Fabrication of Stable Nanofiber Matrices for Tissue Engineering via Electrospinning of Bare Laser-Synthesized Au Nanoparticles in Solutions of High Molecular Weight Chitosan




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
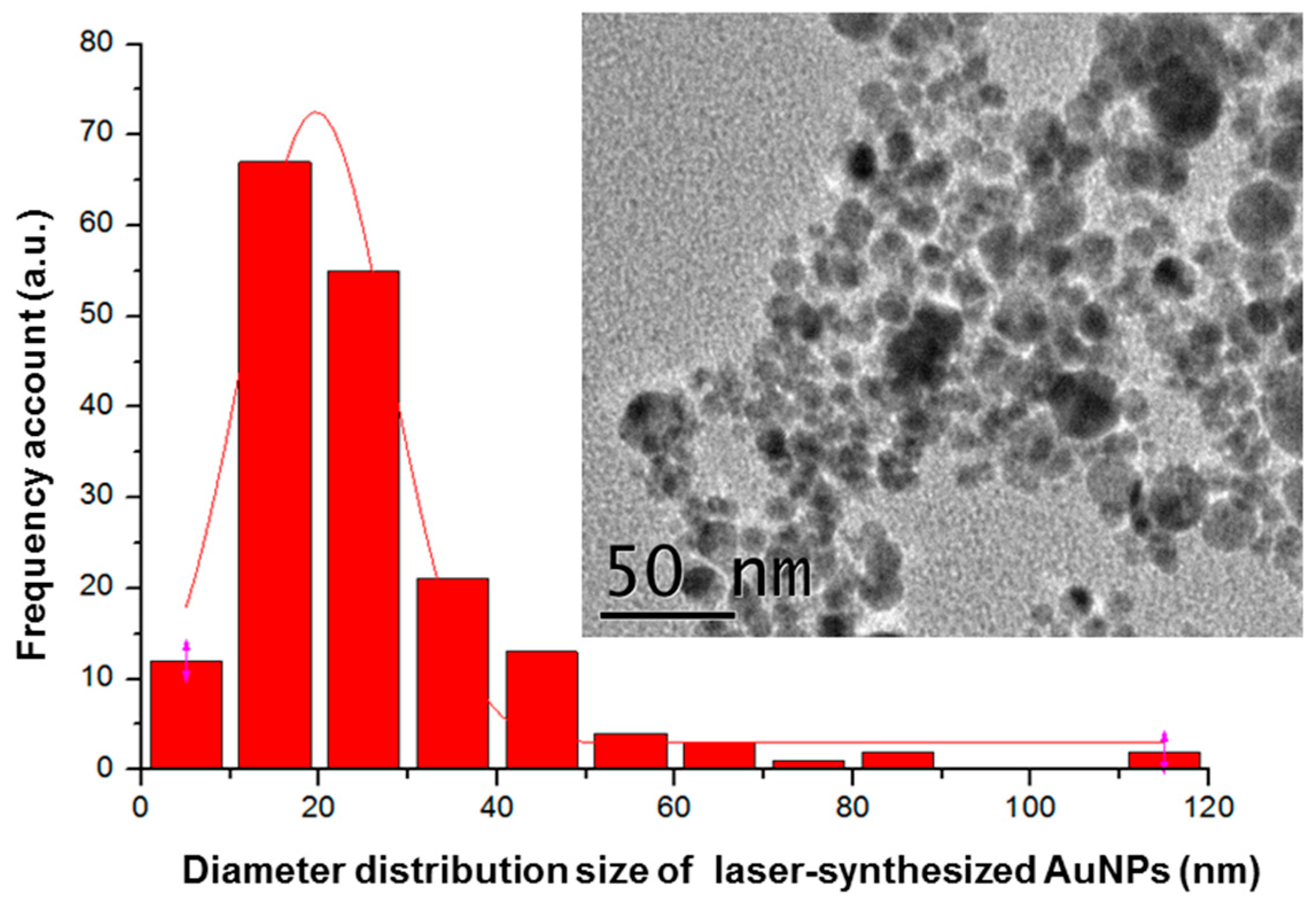
2.2.1. Laser-Ablative Synthesis of Bare AuNPs
2.2.2. Preparation of Electrospinning Solutions
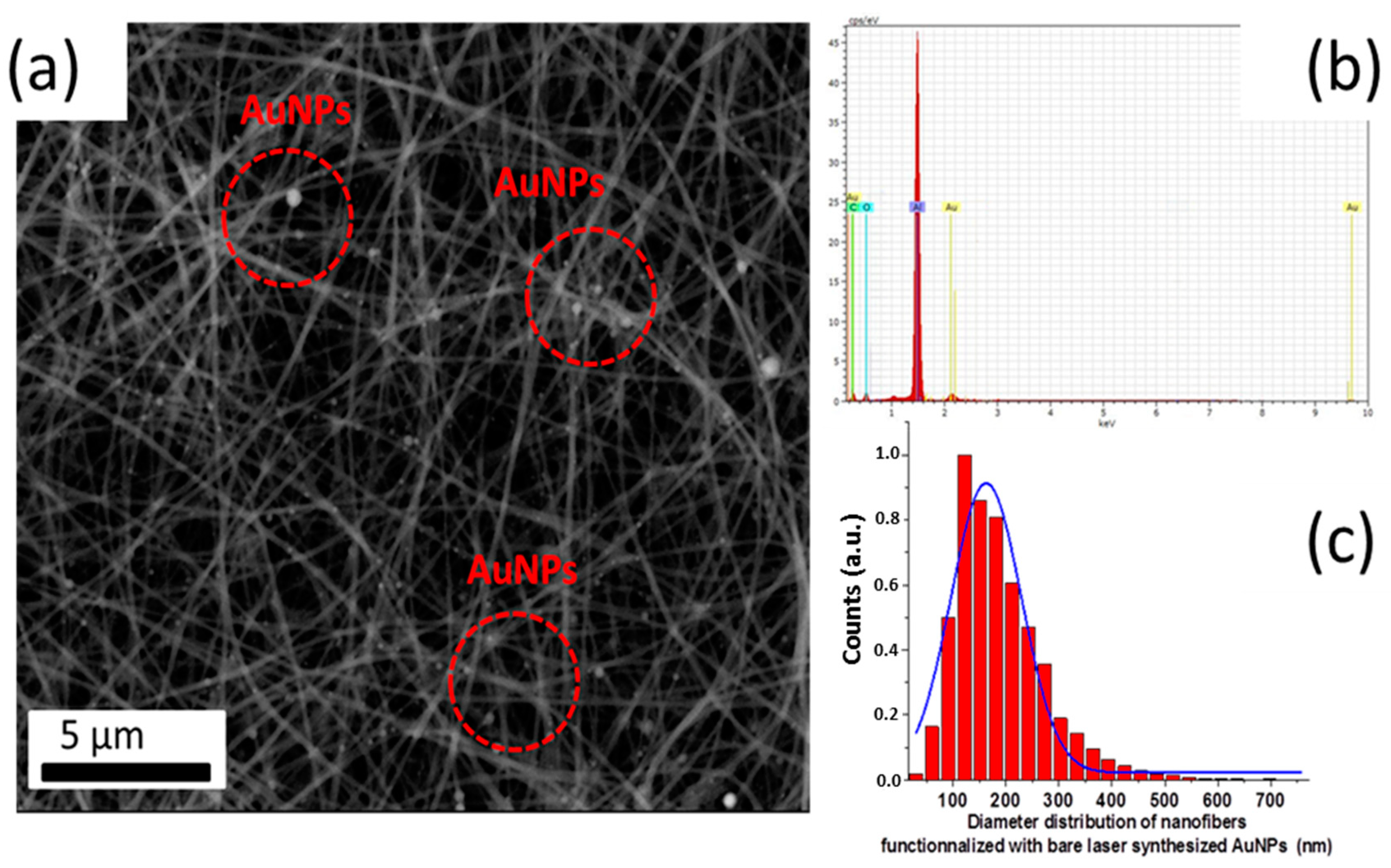
2.2.3. Electrospinning Hybrid Multifunctional Nanofibers
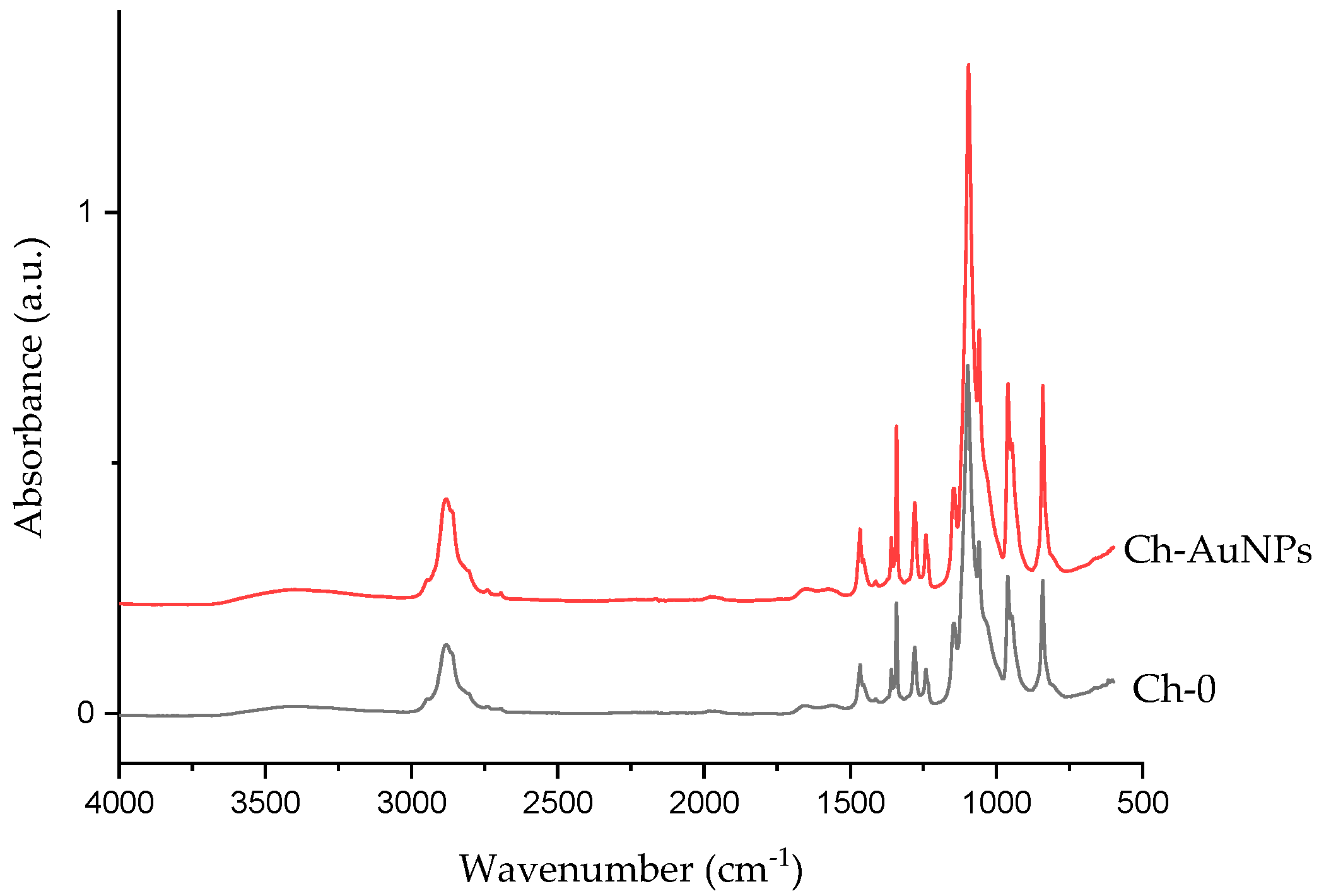
2.3. Morphological and Chemical Analysis
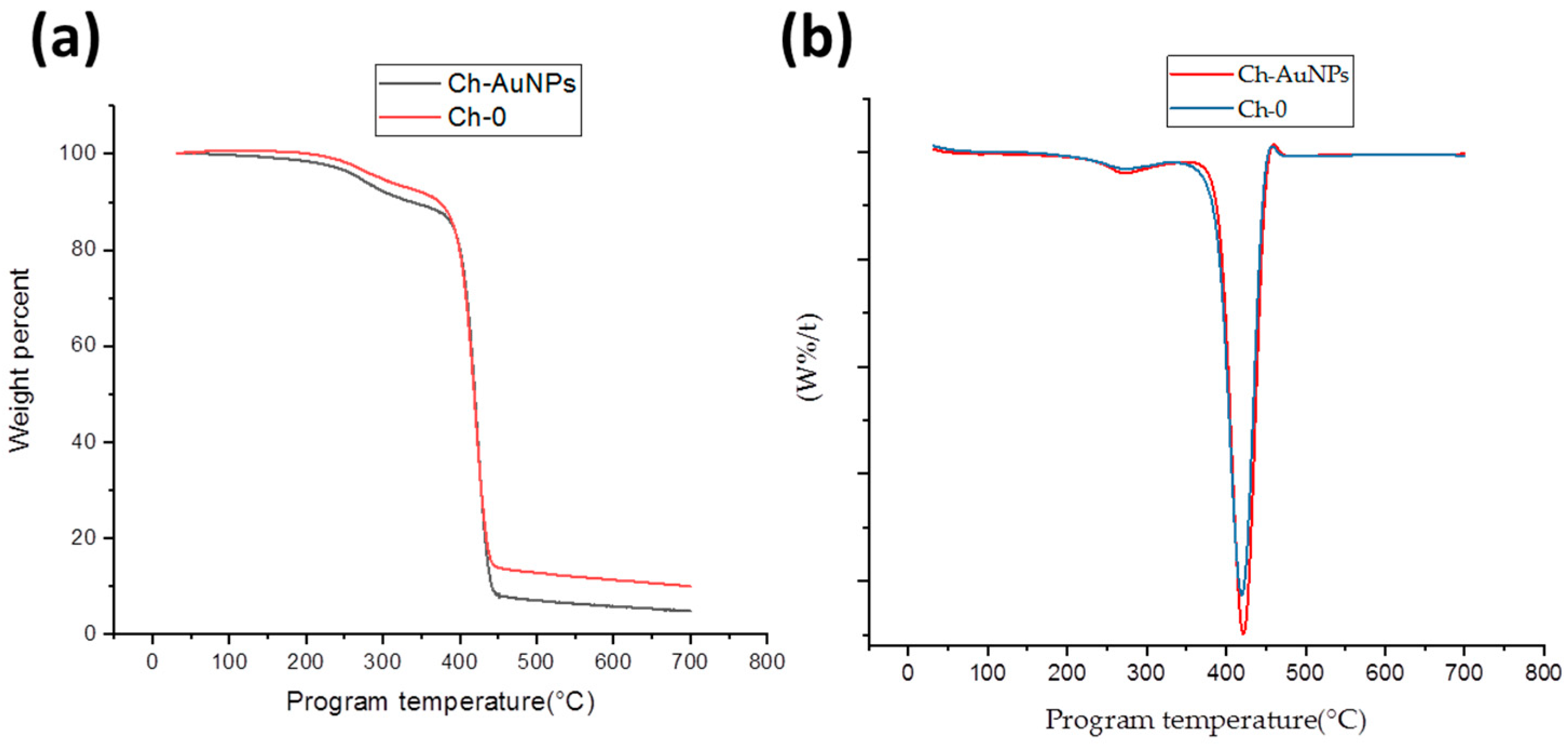
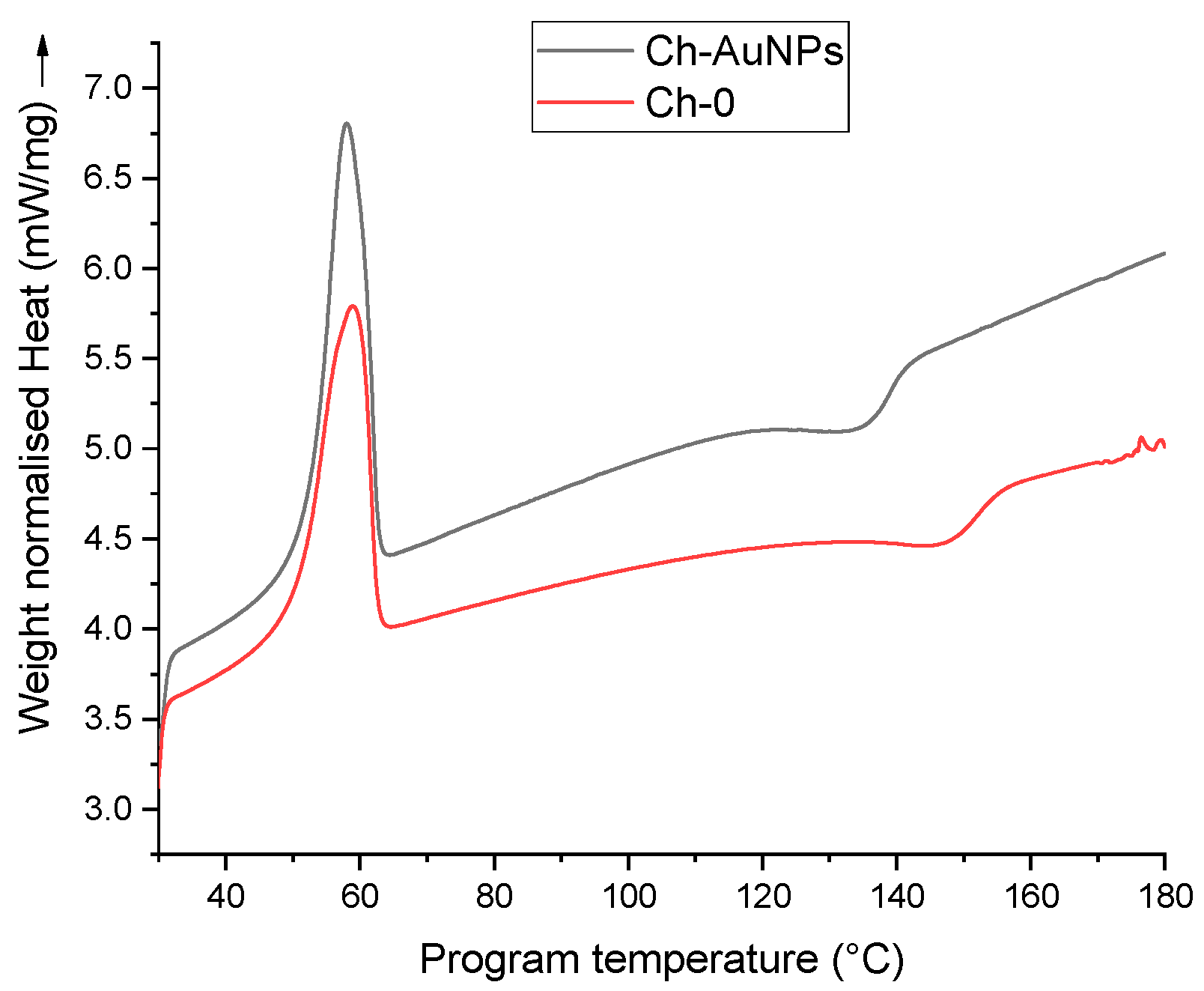
2.3.1. Thermal Analysis
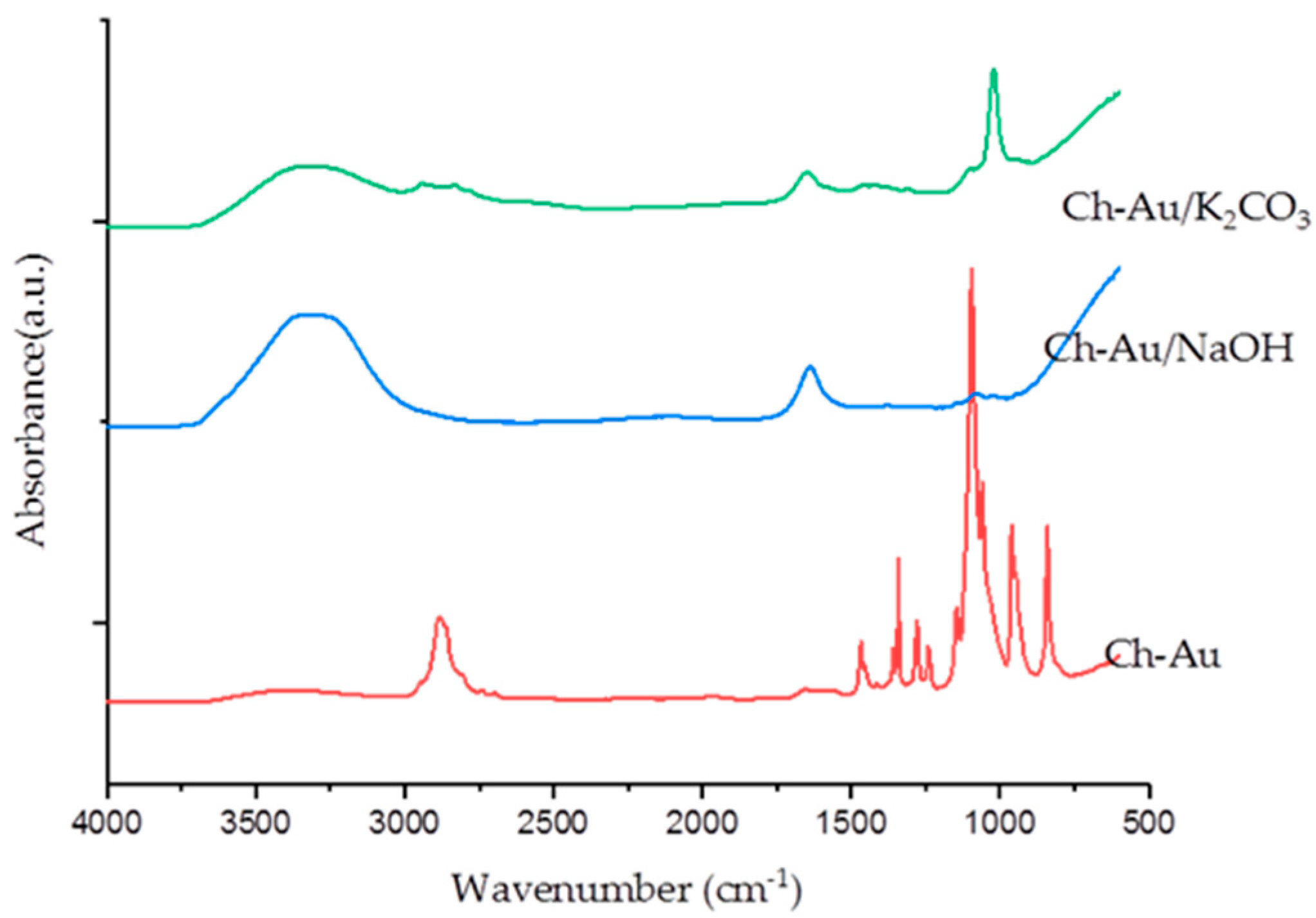
2.3.2. Neutralization of Chitosan/PEO Nanofibers
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kai, D.; Jin, G.; Prabhakaran, M.P.; Ramakrishna, S. Electrospun synthetic and natural nanofibers for regenerative medicine and stem cells. Biotechnol. J. 2013, 8, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, R.; Sundarrajan, S.; Venugopal, J.R.; Ravichandran, R.; Ramakrishna, S. Electrospun inorganic and polymer composite nanofibers for biomedical applications. J. Biomater. Sci. Polym. Ed. 2013, 24, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, T.R.; Yang, X.; Zhang, J.; Cao, X. In situ and ex situ modifications of bacterial cellulose for applications in tissue engineering. Mater. Sci. Eng. C 2018, 82, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Bonde, M.; Srinivasan, G. Biodegradable polymer scaffold for tissue engineering. Trends Biomater. Artif. Organs 2011, 12, 689–693. [Google Scholar]
