3.1. Morphology of PPy-COOH-g-HDI-GO Grafted Samples
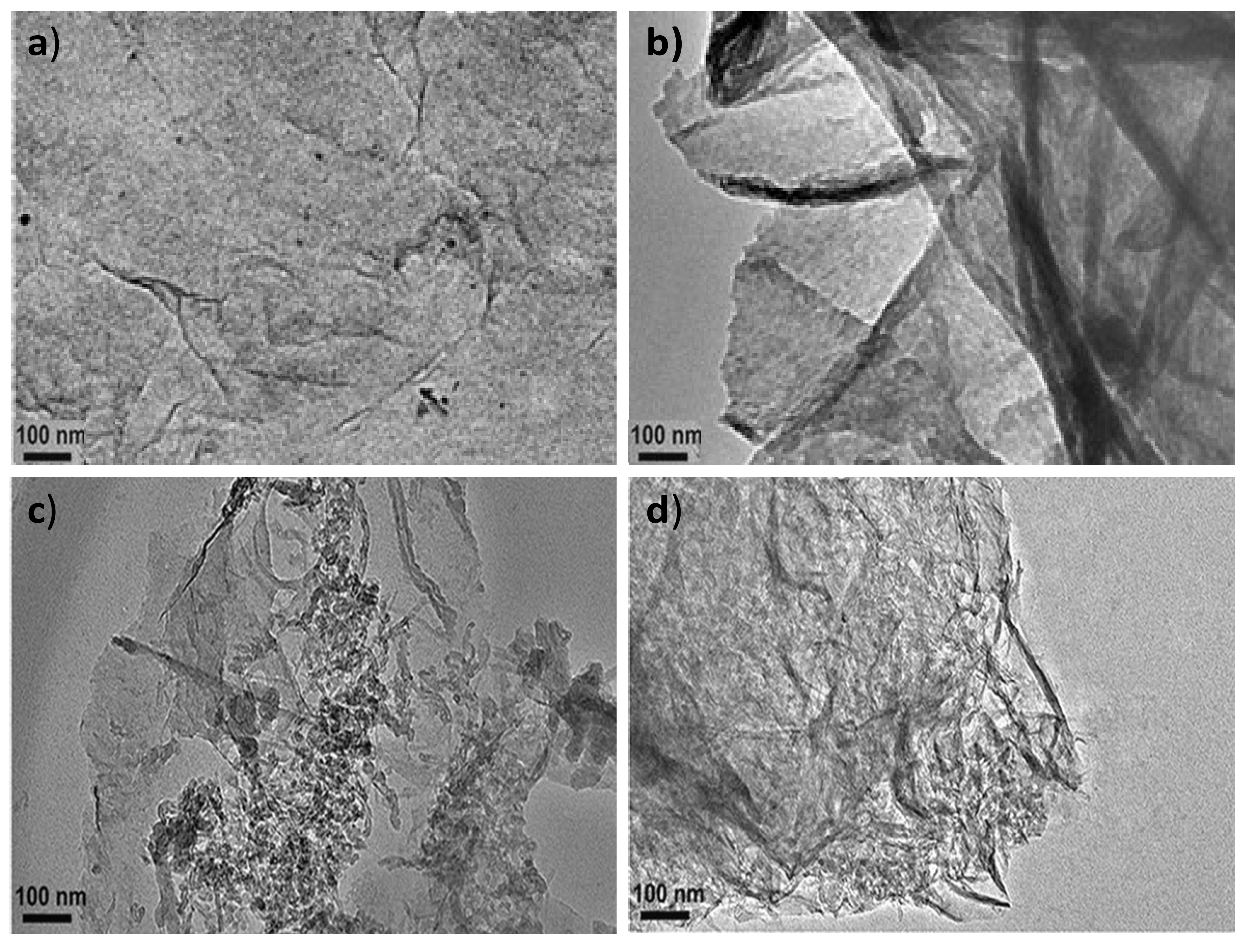
The surface morphology of PPy-COOH, HDI-GO, and the grafted nanocomposites was assessed by TEM, and typical images are compared in
Figure 1. PPy-COOH (
Figure 1a) shows a homogenous somewhat-coarse surface, with good film-forming ability. Nonetheless, its morphology is difficult to be visualized by TEM, due to its amorphous character and the poor contrast of the polymeric functional groups.
On the other hand, the HDI-GO samples display a homogeneous and uniform appearance (
Figure 1b), with a stacked-like structure, comprising flexible and slightly curled flakes with an average thickness of 20 nm. It should be noted that this thickness is significantly higher than that reported for a single monolayer graphene (about 0.34 nm [
24]) due to the presence of oxygen-containing functional groups and the alkyl chains of the organic diisocyanate attached on both sides of the flakes. Further, each sample contains several modified graphene layers. The darker areas in
Figure 1b are probably related to different thicknesses or disposition of the nanosheets due to the presence of the HDI. This is in agreement with former observations described by other authors who anchored alkyl or polymeric chains onto graphene [
25,
26]. In accordance with those works, the grafted chains extend into the solution, but in the solid state the chains collapse onto the GO surface, forming small domains that probably correspond to more-obscure features. It is also important to mention that the HDI-GO samples show a lower degree of folding than typically observed for pristine GO [
27], since the alkyl chains can wrap around the GO nanosheets and shield the wrinkles.
