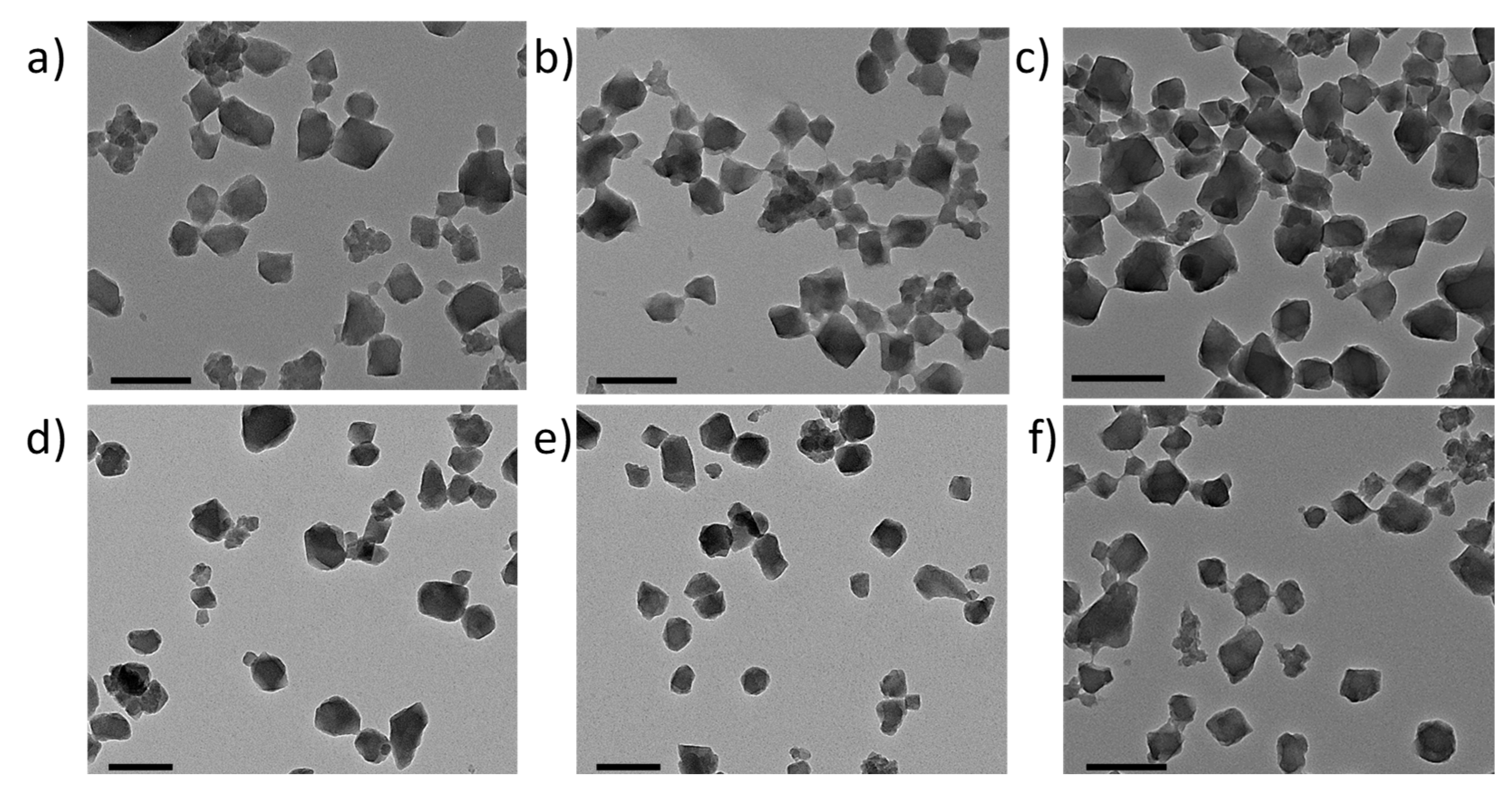
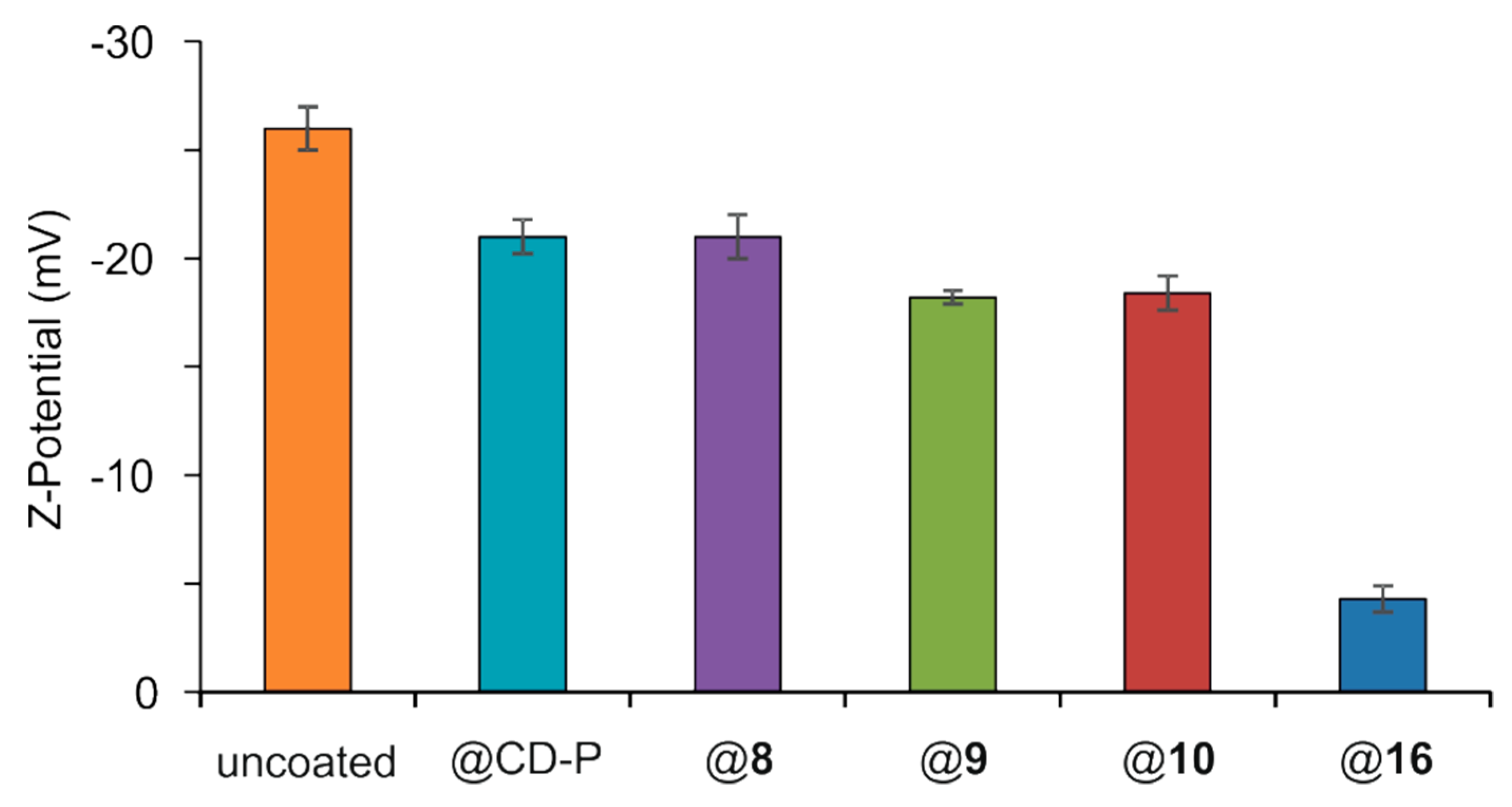
2.14. Surface Modification of MIL-100(Fe) nanoMOFs and Their Characterization
2.15. Quantification of nanoMOFs Uptake by Macrophage Cells
2.16. Surface Modification of Doxorubicin (DOX)-Loaded MIL-100(Fe) Nanomofs with 9
2.17. Isothermal Titration Calorimetry (ITC) Measurements
3. Results and Discussion
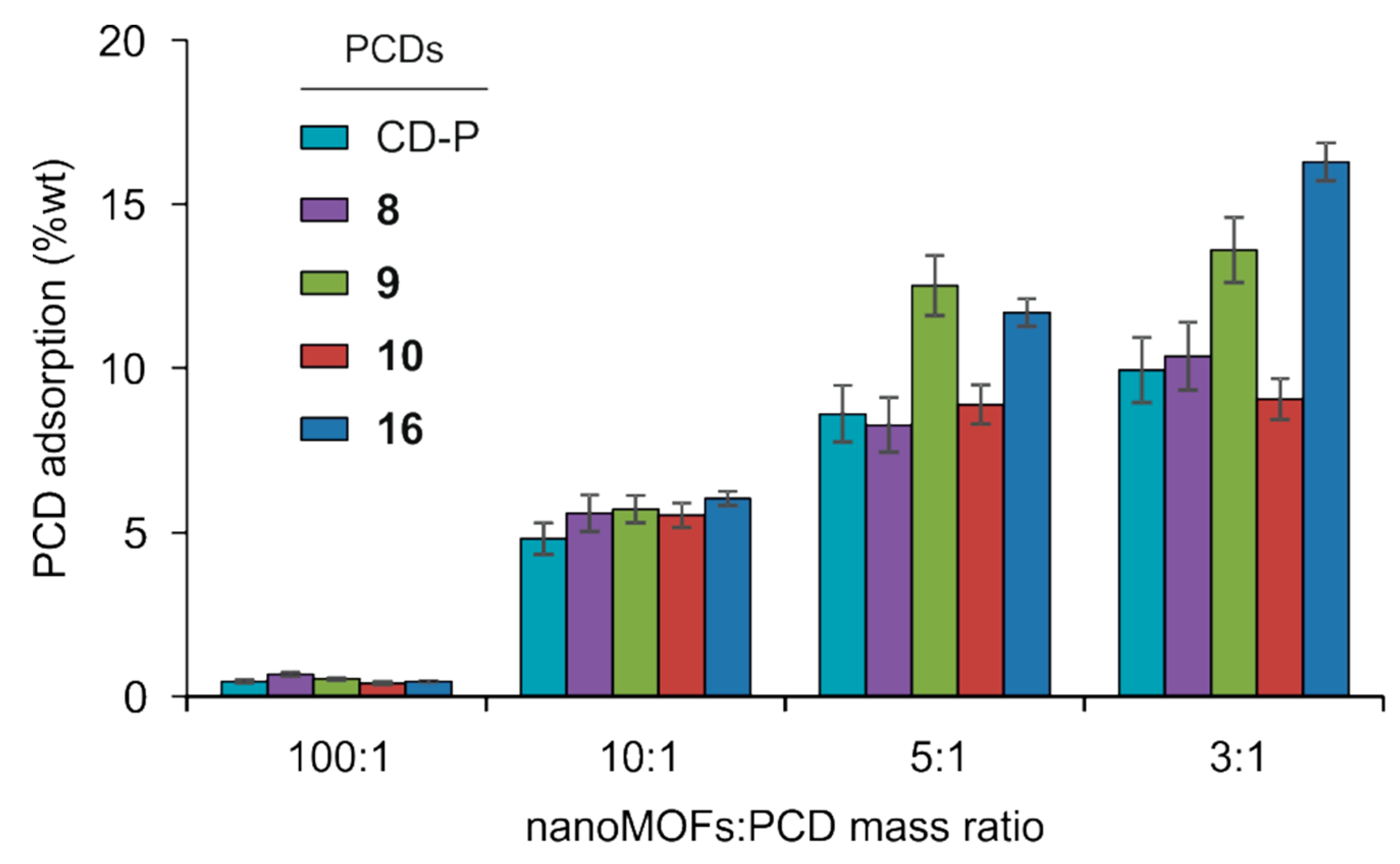
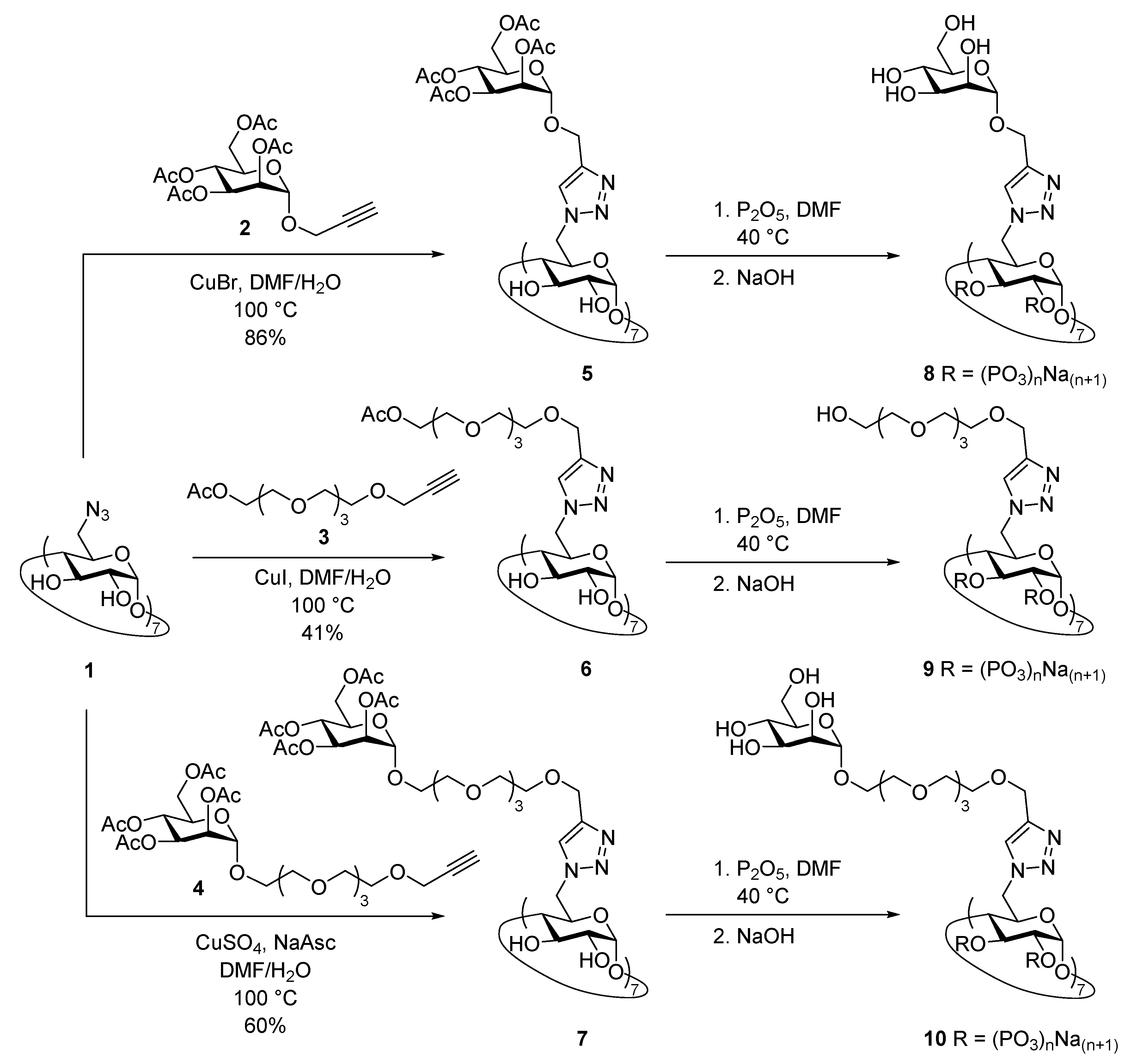
3.1. Synthesis of Phosphate β-CD Derivatives (PCDs)
3.2. MIL-100(Fe) nanoMOFs Synthesis and Surface Modification
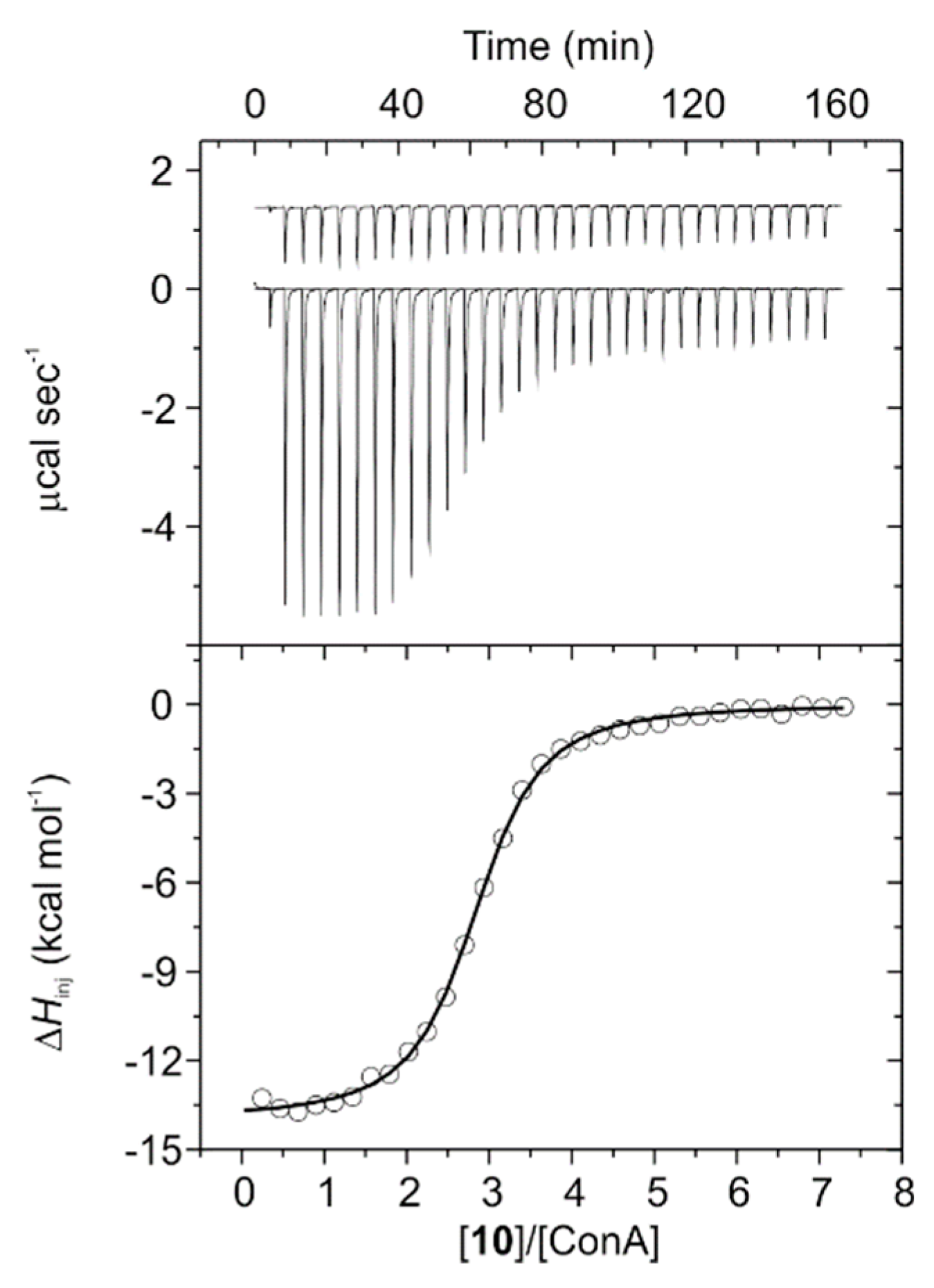
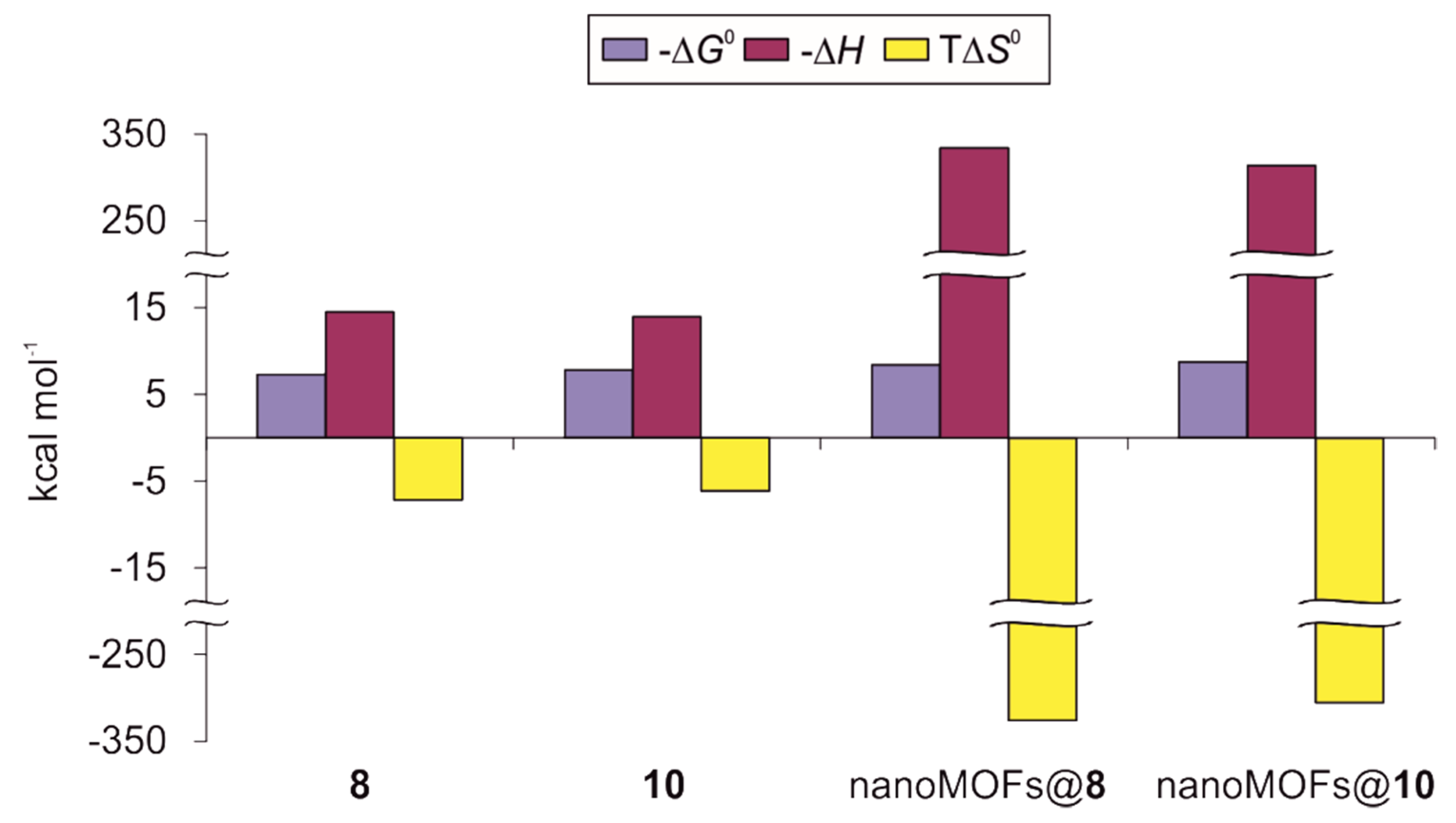
3.3. ITC Experiments on ConA Biorecognition
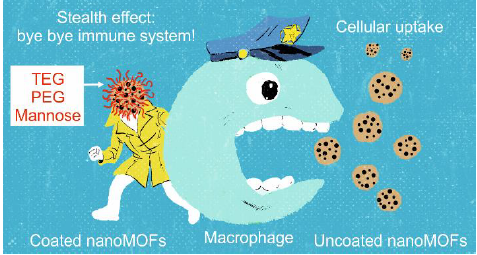
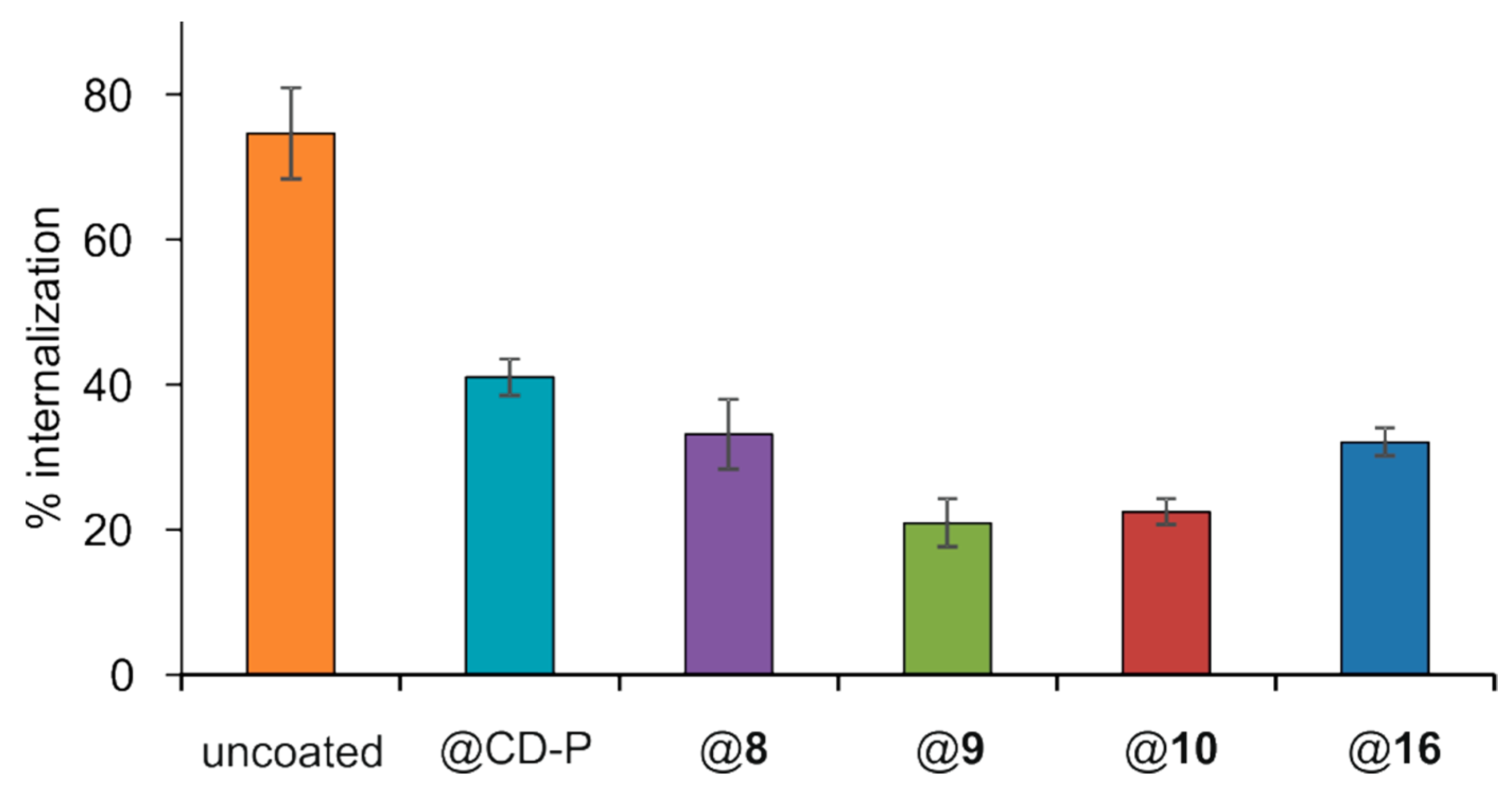
3.4. Interactions of Surface-Modified nanoMOFs with a Macrophage Cell Line
3.5. DOX-Loaded MIL-100(Fe) nanoMOFs Surface Modification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| β-CD | β-cyclodextrin |
| CD-P | β-cyclodextrin phosphate sodium salt |
| ConA | concanavalin A |
| DMAP | 4-dimethylaminopyridine |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMF | dimethylformamide |
| DOX | doxorubicin |
| EtOAc | ethyl acetate |
| FBS | fetal bovine serum |
| HR-ICP-MS | high resolution inductively couples plasma mass spectrometry |
| ITC | isothermal titration calorimetry |
| MALDI-TOF-MS | matrix-assisted laser desorption/ionization time-of-flight mass spectrometry |
| MWCO | molecular weight cut-off |
| NaAsc | (+)-sodium l-ascorbate |
| nanoMOFs | nanosized metal-organic frameworks |
| PCDs | phosphorylated β-CD derivatives |
| PEG | polyethylene glycol |
| TEG | tetraethylene glycol |
| TEM | transmission electron microscopy |
| TLC | thin-layer chromatography |
| XRDP | X-ray diffraction patterns |
| ZP | zeta potential |
References
- Hoskins, B.F.; Robson, R. Infinite polymeric frameworks consisting of three dimensionally linked rod-like segments. J. Am. Chem. Soc. 1989, 111, 5962–5964. [Google Scholar] [CrossRef]
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.T.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Hydrogen storage in microporous metal-organic frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Férey, G.; Serre, C.; Devic, T.; Maurin, G.; Jobic, H.; Llewellyn, P.L.; De Weireld, G.; Vimont, A.; Daturi, M.; Chang, J.-S. Why hybrid porous solids capture greenhouse gases? Chem. Soc. Rev. 2011, 40, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, A.; Garcia, H. Catalysis by metal nanoparticles embedded on metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 5262–5284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-C.; Kitagawa, S. Metal-organic frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, S.; Braig, S.; Preiß, T.; Zimpel, A.; Sicklinger, J.; Bellomo, C.; Rädler, J.O.; Vollmar, A.M.; Bein, T. MOF nanoparticles coated by lipid bilayers and their uptake by cancer cells. Chem. Commun. 2015, 51, 15752–15755. [Google Scholar] [CrossRef] [PubMed]