- Al-Kattan, A.; Nirwan, V.P.; Munnier, E.; Chourpa, I.; Fahmi, A.; Kabashin, A.V. Toward multifunctional hybrid platforms for tissue engineering based on chitosan(PEO) nanofibers functionalized by bare laser-synthesized Au and Si nanoparticles. RSC Adv. 2017, 7, 31759–31766. [Google Scholar] [CrossRef]
- Wu, J.; Xie, L.; Lin, W.Z.Y.; Chen, Q. Biomimetic nanofibrous scaffolds for neural tissue engineering and drug development. Drug Discov. Today 2017, 22, 1357–1384. [Google Scholar] [CrossRef] [PubMed]
- Pierini, F.; Nakielski, P.; Urbanek, O.; Massimiliano, L.; Sio, L.D.; Kowalewski, T.A. Polymer-Based Nanomaterials for Photothermal Therapy: From Light-Responsive to Multifunctional Nanoplatforms for Synergistically Combined Technologies. Biomacromolecules 2018, 11–19, 4147–4167. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.M.; Zhang, Y.Z.; Kotaki, M.; Ramakrishna, S. A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Compos. Sci. Technol. 2003, 63, 2223–2253. [Google Scholar] [CrossRef]
- Sachlos, E.; Czernuszka, J.T.; Gogolewski, S.; Dalby, M. Making tissue engineering scaffolds work. Review on the application ofsolid freeform fabrication technology to the production of tissue engineeringscaffolds. Eur. Cells Mater. 2003, 5, 29–40. [Google Scholar] [CrossRef]