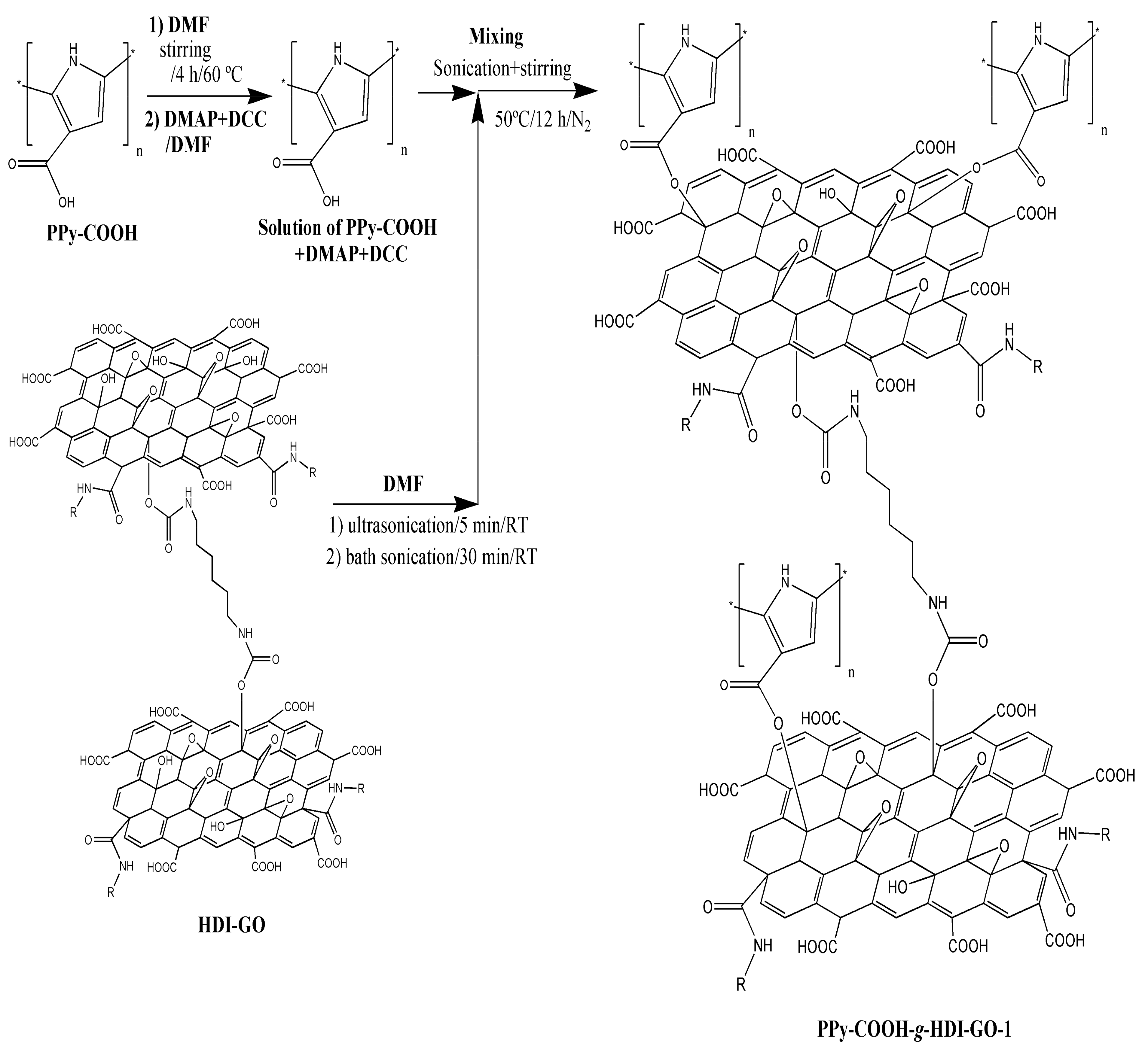
Regarding the PPy-COOH-g-HDI-GO-1 sample (
Figure 1c), it appears as a heterogeneous mixture with continuous clearer areas attributed to the polymer and darker regions corresponding to the HDI-GO. It is important to note that the grafting reactions are not quantitative, due to steric effects, and hence the grafted samples are composed of regions in which the nanofiller is covalently bonded to the monomer matrix material, as well as parts where the nanofiller interacts with the monomers via non-covalent interactions, as will be discussed later. Interestingly, the average flake thickness is slightly decreased compared to HDI-GO, likely because the polymeric chains intercalated between the nanosheets and induced exfoliation. This is in contrast to some previous studies on polymer grafted-graphene samples, in which the thickness increased upon the covalent bonding to the polymeric segments [
28]. This discrepancy can be explained considering two phenomena that can take place upon the PPy-COOH grafting: on the one hand, the presence of polymer chains onto the nanofiller surface can increase layer thickness, while on the other hand the intercalation of the chains within the GO nanosheets can cause exfoliation, thus reducing layer thickness. It seems that in this case the intercalation process predominates, resulting in a lower flake thickness overall.
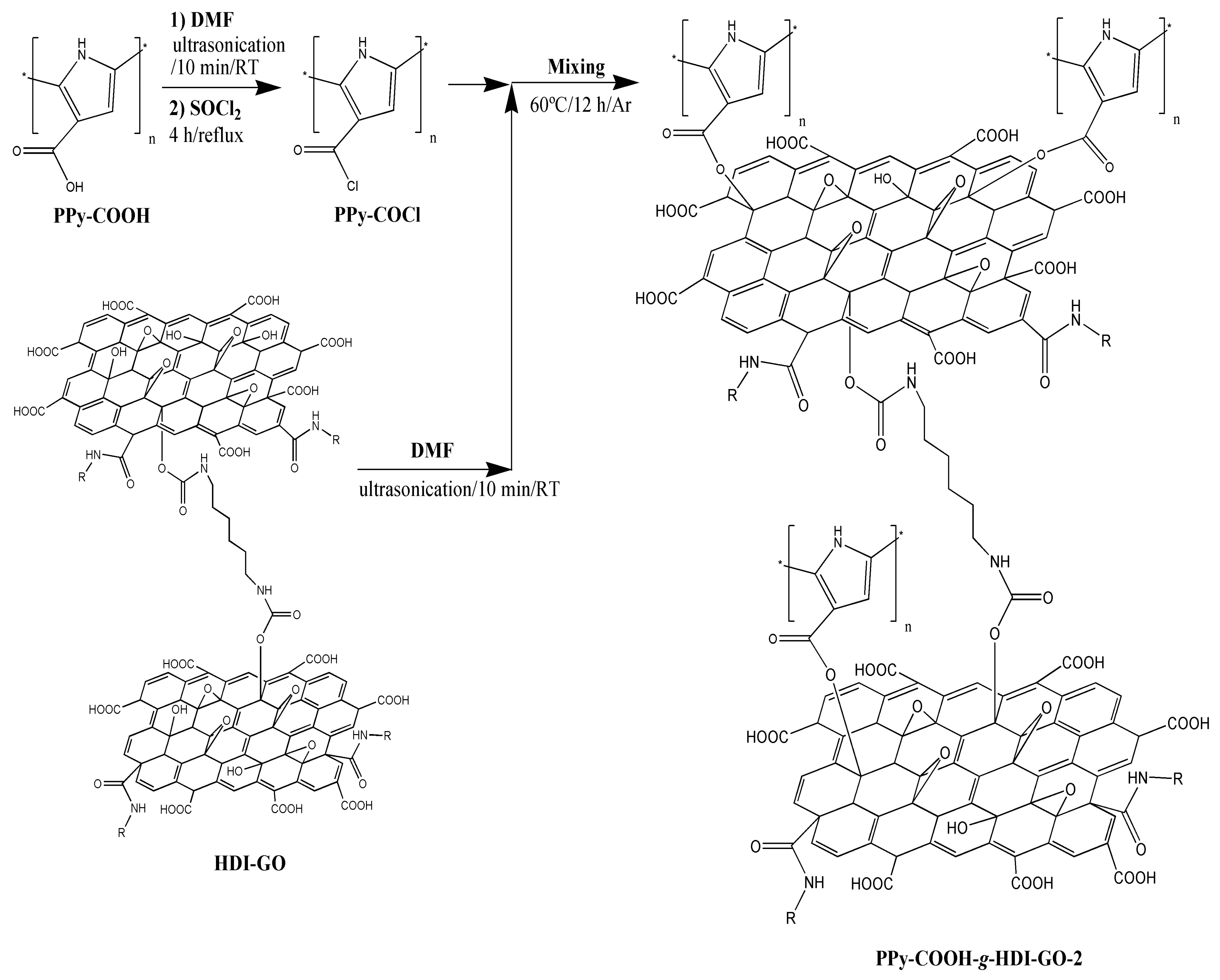
The PPy-COOH-g-HDI-GO-2 (
Figure 1d) appears more homogeneous compared to the other grafted sample, and the darker areas corresponding to the HDI-GO are less visible. It presents a rougher and more wrinkled surface, suggesting the presence of the PPy-COOH wrapping around the GO nanosheets, forming a tight interfacial layer, without voids or discontinuities, indicating good interfacial adhesion and compatibility between the two nanocomposite phases. The average flake thickness is slightly higher than that found for the HDI-GO, suggesting that, in this case, the grafting of the polymer chains onto the GO surface prevails. All these observations confirm that the PPy-COOH is physically and/or chemically bonded to the surface of the HDI-GO.