- Parmar, B.; Patel, P.; Kureshy, R.I.; Khan, N.H.; Suresh, E. Sustainable heterogeneous catalysts for CO2 utilization by using dual ligand ZnII/CdII metal–organic frameworks. Chem. Eur. J. 2018, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, P.L.; Bourrelly, S.; Serre, C.; Vimont, A.; Daturi, M.; Hamon, L.; De Weireld, G.; Chang, J.-S.; Hong, D.-Y.; Hwang, Y.K. High uptakes of CO2 and CH4 in mesoporous metal-organic frameworks MIL-100 and MIL-101. Langmuir 2008, 24, 7245–7250. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal–organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 2010, 9, 172–178. [Google Scholar] [CrossRef]
- Agostoni, V.; Chalati, T.; Horcajada, P.; Willaime, H.; Anand, R.; Semiramoth, N.; Baati, T.; Hall, S.; Maurin, G.; Chacun, H. Towards an improved anti-HIV activity of NRTI via metal-organic frameworks nanoparticles. Adv. Healthc. Mater. 2013, 2, 1630–1637. [Google Scholar] [CrossRef]
- Simon-Yarza, T.; Giménez-Marqués, M.; Mrimi, R.; Mielcarek, A.; Gref, R.; Horcajada, P.; Serre, C.; Couvreur, P. A smart metal-organic framework nanomaterial for lung targeting. Angew. Chem. Int. Ed. 2017, 56, 15565–15569. [Google Scholar] [CrossRef] [PubMed]
- Simon-Yarza, M.T.; Baati, T.; Paci, A.; Lesueur, L.L.; Seck, A.; Chiper, M.; Gref, R.; Serre, C.; Couvreur, P.; Horcajada, P. Antineoplastic busulfan encapsulated in metal organic framework nanocarrier: First in vivo results. J. Mater. Chem. B 2016, 4, 585–588. [Google Scholar] [CrossRef]
- Li, H.; Lv, N.; Li, X.; Botao, L.; Feng, J.; Ren, X.; Guo, T.; Chen, D.; Stoddart, J.F.; Gref, R.; et al. Composite CD-MOF nanocrystals-containing microsphere for sustained drug delivery. Nanoscale 2017, 9, 7454–7463. [Google Scholar] [CrossRef] [PubMed]
- Rojas, S.; Colinet, I.; Cunha, D.; Hidalgo, T.; Salles, F.; Serre, C.; Guillou, N.; Horcajada, P. Toward understanding drug incorporation and delivery from biocompatible metal-organic frameworks in view of cutaneous administration. ACS Omega 2018, 3, 2994–3003. [Google Scholar] [CrossRef] [PubMed]
- Agostoni, V.; Horcajada, P.; Noiray, N.; Malanga, M.; Aykaç, A.; Jicsinszky, L.; Vargas-Berenguel, A.; Semiramoth, N.; Daoud-Mahammed, S.; Nicolas, V.; et al. A “green” strategy to construct non-covalent, stable and bioactive coatings on porous MOF nanoparticles. Sci. Rep. 2015, 5, 7925. [Google Scholar] [CrossRef] [PubMed]