- Al-Kattan, A.; Nirwan, V.P.; Popov, A.A.; Ryabchikov, Y.V.; Tselikov, G.; Sentis, M.; Fahmi, A.; Kabashin, A.V. Recent advances in laser-ablative synthesis of bare Au and Si NPs and assessment of their prospects for tissue engineering applications. Int. J. Mol. Sci. 2018, 19, 1563. [Google Scholar] [CrossRef]
- Abidian, M.R.; Martin, D.C. Multifunctional nanobiomaterials for neural interfaces. Adv. Funct. Mater. 2009, 19, 573–585. [Google Scholar] [CrossRef]
- Agarwal, S.; Greiner, A.; Wendorff, J.H. Functional materials by electrospinning of polymers. Prog. Polym. Sci. 2013, 38, 963–991. [Google Scholar] [CrossRef]
- Klein, K.L.; Melechko, A.V.; McKnight, T.E.; Retterer, S.T.; Rack, P.D.; Fowlkes, J.D.; Joy, D.C.; Simpson, M.L. Surface characterization and functionalization of carbon nanofibers. J. Appl. Phys. 2008, 103, 0631301. [Google Scholar] [CrossRef]
- Ahmed, S.; Ikram, S. Chitosan Based Scaffolds and Their Applications in Wound Healing. Achiev. Life Sci. 2016, 10, 27–37. [Google Scholar] [CrossRef]
- Zhao, L.M.; Shi, L.E.; Zhang, Z.L.; Chen, J.M.; Shi, D.D.; Yang, J.; Tang, Z.X. Preparation and application of chitosan nanoparticles and nanofibers. Braz. J. Chem. Eng. 2011, 28, 353–362. [Google Scholar] [CrossRef]
- Muxika, A.; Etxabide, A.; Uranga, J.; Guerrero, P.; de la Caba, K. Chitosan as a bioactive polymer: Processing, properties and applications. Int. J. Biol. Macromol. 2017, 105, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan–A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Elsabee, M.Z.; Naguib, H.F.; Morsi, R.E. Chitosan based nanofibers, review. Mater. Sci. Eng. C 2012, 32, 1711–1762. [Google Scholar] [CrossRef]