BET specific surface area (SSA) was measured to get further insight about the influence of the esterification reactions on the level of exfoliation of the GO nanosheets in the different samples, and the results obtained are collected in
Table S1 in the Supplementary Materials. A high SSA is interesting for a variety of applications such as supercapacitors [
29]. It is known that the SSA of graphene-based nanomaterials increases as the sheet thickness decreases [
15]. The SSA of GO (about 100 m
2/g) slightly decreased upon functionalization with HDI, which is reasonable considering that the incorporation of the alkyl chains reduces the sp
2 surface area. On the other hand, the SSA of PPy-COOH is close to 25 m
2/g, and increased upon HDI-GO grafting, in agreement with previous studies on PPy/graphene nanocomposites [
29]. This increment was more significant for PPy-COOH-g-HDI-GO-1, in agreement with its thinner flakes and better level of exfoliation, as revealed by TEM.
Further information about the morphology of the samples was attained by SEM, and typical images are shown in
Figure 2.
Figure 2a reveals nano- and meso-particles of PPy-COOH with spherical morphology and a smooth surface arranged in random clusters showing an average particle diameter of 110 nm. Regarding the HDI-GO (
Figure 2b), it is composed of flexible and quite well-exfoliated flakes, with thicknesses between 10 and 50 nm and lateral sizes of 1–2 μm. Conversely, PPy-COOH-
g-HDI-GO-2 (
Figure 2c) appears as a homogenous sample with the graphene nanosheets randomly dispersed within the PPy-COOH particles. The sheets are less bendable and appear with a smoother surface than those in HDI-GO, probably due to the grafting of the polymeric, and appear thicker, with thicknesses of up to 70 nm, as can be observed at a higher magnification (
Figure 2d). Similar morphology, albeit with thinner sheets and uneven HDI-GO dispersion, was found for PPy-COOH-
g-HDI-GO-1. From all the above observations, it can be inferred that the esterification reaction via acylation with thionyl chloride leads to higher grafting yields, hence larger amounts of polymer chains anchored to the nanofiller surface, which will be discussed in the following sections.
3.2. IR and Raman Spectroscopies
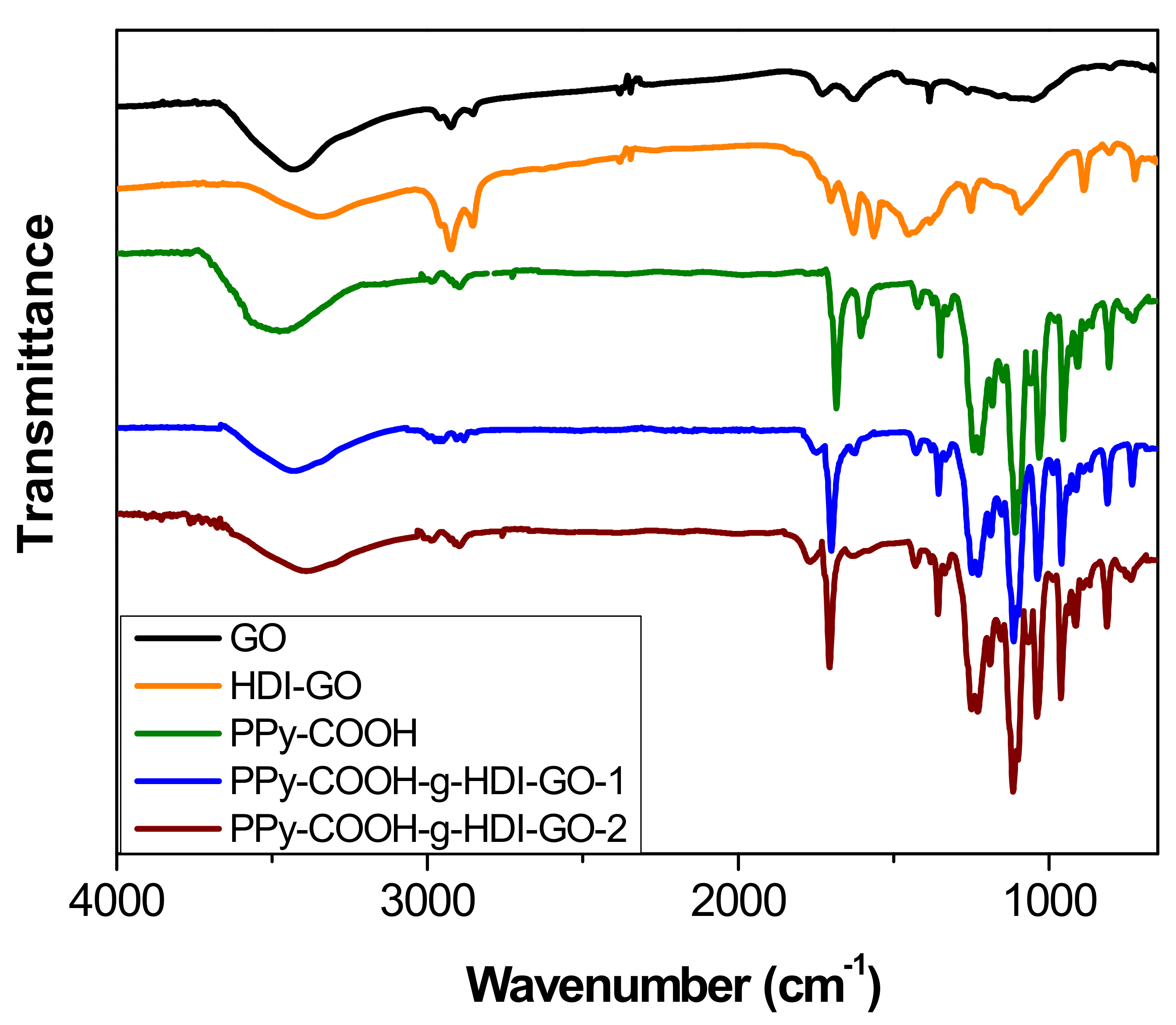
The chemical changes that took place after treatment of PPy-COOH with HDI-GO were examined by FT-IR spectroscopy (
Figure 3). For comparative purposes, the spectrum of pristine GO was also examined. Neat GO encloses epoxide and hydroxyl groups on the basal planes and carboxylic acids at the flake edges [
11,
15]. The most distinctive characteristics in the IR spectrum of GO are found at ~3435 cm
−1, ascribed to the O–H stretching; at about 2930 and 2850 cm
−1, attributed to sp
2 and sp
3 C–H stretching vibrations (formed at defect sites of the aromatic system); at 1730 cm
−1 due to the C=O stretching of the carboxylic group; at ~1620 cm
−1, ascribed to the C–C stretching of the aromatic network; at around 1400 cm
−1, corresponding to the O–H deformation; and at 1260 cm
−1, due to the C–OH stretching [
30].