- Baati, T.; Njim, L.; Neffati, F.; Kerkeni, A.; Bouttemi, M.; Gref, R.; Fadhel Najjar, M.; Zakhama, A.; Couvreur, P.; Serre, C.; et al. In depth analysis of the in vivo toxicity of nanoparticles of porous iron(III) metal–organic frameworks. Chem. Sci. 2013, 4, 1597–1607. [Google Scholar] [CrossRef]
- Aykaç, A.; Noiray, M.; Malanga, M.; Agostoni, V.; Casas-Solvas, J.M.; Fenyvesi, É.; Gref, R.; Vargas-Berenguel, A. A non-covalent “click chemistry” strategy to efficiently coat highly porous MOF nanoparticles with a stable polymeric shell. Biochim. Biophys. Acta 2017, 1861, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, G.M.; Korneeva, E.V.; Smolina, N.A.; Schubert, U.S. Hydrodynamic properties of cyclodextrin molecules in dilute solutions. Eur. Biophys. J. 2010, 39, 371–379. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nguyen, V.H.; Lee, B.-J. Protein corona: A new approach for nanomedicine design. Int. J. Nanomed. 2017, 12, 3137–3151. [Google Scholar] [CrossRef]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef]
- Gimenez-Marques, M.; Bellido, E.; Berthelot, T.; Simon-Yarza, T.; Hidalgo, T.; Simon-Vazquez, R.; Gonzalez-Fernandez, A.; Avila, J.; Asensio, M.C.; Gref, R.; et al. GraftFast surface engineering to improve MOF nanoparticles furtiveness. Small 2018, 14, 1801900–1801911. [Google Scholar] [CrossRef] [PubMed]
- Casas-Solvas, J.M.; Vargas-Berenguel, A. Glycoclusters and their applications as anti-infective agents, vaccines, and targeted drug delivery systems. In Carbohydrate Nanotechnology; Stine, K.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Chapter 7; pp. 175–210. [Google Scholar] [CrossRef]
- Kang, B.; Opatz, T.; Landfester, K.; Wurm, F.R. Carbohydrate nanocarriers in biomedical applications: Functionalization and construction. Chem. Soc. Rev. 2015, 44, 8301–8325. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chaix, A.; Gary-Bobo, M.; Angeletti, B.; Masion, A.; Da Silva, A.; Daurat, M.; Lichon, L.; Garcia, M.; Morère, A.; et al. Stealth biocompatible Si-based nanoparticles for niomedical applications. Nanomaterials 2017, 7, 288. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Okwieka, P.; Schöttler, S.; Winzen, S.; Langhanki, J.; Mohr, K.; Opatz, T.; Mailänder, V.; Landfester, K.; Wurm, F.R. Carbohydrate-based nanocarriers exhibiting specific cell targeting with minimum influence from the protein corona. Angew. Chem. Int. Ed. 2015, 54, 7436–7440. [Google Scholar] [CrossRef] [PubMed]
