Nanohybrid Assemblies of Porphyrin and Au10 Cluster Nanoparticles

 ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
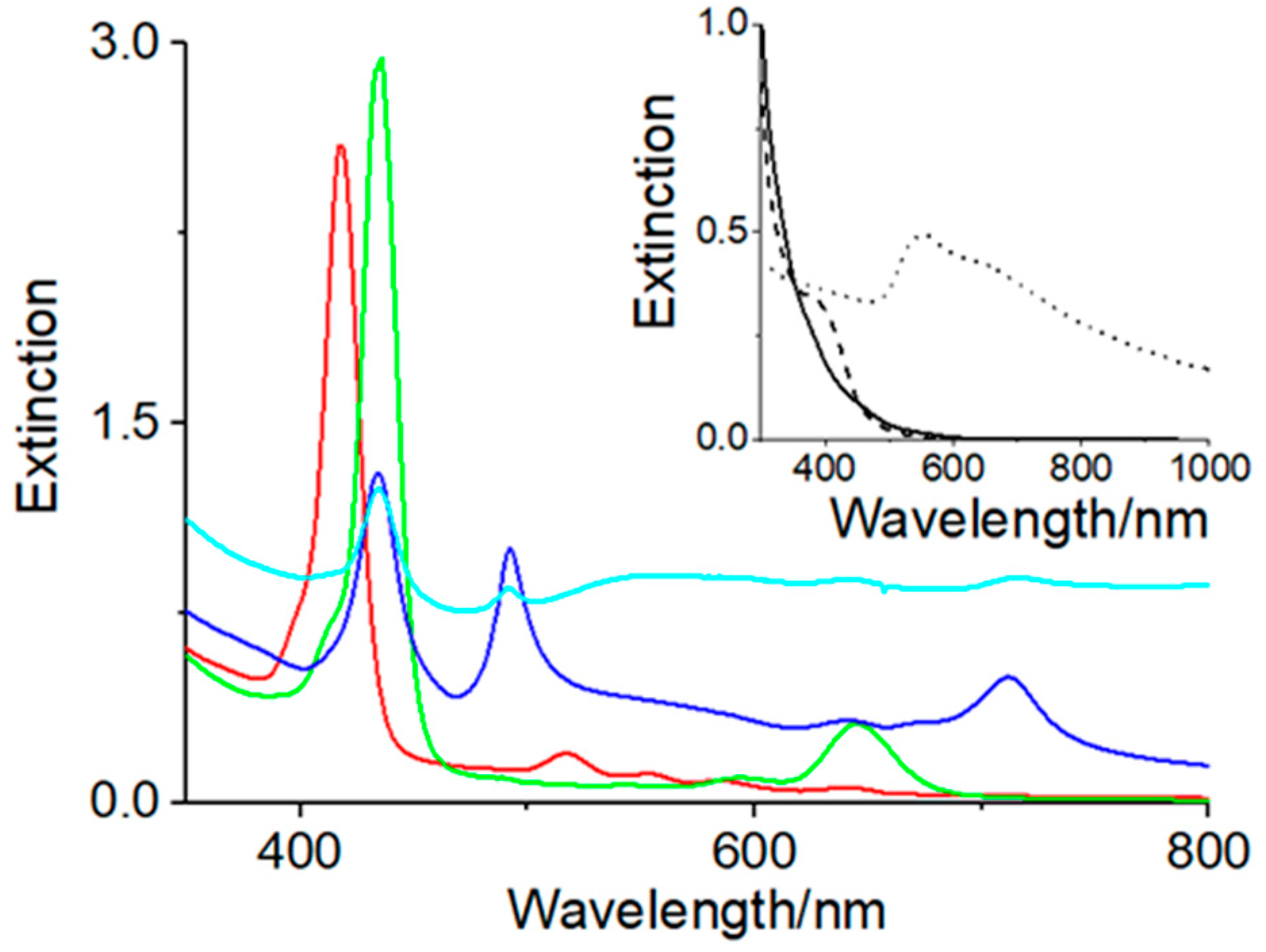