- Al-kattan, A.; Nirwan, V.P.; Munnier, E.; Chourpa, I.; Popov, A.A.; Al-Kattan, A.; Nirwan, V.P.; Munnier, E.; Tselikov, G.; Ryabchikov, Y.V.; et al. Bare laser-synthesized Si nanoparticles as functional elements for chitosan nanofiber-based tissue. Proc. SPIE. Synth. Photonics Nanoscale Mater. XV 2018, 9, 10521. [Google Scholar]
- Jayakumar, R.; Menon, D.; Manzoor, K.; Nair, S.V.; Tamura, H. Biomedical applications of chitin and chitosan based nanomaterials-A short review. Carbohydr. Polym. 2010, 82, 227–232. [Google Scholar] [CrossRef]
- Torres-Giner, S.; Ocio, M.J.; Lagaron, J.M. Development of active antimicrobial fiber based chitosan polysaccharide nanostructures using electrospinning. Eng. Life Sci. 2008, 8, 303–314. [Google Scholar] [CrossRef]
- Adrienne, H.; Nanofibers, E.C. Electrospinning chitosan-based nanofibers for biomedical application. In Degree of Master of Science; Graduate Faculty of North Carolina State University: Raleigh, NC, USA, 2006. [Google Scholar]
- Lemma, S.M.; Bossard, F.; Rinaudo, M. Preparation of pure and stable chitosan nanofibers by electrospinning in the presence of poly(ethylene oxide). Int. J. Mol. Sci. 2016, 17, 2710–2714. [Google Scholar]
- Wendorff, J.H.; Agarwal, S.; Greiner, A. Electrospinning: Materials, Processing, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9783527320806. [Google Scholar]