- Lis, H.; Sharon, N. Lectins: Carbohydrate-specific proteins that mediate cellular recognition. Chem. Rev. 1998, 98, 637–674. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.T.; Lee, Y.C. Affinity enhancement by multivalent lectin–carbohydrate interaction. Glycoconj. J. 2000, 17, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Neranon, K.; Ramström, O.; Yan, M. Glyconanomaterials for biosensing applications. Biosens. Bioelectron. 2016, 76, 113–130. [Google Scholar] [CrossRef]
- Reina, J.J.; Rojo, J. Carbohydrate multivalent systems: Synthesis and therapeutic opportunities. In Carbohydrate Chemistry: State of the Art and Challenges for Drug Development; Cipolla, L., Ed.; Imperial College Press: London, UK, 2015; pp. 419–439, Chapter 17. [Google Scholar] [CrossRef]
- Swierzko, A.S.; Kilpatrick, D.C.; Cedzynski, M. Mannan-binding lectin in malignancy. Mol. Immunol. 2013, 55, 16–21. [Google Scholar] [CrossRef]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wojnar, J. Galectin targeted therapy in oncology: Current knowledge and perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef]
- Cutrone, G.; Casas-Solvas, J.M.; Vargas-Berenguel, A. Cyclodextrin-modified inorganic materials for the construction of nanocarriers. Int. J. Pharm. 2017, 531, 621–639. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, G.; Huang, H. The glyconanoparticle as carrier for drug delivery. Drug Deliv. 2018, 25, 1840–1845. [Google Scholar] [CrossRef] [PubMed]
- Scharenberg, M.; Schwardt, O.; Rabbani, S.; Ernst, B. Target selectivity of FimH antagonists. J. Med. Chem. 2012, 55, 9810–9816. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.K.; Rajaram, M.V.S.; Schlesinger, L.S. Exploitation of the macrophage mannose receptor (CD206) in infectious disease diagnostics and therapeutics. J. Cytol. Mol. Biol. 2014, 1, 1000003. [Google Scholar] [CrossRef] [PubMed]
- Ladaviere, C.; Gref, R. Toward an optimized treatment of intracellular bacterial infections: Input of nanoparticulate drug delivery systems. Nanomedicine 2015, 10, 3033–3055. [Google Scholar] [CrossRef] [PubMed]
- Rouzes, C.; Gref, R.; Leonard, M.; De Sousa Delgado, A.; Dellacherie, E. Surface modification of poly(lactic acid) nanospheres using hydrophobically modified dextrans as stabilizers in an o/w emulsion/evaporation technique. J. Biomed. Mater. Res. 2000, 50, 557–565. [Google Scholar] [CrossRef]
- Warther, D.; Jimenez, C.M.; Raehm, L.; Gérardin, C.; Durand, J.-O.; Morère, A.; El Cheikh, K.; Gallud, A.; Gary-Bobo, M.; Maynadier, M. Small sized mesoporous silica nanoparticles functionalized with mannose for retinoblastoma cell imaging. RSC Adv. 2014, 4, 37171–37179. [Google Scholar] [CrossRef]
- Chaix, A.; El Cheikh, K.; Bouffard, E.; Maynadier, M.; Aggad, D.; Stojanovic, V.; Knezevic, N.; Garcia, M.; Maillard, P.; Morère, A.; et al. Mesoporous silicon nanoparticles for targeted two-photon theranostics of prostate cancer. J. Mater. Chem. B 2016, 4, 3639–3642. [Google Scholar] [CrossRef]