Upon treatment with the HDI, the strength of the O–H stretching decreased and the band shifted to lower wavenumbers, due to the formation of H bonds between the residual OH moieties and other oxygenated groups close to them. Further, the shift should also be a result of the overlapping with the N–H stretching of the carbamate groups. Nonetheless, it should be taken into account that the intensity of the O–H stretching peak can also be influenced by the presence of environmental humidity, and hence cannot be used as a conclusive proof of HDI grafting. Moreover, the peaks at ~2930 and 2850 cm
−1 originated from asymmetrical and symmetrical stretching vibrations of the methylene chains of the HDI, respectively. Further, instead of the peak at 1730 cm
−1 corresponding to the C=O stretching of carboxylic acids, a new band appears at ~1710 cm
−1, ascribed to the C=O of the carbamate esters [
21]. In addition, strong novel bands arising from the carbamate ester groups can be observed at about 1650 and 1580 cm
−1, the first due to the coupling of the C=O stretching with the N–H bending, and the second due to the coupling of the N–H bending with the C–N stretching [
31]. Other peaks can also be found at about 1110 cm
−1, related to –C(=O)–O and C–N stretching vibrations of the carbamate groups; at 880 cm
−1, attributed to C–H out-of-plane bending vibrations of the substituted aromatic rings; and at around 720 cm
−1, due to the rocking of the methylene groups of HDI.
Regarding the PPy-COOH, an intense and broad band can be found centered at 3482 cm
−1, due to the overlapping of O–H and N–H stretching vibrations. The peak at 1673 cm
−1 corresponds to the C=O stretching of the carboxylic acid, the peaks at 1580 and 1430 cm
−1 are assigned to fundamental vibrations of the pyrrole rings, and those at about 1200 and 920 cm
−1 correspond to the bipolaron bands [
32], indicating that the synthesized PPy-COOH is in an oxidized state.
The peaks of HDI-GO cannot be observed in the spectra of PPy-COOH-g-HDI-GO-1 or PPy-COOH-g-HDI-GO-2, which is reasonable considering that the total amount of this modified GO is only 9% of the weight of the grafted samples, hence its bands should be very weak and masked by the polymer ones. Thus, the spectra of both samples is quite similar to that of the polymer, albeit with decreased intensity of the O–H stretching band due to the esterification of part the hydroxyl groups with the carboxylic acids of PPy-COOH. Nonetheless, as mentioned earlier, this reduction should be considered only as indicative since the peak intensity could have been affected by the stretching vibration of the water molecules in the environment. Further, the band is significantly shifted to lower wavenumbers, and appears at 3430 and 3388 cm
−1 for PPy-COOH-g-HDI-GO-1 and PPy-COOH-g-HDI-GO-2, respectively. Besides, the band corresponding to the carboxylic acid shifts up to 1699 and 1708 cm
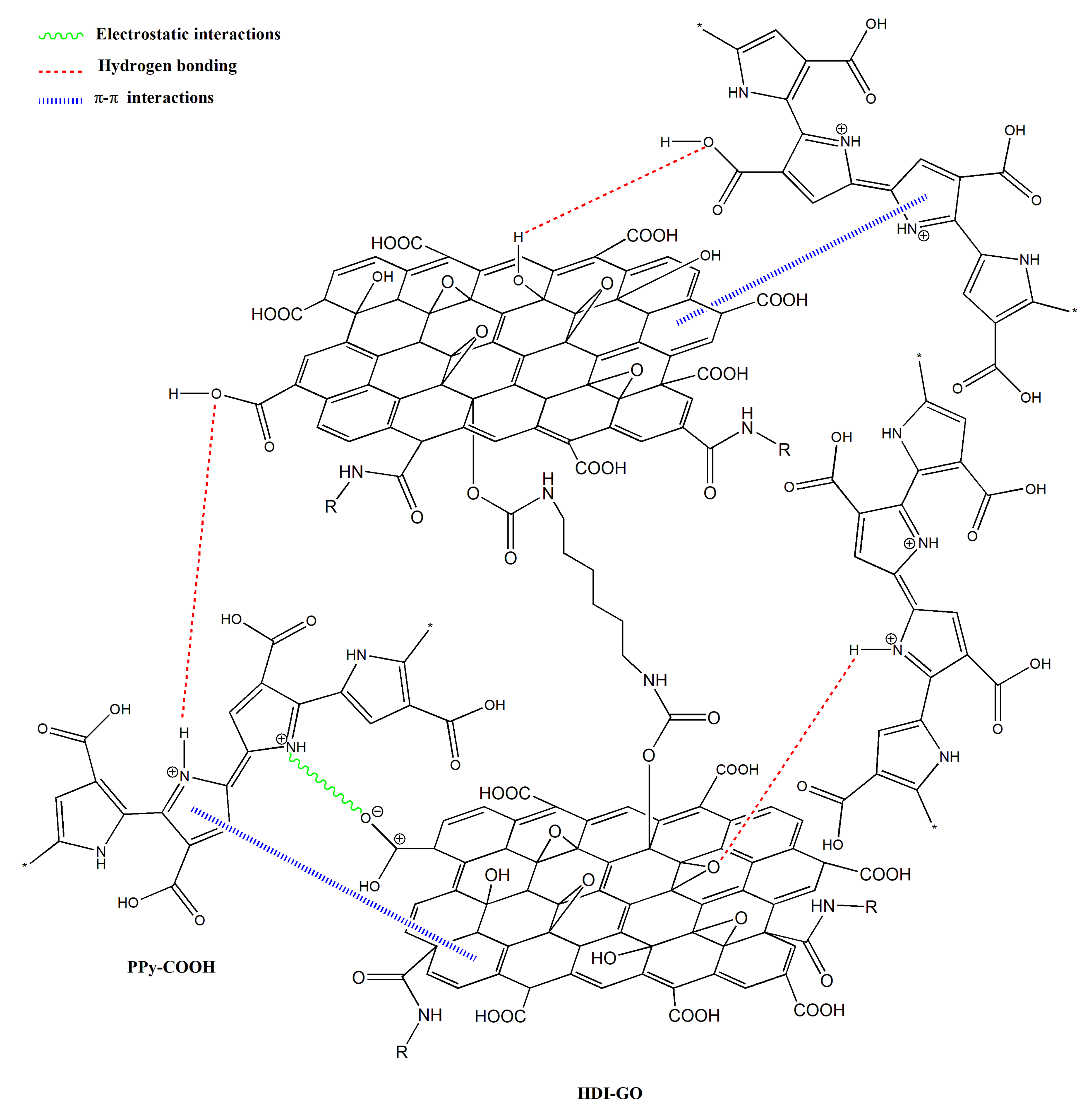
−1, respectively, and this behavior might be an indication of H-bonding interactions between the OH and NH moieties of PPy-COOH and the oxygenated functional groups of the GO surface [
33,
34] (see
Scheme 3). In addition, strong π-π stacking interactions take place between the aromatic rings of pyrrole and those of GO, as well as electrostatic interactions between the negatively charged carboxylic acid groups of the HDI-GO surface and the positively charged PPy-GOOH rings in the oxidized state (bipolaron structure), which would also induce a shift in the band positions. More importantly, after the grafting reactions, a new band appears at 1743 and 1760 cm
−1 for PPy-COOH-g-HDI-GO-1 and PPy-COOH-g-HDI-GO-2, respectively, related to the C=O stretching of the ester group. The appearance of this band in the spectra of both grafted samples is one of the outmost outcomes derived from the IR analysis, given that it corroborates that part of the carboxylic acids of PPy-COOH have reacted with the hydroxyl moieties of the HDI-GO.