- Ohkawa, K.; Cha, D.; Kim, H.; Nishida, A.; Yamamoto, H. Electrospinning of Chitosan. Macromol. Rapid Commun. 2004, 25, 1600–1605. [Google Scholar] [CrossRef]
- Vasconcelos, C.L.; Bezerril, P.M.; dos Santos, D.E.S.; Dantas, T.N.C.; Pereira, M.R.; Fonseca, J.L.C. Effect of molecular weight and ionic strength on the formation of polyelectrolyte complexes based on poly(methacrylic acid) and chitosan. Biomacromolecules 2006, 7, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Kabashin, V.K.; Meunier, M. Synthesis of Colloidal Nanoparticles during Femtosecond Laser Ablation of Gold in Water. J. Appl. Phys. 2003, 94, 7941. [Google Scholar] [CrossRef]
- Kabashin, A.V.; Meunier, M. Laser-Induced Treatment of Silicon in Air and Formation of Si/SiOx Photoluminescent Nanostructured Layers. Mater. Sci. Eng. B 2003, 101, 60–64. [Google Scholar] [CrossRef]
- Gongalsky, M.B.; Osminkina, L.A.; Pereira, A.; Manankov, A.A.; Fedorenko, A.A.; Vasiliev, A.N.; Solovyev, V.V.; Kudryavtsev, A.A.; Sentis, M.; Kabashin, A.V.; et al. Laser-Synthesized Oxide-Passivated Bright Si Quantum Dots for Bioimaging. Sci. Rep. 2016, 6, 24732. [Google Scholar] [CrossRef] [PubMed]
- Maximova, K.; Aristov, A.; Sentis, M.; Kabashin, A.V. Size-Controllable Synthesis of Bare Gold Nanoparticles by Femtosecond Laser Fragmentation in Water. Nanotechnology 2015, 26, 65601. [Google Scholar] [CrossRef] [PubMed]