- Müller, C.; Despras, G.; Lindhorst, T.K. Organizing multivalency in carbohydrate recognition. Chem. Soc. Rev. 2016, 45, 3275–3302. [Google Scholar] [CrossRef]
- Poláková, M.; Beláňová, M.; Mikušová, K.; Lattová, E.; Perreault, H. Synthesis of 1,2,3-triazolo-linked octyl (1→6)-α-d-oligomannosides and their evaluation in mycobacterial mannosyltransferase assay. Bioconj. Chem. 2011, 22, 289–298. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, Y.; Park, H.-J.; Boggs, J.M.; Basu, A. Carbohydrate-coated fluorescent silica nanoparticles as probes for the galactose/3-sulfogalactose carbohydrate-carbohydrate interaction using model systems and cellular binding studies. Bioconj. Chem. 2012, 23, 1166–1173. [Google Scholar] [CrossRef]
- van der Peet, P.; Gannon, C.T.; Walker, I.; Dinev, Z.; Angelin, M.; Tam, S.; Ralton, J.E.; McConville, M.J.; Williams, S.J. Use of click chemistry to define the substrate specificity of Leishmania β-1,2-mannosyltransferases. ChemBioChem 2006, 7, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.R.; Han, S.C.; Jin, S.; Lee, J.W. Convergent synthesis and characterization of dumbbell type dendritic materials by click chemistry. Bull. Korean Chem. Soc. 2011, 32, 3933–3940. [Google Scholar] [CrossRef][Green Version]
- Park, K.D.; Morieux, P.; Salomé, C.; Cotten, S.W.; Reamtong, O.; Eyers, C.; Gaskell, S.J.; Stables, J.P.; Liu, R.; Kohn, H. Lacosamide isothiocyanate-based agents: Novel agents to target and identify lacosamide receptors. J. Med. Chem. 2009, 52, 6897–6911. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Monti, D.; Caneva, E.; Delnevo, E.; Russo, G.; Prosperi, D. One-step bioengineering of magnetic nanoparticles via a surface diazo transfer/azide-alkyne click reaction sequence. Chem. Commun. 2008, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.; Ling, C.-C. Efficient and versatile modification of the secondary face of cyclodextrins through copper-catalyzed Huisgen 1,3-dipolar cycloaddition. Eur. J. Org. Chem. 2011, 2011, 4853–4861. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.F.L. Purification of Laboratory Chemicals, 3rd ed.; Pergamon: Oxford, UK, 1989. [Google Scholar]
- Fügedi, P. Synthesis of heptakis (6-O-tert-butyldimethylsilyl) cyclomaltoheptaose and octakis (6-O-tert-butyldimethylsilyl) cyclomaltooctaose. Carbohydr. Res. 1989, 192, 366–369. [Google Scholar] [CrossRef]