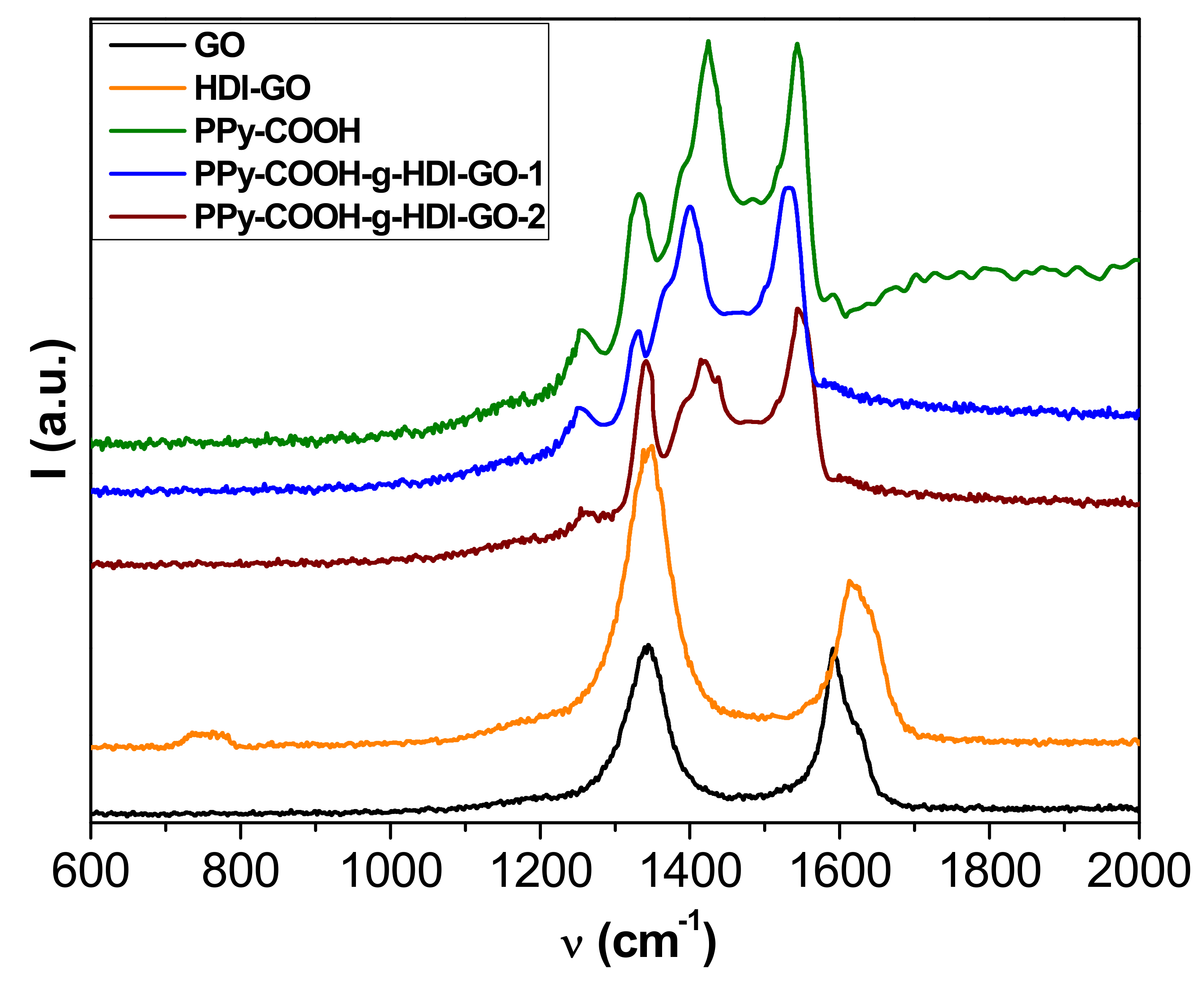
More information about the interactions between the polymer and the nanofillers was obtained from Raman Spectroscopy, and the spectra obtained from the different samples are depicted in
Figure 4. The Raman spectrum of GO shows two prominent bands: the defect D band that accounts for the structural disorder at 1354 cm
−1, and the tangential G band arising from graphitized structures at about 1600 cm
−1 [
29]. An analogous spectrum is found for HDI-GO, although it shows a widening and an upshift of the G-band, attributed to a modification in the electronic structure of GO when electron-acceptor groups are incorporated into the nanosheets [
21]. Moreover, this upshift can be linked to a rise in the concentration of defects in the graphene sheets. It is important to note that the D to G band intensity ratio (
ID/
IG) gives quantitative information on the concentration of defects in graphene flakes—the higher the ratio, the larger the amount of defects [
22]. This ratio increases by 1.74 from GO to HDI-GO, which demonstrates a fall in the structural order after anchoring the GO to the HDI chains.
The spectrum of PPy-COOH exhibits five main features [
35]: the C–C inter-ring stretching at 1260 cm
−1, the single C–C stretching at 1330 cm
−1, the C-N stretching at 1390 cm
−1, the C=C symmetrical stretching at 1430 cm
−1, and the C=C antisymmetrical stretching at 1550 cm
−1. Regarding the spectra of the grafted samples, the D and G bands of HDI-GO cannot be observed because they are likely very weak and masked by the polymer bands. On the other hand, a drop in the intensity of the bands of the polymer and a shift in their position is detected. For the PPy-COOH-g-HDI-GO-1, the bands are shifted towards lower wavenumbers, which could be due to the rupture and deformation of bonds induced by the grafting process [
36]. In contrast, the bands in the PPy-COOH-g-HDI-GO-2 are slightly upshifted, likely because the grafting of polymeric chains onto the HDI-GO surface prevails (as revealed by TEM images), together with the adsorption of the PPy-COOH segments onto the nanofiller via π-π stacking and H-bonding interactions, which can lead to the formation of a tightly coated polymeric layer on the nanomaterial surface. These shifts in the peaks confirm again the intense interactions between the polymer derivative and the modified GO. Analogous behavior of the shift of the Raman bands has previously been reported for poly(N-vinyl carbazole)/PPy/rGO composites [
37].