- Hebié, S.; Holade, Y.; Maximova, K.; Sentis, M.; Delaporte, P.; Kokoh, K.B.; Napporn, T.W.; Kabashin, A.V. Advanced Electrocatalysts on the Basis of Bare Au Nanomaterials for Biofuel Cell Applications. ACS Catal. 2015, 5, 6489–6496. [Google Scholar] [CrossRef]
- Al-Kattan, A.; Ryabchikov, Y.V.; Baati, T.; Chirvony, V.; Sánchez-Royo, J.F.; Sentis, M.; Braguer, D.; Timoshenko, V.Y.; Estève, M.-A.; Kabashin, A.V. Ultrapure Laser-Synthesized Si Nanoparticles with Variable Oxidation States for Biomedical Applications. J. Mater. Chem. B 2016, 4, 7852–7858. [Google Scholar] [CrossRef]
- Correard, F.; Maximova, K.; Estève, M.-A.; Villard, C.; Roy, M.; Al-Kattan, A.; Sentis, M.; Gingras, M.; Kabashin, A.V.; Braguer, D. Gold Nanoparticles Prepared by Laser Ablation in Aqueous Biocompatible Solutions: Assessment of Safety and Biological Identity for Nanomedicine Applications. Int. J. Nanomed. 2014, 9, 5415–5430. [Google Scholar]
- Baati, T.; Al-kattan, A.; Esteve, M.; Njim, L.; Ryabchikov, Y.; Chaspoul, F.; Hammami, M.; Sentis, M.; Kabashin, A.V.; Braguer, D. Ultrapure Laser-Synthesized Si-Based Nanomaterials for Biomedical Applications : In Vivo Assessment of Safety and Biodistribution. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kabashin, A.V.; Timoshenko, V.Y. What Theranostic Applications Could Ultrapure Laser-Synthesized Si Nanoparticles Have in Cancer? Nanomedicine 2016, 11, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Kögler, M.; Ryabchikov, Y.V.; Uusitalo, S.; Popov, A.; Popov, A.; Tselikov, G.; Välimaa, A.