- Casas-Solvas, J.M.; Ortiz-Salmerón, E.; García-Fuentes, L.; Vargas-Berenguel, A. Ferrocene–mannose conjugates as electrochemical molecular sensors for concanavalin A lectin. Org. Biomol. Chem. 2008, 6, 4230–4235. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Muñoz, M.; Morales-Sanfrutos, J.; Perez-Balderas, F.; Hernandez-Mateo, F.; Giron-Gonzalez, M.D.; Sevillano-Tripero, N.; Salto-Gonzalez, R.; Santoyo-Gonzalez, F. Click multivalent neoglycoconjugates as synthetic activators in cell adhesion and stimulation of monocyte/machrophage cell lines. Org. Biomol. Chem. 2007, 5, 2291–2301. [Google Scholar] [CrossRef]
- Martínez, Á.; Ortiz Mellet, C.; García Fernández, J.M. Cyclodextrin-based multivalent glycodisplays: Covalent and supramolecular conjugates to assess carbohydrate–protein interactions. Chem. Soc. Rev. 2013, 42, 4746–4773. [Google Scholar] [CrossRef]
- Gallego-Yerga, L.; Benito, J.M.; Blanco-Fernández, L.; Martínez-Negro, M.; Vélaz, I.; Aicart, E.; Junquera, E.; Ortiz Mellet, C.; Tros de Ilarduya, C.; García Fernández, J.M. Plasmid-templated control of DNA–cyclodextrin nanoparticle morphology through molecular vector design for effective gene delivery. Chem. Eur. J. 2018, 24, 3825–3835. [Google Scholar] [CrossRef]
- Twyman, R.M. NMR spectroscopy-applicable elements/phosphorous-31. In Encyclopedia of Analytical Science, 2nd ed.; Worsfold, P., Townshend, A., Poole, C., Eds.; Elsevier: Oxford, UK, 2005; Volume 6, pp. 278–286. [Google Scholar] [CrossRef]
- Semple, J.E.; Sullivan, B.; Vojkovsky, T.; Sill, K.N. Synthesis and facile end-group quantification of functionalized PEG azides. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2888–2895. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Moscoso, A.; Guilloteau, N.; Bienvenu, C.; Méndez-Ardoy, A.; Jiménez Blanco, J.L.; Benito, J.M.; Le Gourriérec, L.; Di Giorgio, C.; Vierling, P.; Defaye, J.; et al. Mannosyl-coated nanocomplexes from amphiphilic cyclodextrins and pDNA for site-specific gene delivery. Biomaterials 2011, 32, 7263–7273. [Google Scholar] [CrossRef] [PubMed]
- François-Heude, M.; Méndez-Ardoy, A.; Cendret, V.; Lafite, P.; Daniellou, R.; Ortiz Mellet, C.; García Fernández, J.M.; Moreau, V.; Djedaïni-Pilard, F. Synthesis of high-mannose oligosaccharide analogues through click chemistry: True functional mimics of their natural counterparts against lectins? Chem. Eur. J. 2015, 21, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Cutrone, G.; Benkovics, G.; Malanga, M.; Casas-Solvas, J.M.; Fenyvesi, É.; Sortino, S.; García-Fuentes, L.; Vargas-Berenguel, A. Mannoside and 1,2-mannobioside β-cyclodextrin-scaffolded NO-photodonors for targeting antibiotic resistant bacteria. Carbohydr. Polym. 2018, 199, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Baussanne, I.; Benito, J.M.; Ortiz Mellet, C.; García Fernández, J.M.; Defaye, J. Dependence of concanavalin A binding on anomeric configuration, linkage type, and ligand multiplicity for thiourea-bridged mannopyranosyl–β-cyclodextrin conjugates. ChemBioChem 2001, 2, 777–783. [Google Scholar] [CrossRef]