3.3. Thermal Stability and Yield of the Grafting Reaction
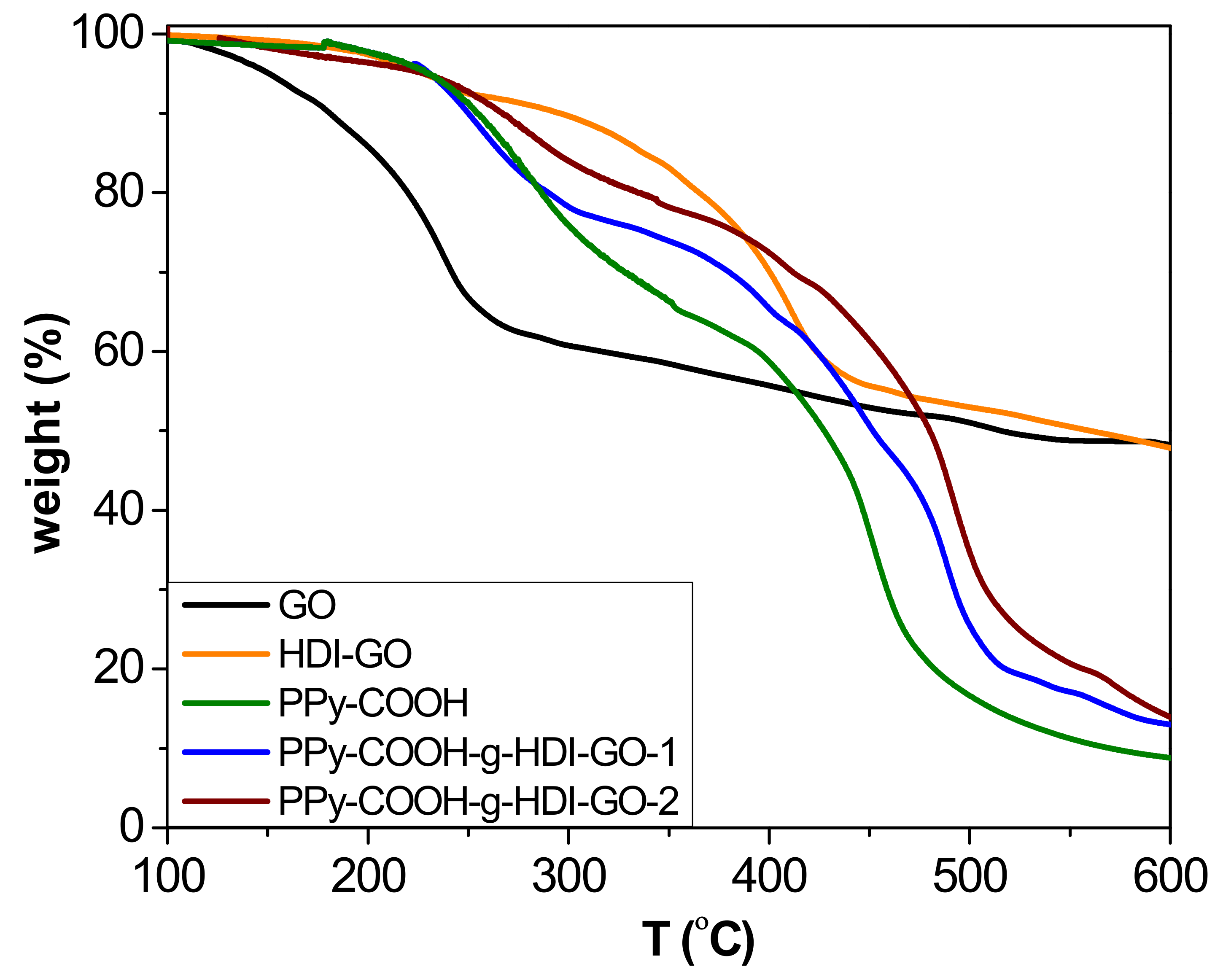
TGA analysis under a nitrogen atmosphere was carried out to investigate the thermal stability of the synthesized PPy-COOH and the grafted samples (
Figure 5), and to obtain information about the yield of the grafting reactions. The esterification reactions carried out in this work do not proceed in quantitative yields due to steric effects, and the efficiency depends on the esterification route. The thermobalance was coupled to a mass spectrometer, enabling identification of the degradation products. All the results derived from TGA analysis are collected in
Table S1 (
Supplementary Materials). Pristine GO shows a one-step degradation process, with the most important weight loss at temperatures below 240 °C attributed to the removal of epoxide, hydroxyl, and carboxylic acid surface groups [
21]. In addition, a small weight loss is observed at higher temperatures due to the elimination of more functional groups. In contrast, the HDI-GO presents two decomposition steps, the former arising from the removal of remaining surface oxygen-containing groups, and the other to the degradation of the HDI moieties connected to the GO surface. The incorporation of the alkyl chains onto the nanomaterial surface results in a significant improvement in thermal stability, leading to an increase in the initial degradation temperature (T
i) of about 70 °C. On the other hand, the PPy-COOH exhibits a weight loss in the range of 200–380 °C that is associated with a fragment of m/z 45 (corresponding to elimination of the carboxylic acids [
38]), as well as a second weight loss between 380 and 500 °C related to the decomposition of the polymeric backbone, which exhibits the maximum rate (T
max) at 444 °C. Regarding the grafted samples, two major weight losses can also be observed that are similar to the parent polymer, the first taking place in the same temperature range while the second is shifted to higher temperatures. Taking into account the weight loss of the first stage, the amount of COOH groups chemically bound to the OH moieties of the HDI could be predicted (considering the experimental error). The difference between the weight loss of this step for the polymer derivative and the grafted samples obtained after the esterification process corresponds to the yield of the grafting reaction [
16]: ~31% and 42% for PPy-COOH-g-HDI-GO-1 and PPy-COOH-g-HDI-GO-2, respectively. Thus, it is confirmed that the esterification approach via reaction of the carboxylic groups with SOCl
2 was more effective, leading to higher reaction yields.
The second step is associated with the decomposition of the PPy-COOH backbone, which occurs at higher temperatures in the grafted samples than the PPy-COOH. Thus, for PPy-COOH-g-HDI-GO-1 and PPy-COOH-g-HDI-GO-2, T
max of this second stage occurs about 30 and 40 °C above that of the neat polymer, respectively. The higher thermal stability of the second sample is ascribed to its higher level of grafting. This enhancement in the maximum degradation rate is associated with the HDI-GO alkyl chains covalently bonded to the polymeric segments, which successfully hinder the transport of the degradation products from the interior of the matrix to the gas phase. In addition, the higher decomposition temperatures indicate that chemical interactions exist between the polymer and the nanofillers [
39]. Furthermore, it has been reported [
40] that the thermal resistance at the interface between carbon nanomaterials and polymers decreases with the creation of chemical bonds, resulting in higher thermal conductivity that promotes heat dissipation within the sample. Thus, the increments found herein are significantly higher than those reported for PPy/rGO and PPy/graphene nanosheet nanocomposites prepared via in situ polymerization with nanofiller contents up to 20 and 40 wt%, respectively [
41,
42], which further corroborates the efficiency of the polymer-nanofiller covalent bonding to improve the thermal stability. A similar behavior of thermal stability improvement, with increments in T
max in the range of 40–65 °C, has been described for modified poly(ether ether ketone) (PEEK) derivatives covalently anchored to single-walled carbon nanotubes (SWCNTs) functionalized with carboxylic acids [
16]. In this study, the superior increment observed for the sample prepared via conversion of the carboxylic groups to acyl chloride should be related to its greater number of covalent linkages. Residue data of the different samples at 600 °C are collected in
Table S1. The values obtained for the grafted samples are consistent with their HDI-GO percentage taking into account that the HDI-GO loses about 48% of its weight below 600 °C, and that the polymer is not completely decomposed at such temperature.