-L.; Al-Kattan, A.; Hiltunen, J.; Laitinen, R.; et al. Bare laser-synthesized Au-based nanoparticles as non-disturbing SERS probes for Bacteria Detection. J. Biophotonics 2018, 11, e201700225. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.K.; Park, S.J.; Kim, M.S.; Kang, C.M.; Kim, J.I.; Kim, C.H. Fabrication of sonicated chitosan nanofiber mat with enlarged porosity for use as hemostatic materials. Carbohydr. Polym. 2013, 97, 65–73. [Google Scholar] [CrossRef]
- Sangsanoh, P.; Supaphol, P. Stability improvement of electrospun chitosan nanofibrous membranes in neutral or weak basic aqueous solutions. Biomacromolecules 2006, 7, 2710–2714. [Google Scholar] [CrossRef]
- Pawlak, A.; Mucha, M. Thermogravimetric and FTIR studies of chitosan blends. Proc. Thermochim. Acta 2003, 396, 153–166. [Google Scholar] [CrossRef]
- Osman, Z.; Arof, A.K. FTIR studies of chitosan acetate based polymer electrolytes. Electrochim. Acta 2003, 48, 993–999. [Google Scholar] [CrossRef]
- Zivanovic, S.; Li, J.; Davidson, P.M.; Kit, K. Physical, mechanical, and antibacterial properties of chitosan/PEO blend films. Biomacromolecules 2007, 8, 1505–1510. [Google Scholar] [CrossRef]
- Pucić, I.; Jurkin, T. FTIR assessment of poly(ethylene oxide) irradiated in solid state, melt and aqeuous solution. Radiat. Phys. Chem. 2012, 81, 1426–1429. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Sample Ch-AuNPs | Reference Sample Ch-0 |
|---|---|
| 3% (w/v) chitosan in 10% (v/v) acetic acid | 3% (w/v) chitosan in 10% (v/v) acetic acid |