- Smiljanic, N.; Moreau, V.; Yockot, D.; Benito, J.M.; García Fernández, J.M.; Djedaïni-Pilard, F. Supramolecular control of oligosaccharide–protein interactions: Switchable and tunable ligands for concanavalin A based on β-cyclodextrin. Angew. Chem. Int. Ed. 2006, 45, 5465–5468. [Google Scholar] [CrossRef] [PubMed]
- Dam, T.K.; Brewer, C.F. Thermodynamic studies of lectin-carbohydrate interactions by isothermal titration calorimetry. Chem. Rev. 2002, 102, 387–430. [Google Scholar] [CrossRef]
- Wang, X.; Matei, E.; Gronenborn, A.M.; Ramström, O.; Yan, M. Direct measurement of glyconanoparticles and lectin interactions by isothermal titration calorimetry. Anal. Chem. 2012, 84, 4248–4252. [Google Scholar] [CrossRef]
- Mangold, S.L.; Cloninger, M.J. Binding of monomeric and dimeric Concanavalin A to mannose-functionalized dendrimers. Org. Biomol. Chem. 2006, 4, 2458–2465. [Google Scholar] [CrossRef]
- Li, X.; Semiramoth, N.; Hall, S.; Tafani, V.; Josse, J.; Laurent, F.; Salzano, G.; Foulkes, D.; Brodin, P.; Majlessi, L.; et al. Compartmentalized encapsulation of two antibiotics in porous nanoparticles: An efficient strategy to treat intracellular infections. Part. Part. Syst. Char. 2019, 36, 1800360–1800369. [Google Scholar] [CrossRef]
- Gref, R.; Lu, M.; Quellec, P.; Marchand, M.; Dellacheriec, E.; Harnisch, S.; Blunk, T.; Muller, R.H. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Coll. Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Pei, Y.; Yeo, Y. Drug delivery to macrophages: Challeges and opportunities. J. Control. Release 2016, 240, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)-and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, Y.; Li, Z.; Yang, G.; Kochovski, Z.; Chen, G.; Jiang, M. “Sweet” architecture-dependent uptake of glycocalyx-mimicking nanoparticles based on biodegradable aliphatic polyesters by macrophages. J. Am. Chem. Soc. 2017, 139, 14684–14692. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.C.C.; Chaves, L.L.; Pinheiro, M.; Costa Lima, S.A.; Ferreira, D.; Sarmento, B.; Reis, S. Mannosylated solid lipid nanoparticles for the selective delivery of rifampicin to macrophages. Artif. Cells Nanomed. Biotechnol. 2019, 46 (Suppl. 1), 653–663. [Google Scholar] [CrossRef] [PubMed]
- Shibaguchi, K.; Tamura, A.; Terauchi, M.; Matsumura, M.; Miura, H.; Yui, N. Mannosylated polyrotaxanes for increasing cellular uptake efficiency in macrophages through receptor-mediated endocytosis. Molecules 2019, 24, 439. [Google Scholar] [CrossRef] [PubMed]