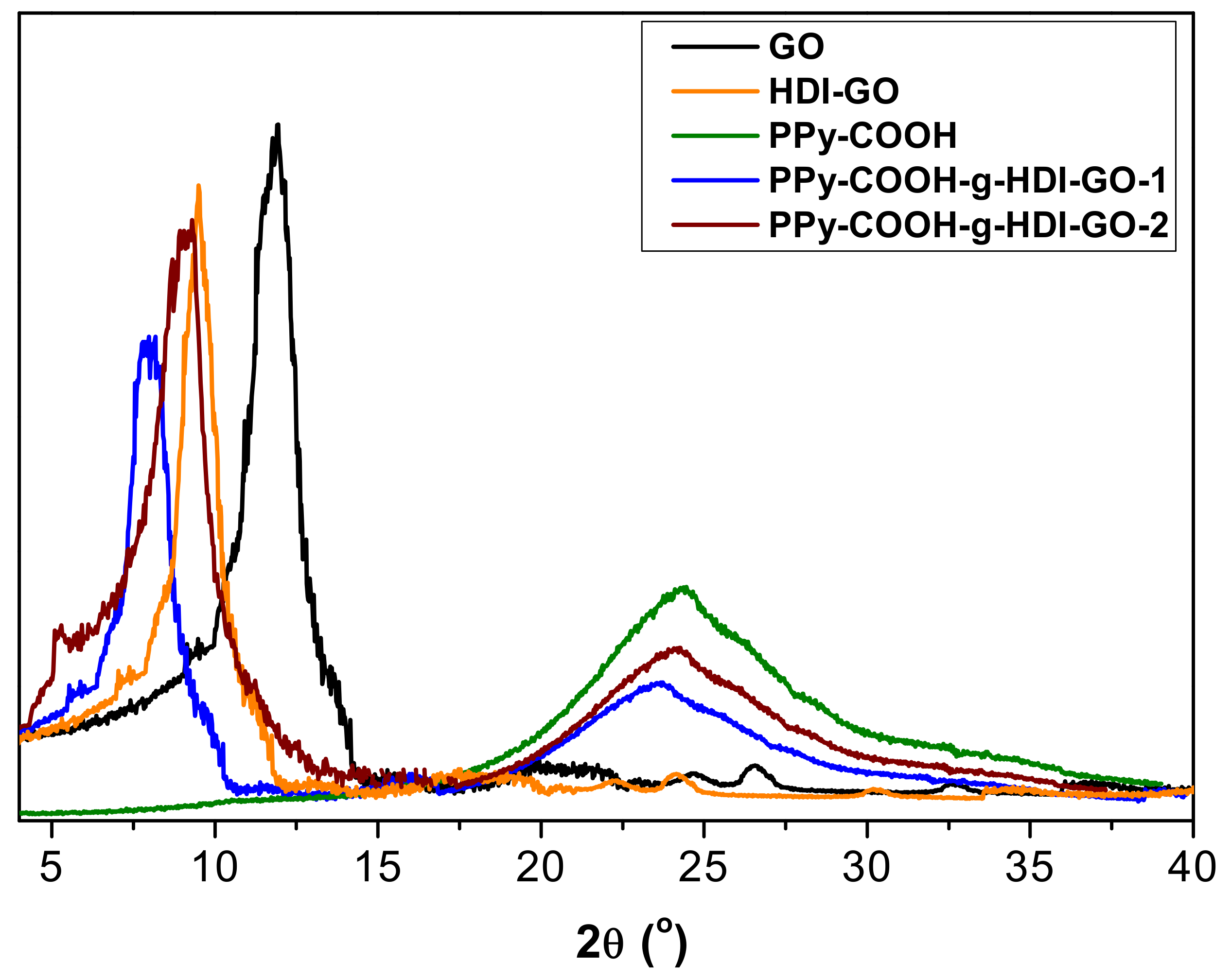
3.4. XRD Analysis
The different samples were also analyzed by XRD measurements, and representative diffractograms of PPy-COOH, GO, HDI-GO and the two grafted samples are compared in
Figure 6. GO shows a typical peak at 2θ = 11.8° ascribed to the (002) crystalline plane [
11], which corresponds to an interlayer
d spacing of 0.748 nm according to the Bragg’s equation [
43]. In the HDI-GO, the diffraction peak shows lower intensity and appears at 2θ = 9.2°, which corresponds to a
d value of 0.961 nm. This increase in the
d spacing is ascribed to the intercalation of the alkyl chains between the GO layers, as reported previously for other polymer/GO nanocomposites [
44,
45].
Regarding PPy-COOH, a broad peak is found in the region 17° < 2θ < 30°, revealing its amorphous nature. Such a wide peak indicates the absence of ordered arrangement of the chains, and is consistent with the structure reported in the literature [
46]. This broad reflection is also found in the patterns of the grafted samples, although it appears at lower 2θ values, suggesting that the chains are more separated. Further, its intensity decreases, which is consistent with the reduction in the percentage of PPy-COOH upon HDI-GO grafting. These results are in contrast with those reported for PPy/graphene nanosheets, where the polymer grew along the nanomaterial surface during the in situ polymerization, resulting in a more-ordered structure [
42]. On the other hand, a downshift of the (002) peak is also observed here in comparison with both GO and HDI-GO (
Figure 6), corroborating the rise in the interlayer spacing of the carbon nanomaterial. The increment is higher for PPy-COOH-g-HDI-GO-1, in agreement with its larger degree of exfoliation and lower flake thickness (as indicated by TEM) and its higher SSA (calculated from BET measurements). Overall, XRD patterns confirm the effectiveness of the grafting process developed herein to aid the dispersion of the PPy-COOH chains within the interlayer spacing of the HDI-GO sheets.
3.5. Sheet Resistance
The influence of HDI-GO on the electrical properties of PPy-COOH was investigated by measuring the sheet resistance (R
S), and the results are collected in
Table S1. Raw GO and HDI-GO were found to be electrically insulating, since the functional groups on the GO surface disrupted the conjugated π-electron system of the graphene sheets and hence it was not possible to measure their R
S value. In contrast, PPy-COOH displayed a low sheet resistance of about 180 Ω/sq, in agreement with the high electrical conductivity reported in previous studies [
47]. This good electrical performance is attributed to the compact and smooth surface morphologies of the polymer nano- and meso-particles, as revealed by SEM analysis. As expected, the grafted samples show higher R
S than neat PPy-COOH, due to the anchoring of an insulating nanomaterial to the polymeric chains. Nonetheless, the values obtained (330 and 253 Ω/sq for PPy-COOH-g-HDI-GO-1 and PPy-COOH-HCI-GO-2, respectively) are lower than those estimated when considering the percentage of HDI-GO in the nanocomposites by a simple rule of mixtures. The discrepancy could be explained by taking into account the large number of factors that affect the conduction mechanism in this polymer, such as level of crystallinity, grain size, changes in the conformation of the polymeric chains, and doping and screening effects [
48]. PPy-COOH possesses π electrons, since its structure contains alternate double and single bonds. Electrical conductivity in this polymer originates from the motion of electrons, or from positively charged carriers along the chains and the hopping of these carriers among chains [
48]. Throughout the chemical polymerization, partial oxidation of the chains results in the formation of positive charges on the polymer backbone. The electrons of the π cloud of GO could settle near the positive charges to maintain neutrality of PPy backbone. Thus, the HDI-GO could act as a counterion of the oxidized form of PPy-COOH. This would result in the formation of PPy-COOH-HDI-GO charge-transfer complexes via strong donor–acceptor interactions, hence improving charge carrier transport. Further, the large SSA of HDI-GO could facilitate the charge transfer along the chains. All these facts could account for the relatively high electrical conductivity found for the grafted samples, despite the high electrical resistance of HDI-GO.