| 8% (w/v) PEO in 2 mL conc. AuNPs in deionized water + 2mL deionized water | 8% (w/v) PEO in 4 mL deionized water |
| 1:3 final ratio by volume | 1:3 final ratio by volume. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nirwan, V.P.; Al-Kattan, A.; Fahmi, A.; Kabashin, A.V. Fabrication of Stable Nanofiber Matrices for Tissue Engineering via Electrospinning of Bare Laser-Synthesized Au Nanoparticles in Solutions of High Molecular Weight Chitosan. Nanomaterials 2019, 9, 1058. https://doi.org/10.3390/nano9081058
Nirwan VP, Al-Kattan A, Fahmi A, Kabashin AV. Fabrication of Stable Nanofiber Matrices for Tissue Engineering via Electrospinning of Bare Laser-Synthesized Au Nanoparticles in Solutions of High Molecular Weight Chitosan. Nanomaterials. 2019; 9(8):1058. https://doi.org/10.3390/nano9081058
Chicago/Turabian StyleNirwan, Viraj P., Ahmed Al-Kattan, Amir Fahmi, and Andrei V. Kabashin. 2019. "Fabrication of Stable Nanofiber Matrices for Tissue Engineering via Electrospinning of Bare Laser-Synthesized Au Nanoparticles in Solutions of High Molecular Weight Chitosan" Nanomaterials 9, no. 8: 1058. https://doi.org/10.3390/nano9081058
APA StyleNirwan, V. P., Al-Kattan, A., Fahmi, A., & Kabashin, A. V. (2019). Fabrication of Stable Nanofiber Matrices for Tissue Engineering via Electrospinning of Bare Laser-Synthesized Au Nanoparticles in Solutions of High Molecular Weight Chitosan. Nanomaterials, 9(8), 1058. https://doi.org/10.3390/nano9081058