- Moros, M.; Hernáez, B.; Garet, E.; Dias, J.T.; Sáez, B.; Grazú, V.; González-Fernández, Á.; Alonso, C.; de la Fuente, J.M. Monosaccharides versus PEG-functionalized NPs: Influence in the celular uptake. ACS Nano 2012, 6, 1565–1577. [Google Scholar] [CrossRef]
- Anand, R.; Borghi, F.; Manoli, F.; Manet, I.; Agostoni, V.; Reschiglian, P.; Gref, R.; Monti, S. Host–guest interactions in Fe(III)-trimesate MOF nanoparticles loaded with doxorubicin. J. Phys. Chem. B 2014, 118, 8532–8539. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugate | N a | K × 10−5 (M−1) | ΔG0 (kcal mol−1) | ΔH (kcal mol−1) | TΔS0 (kcal mol−1) |
|---|---|---|---|---|---|
| 8b | 2.44 ± 0.01 | 2.25 ± 0.08 | −7.30 ± 0.02 | −14.49 ± 0.06 | −7.19 |
| 10b | 2.79 ± 0.01 | 5.12 ± 0.25 | −7.79 ± 0.03 | −13.96 ± 0.07 | −6.17 |
| nanoMOFs@8 c | 0.90 ± 0.01 | 14.20 ± 1.64 | −8.39 ± 0.07 | −334.20 ± 6.91 | −325.81 |
| nanoMOFs@10 c | 1.06 ± 0.01 | 24.50 ± 1.17 | −8.72 ± 0.03 | −314.10 ± 3.37 | −305.38 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cutrone, G.; Li, X.; Casas-Solvas, J.M.; Menendez-Miranda, M.; Qiu, J.; Benkovics, G.; Constantin, D.; Malanga, M.; Moreira-Alvarez, B.; Costa-Fernandez, J.M.; et al. Design of Engineered Cyclodextrin Derivatives for Spontaneous Coating of Highly Porous Metal-Organic Framework Nanoparticles in Aqueous Media. Nanomaterials 2019, 9, 1103. https://doi.org/10.3390/nano9081103
Cutrone G, Li X, Casas-Solvas JM, Menendez-Miranda M, Qiu J, Benkovics G, Constantin D, Malanga M, Moreira-Alvarez B, Costa-Fernandez JM, et al. Design of Engineered Cyclodextrin Derivatives for Spontaneous Coating of Highly Porous Metal-Organic Framework Nanoparticles in Aqueous Media. Nanomaterials. 2019; 9(8):1103. https://doi.org/10.3390/nano9081103
Chicago/Turabian StyleCutrone, Giovanna, Xue Li, Juan M. Casas-Solvas, Mario Menendez-Miranda, Jingwen Qiu, Gábor Benkovics, Doru Constantin, Milo Malanga, Borja Moreira-Alvarez, José M. Costa-Fernandez, and et al. 2019. "Design of Engineered Cyclodextrin Derivatives for Spontaneous Coating of Highly Porous Metal-Organic Framework Nanoparticles in Aqueous Media" Nanomaterials 9, no. 8: 1103. https://doi.org/10.3390/nano9081103
APA StyleCutrone, G., Li, X., Casas-Solvas, J. M., Menendez-Miranda, M., Qiu, J., Benkovics, G., Constantin, D., Malanga, M., Moreira-Alvarez, B., Costa-Fernandez, J. M., García-Fuentes, L., Gref, R., & Vargas-Berenguel, A. (2019). Design of Engineered Cyclodextrin Derivatives for Spontaneous Coating of Highly Porous Metal-Organic Framework Nanoparticles in Aqueous Media. Nanomaterials, 9(8), 1103. https://doi.org/10.3390/nano9081103