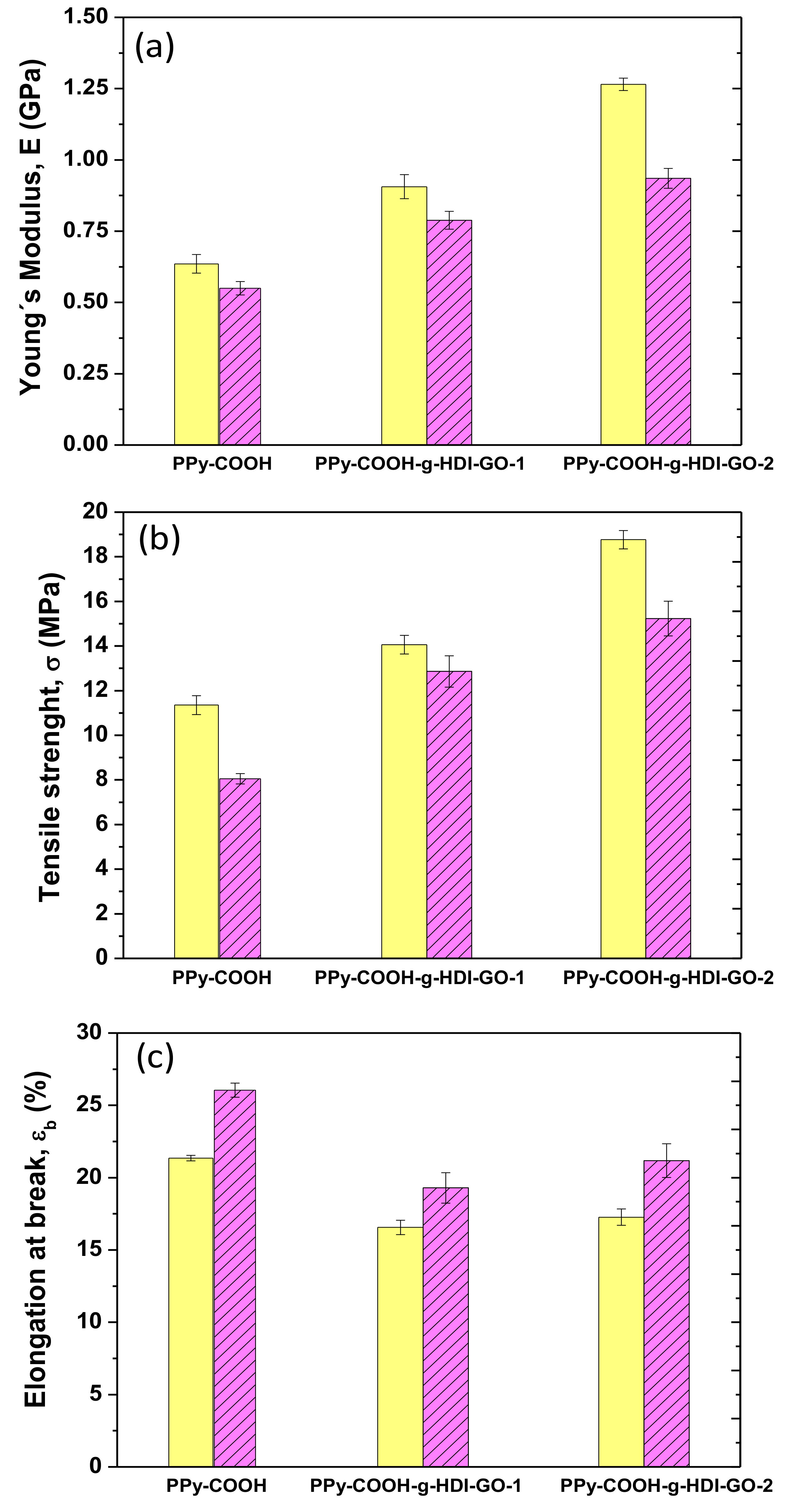
3.6. Mechanical Properties
The mechanical properties of PPy-COOH and the grafted samples were explored by tensile measurements at 25 and 100 °C, and their Young´s (or elastic) modulus, tensile strength, and elongation (or strain) at break are plotted in
Figure 7. At 25 °C, PPy-COOH exhibits elastic modulus and tensile strength values of 0.62 GPa and 11 MPa, respectively. The grafting of the HDI-GO causes large enhancements in both modulus and strength (
Figure 7a,b) by about two-fold and 73%, respectively, for the sample with the highest grafting efficiency. An analogous tendency, though with somewhat lower increases, is found for the PPy-COOH-g-HDI-GO-1. The exceptional modulus and strength improvements attained demonstrate the high reinforcing efficiency of HDI-GO, likely arising from the strong PPy-COOH-HDI-GO interface adhesion achieved by the covalent bonding, therefore very effective load transfer from the PPy-COOH to the nanofiller sheets. A similar percentage of modulus increment was reported for a PEEK derivative covalently anchored to acid-functionalized SWCNTs [
16]. The higher increment observed for PPy-COOH-g-HDI-GO-2 in comparison with the other grafted sample is likely associated with its higher yield of grafting. Furthermore, it is important to note that these samples contain a high GO content (~10 wt%), higher than the quantity typically incorporated in polymeric composites [
49].
Focusing on the elongation at the break (
Figure 7c), PPy-COOH presents a value close to 21% at 25 °C, which decreases upon the grafting of HDI-GO, the drop being ~20% for both grafted samples. This is the characteristic behavior reported for polymer/nanofiller composites [
50], given that the fillers, especially those covalently bonded to the polymer, confine the ductile flow of the polymeric segments. Further, the strong adhesion between PPy-COOH and HDI-GO achieved by means of hydrogen bonding and electrostatic and π-π stacking interactions also promotes the decreased plasticity. Interestingly, the fall in ductility is similar for PPy-COOH-g-HDI-GO-1 and PPy-COOH-g-HDI-GO-2, despite the second exhibiting a higher grafting yield; hence PPy-COOH-g-HDI-GO-2 would be expected to show lower elongation at break. This could be due to the more-homogenous dispersion of the HDI-GO within the PPy-COOH polymer matrix, as suggested by SEM, since the presence of small GO agglomerates would limit the deformation of the matrix chains [
22]. Thus, the grafting of carbon nanomaterials to polymer derivatives through in situ polymerization reactions leads to nanocomposites with more homogenously dispersed nanofillers than those prepared via direct integration using conventional methods such as melt blending or solution casting, and the higher the efficiency of the grafting reaction, the more homogeneous the resulting nanocomposites [
51].
For certain applications, like photovoltaics [
52] or high-temperature electronics, it is interesting to analyze the effect of temperature on the mechanical properties, stiffness in particular. Thus, the values of the modulus, strength, and ductility were also measured at 100° C, and the results are shown in
Figure 7. Systematically, the stiffness and strength decrease with increasing temperature, due to the plasticization effect of the polymeric chains at high temperatures [
53]. Nonetheless, the overall reinforcing efficiency of the HDI-GO seems to be comparable at both temperatures. Accordingly, the maximum improvements in stiffness and strength compared to PPy-COOH are about 80% and 90%, respectively, for the PPy-COOH-g-HDI-GO-1 sample. This confirms that the chain mobility constraint imposed by the grafted HDI-GO nanosheets is maintained at high temperatures. These results are in agreement with those described for polypropylene filled with similar loadings (~8.0 wt%) of inorganic nanoparticles, in which the reinforcing efficiency hardly changes with increasing temperature [
54]. Conversely, a rise in the elongation at break is found with increasing temperature, which is the typical behavior expected for amorphous and semicrystalline polymeric materials. As the temperature increases, the amorphous regions become more mobile and this higher mobility is reflected in higher ductility. Compared to the neat polymer, a drop in ductility is again found for the grafted samples, the extent of the diminution being about 21% and 17% for PPy-COOH-g-HDI-GO-1 and PPy-COOH-g-HDI-GO-2, respectively. The drop in percentage is of the same magnitude to that found at 25 °C, being in this case slightly higher for the sample with the lower grafting yield, which is likely attributed to its less-homogenous HDI-GO dispersion, as discussed above. Overall, results confirm that mechanical properties that are dominated by the matrix, such as tensile strength, are strongly improved by the covalent grafting of a GO derivative, and that the reinforcing effect is preserved at high temperatures.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