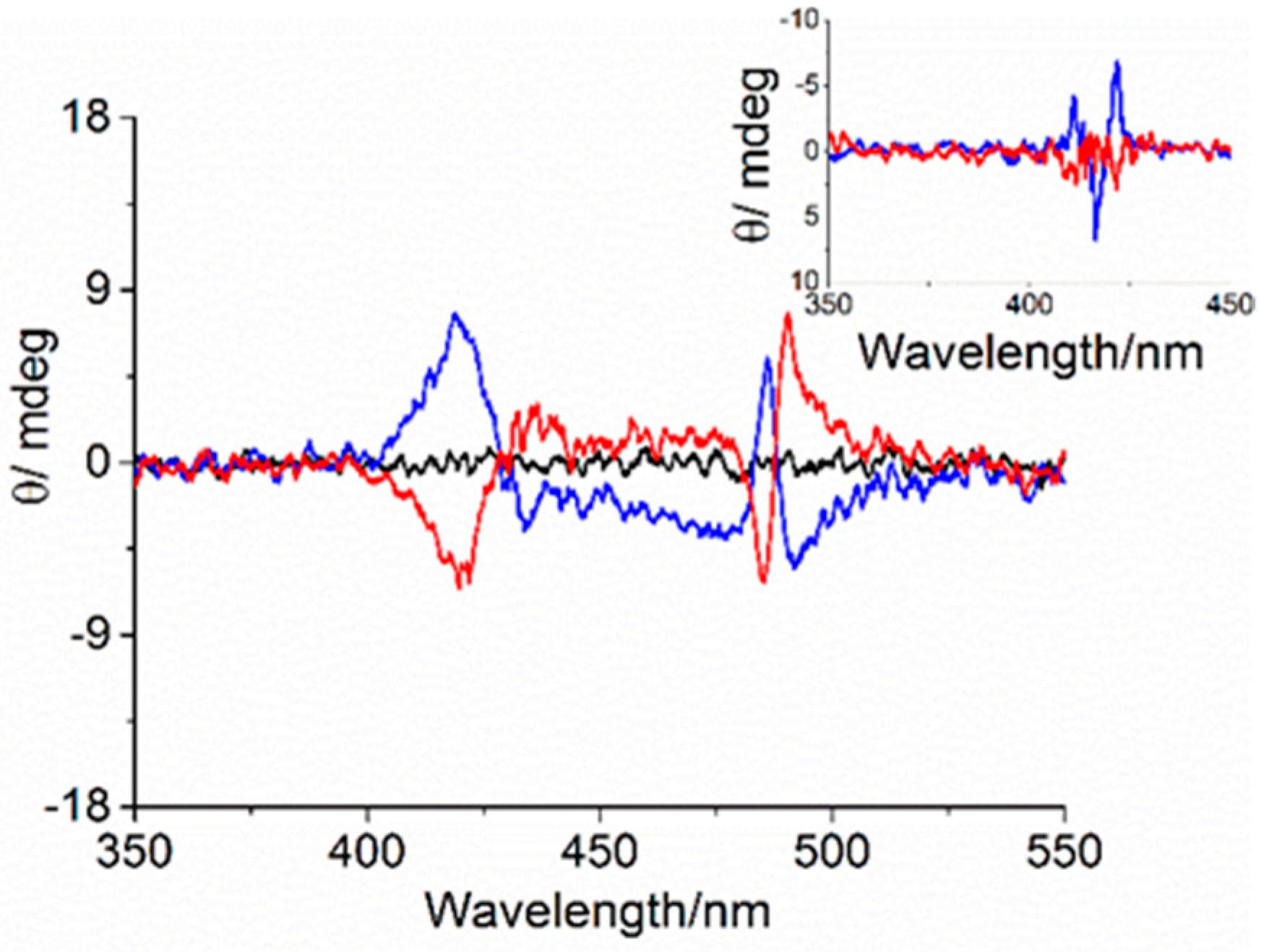
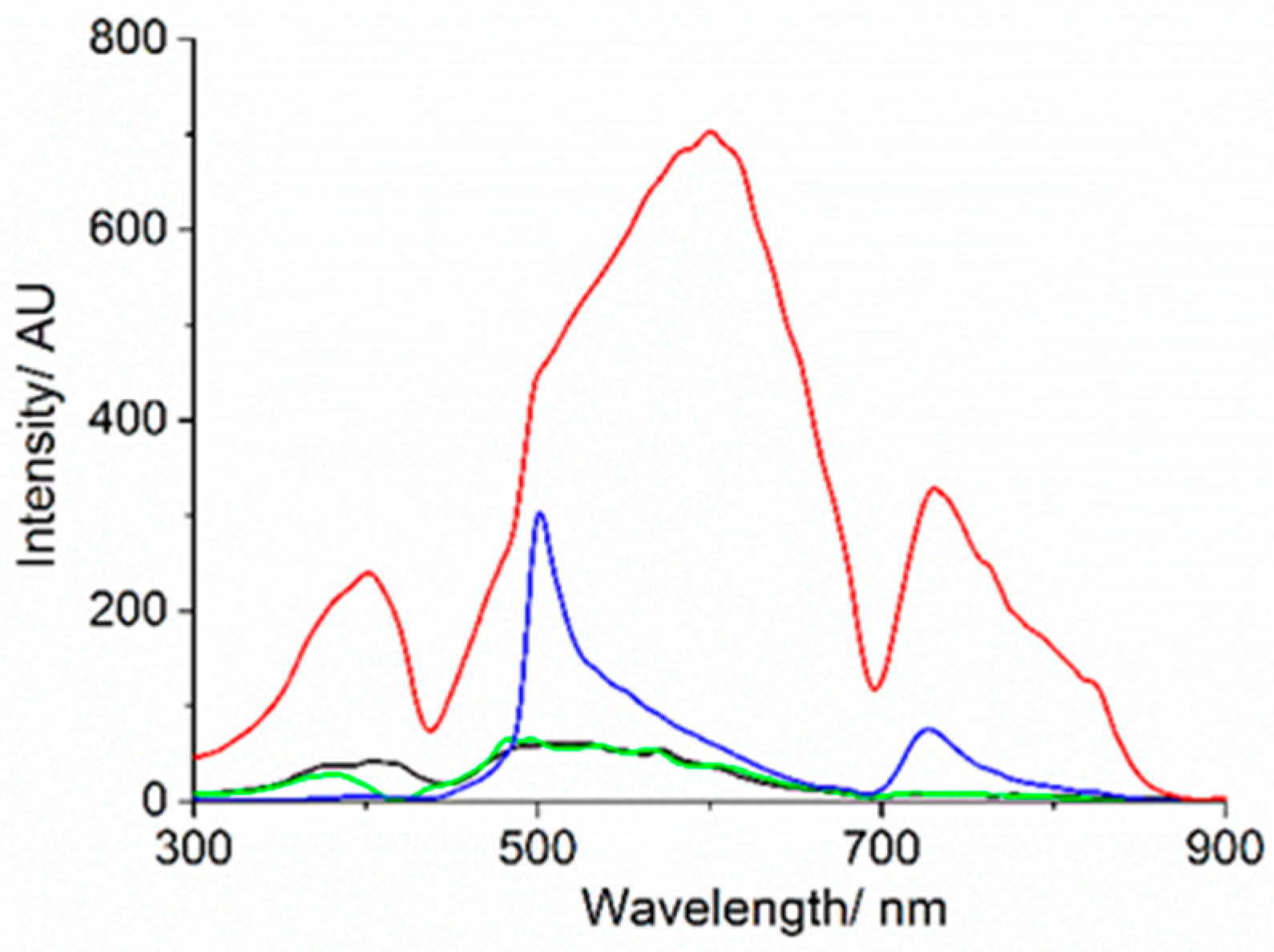
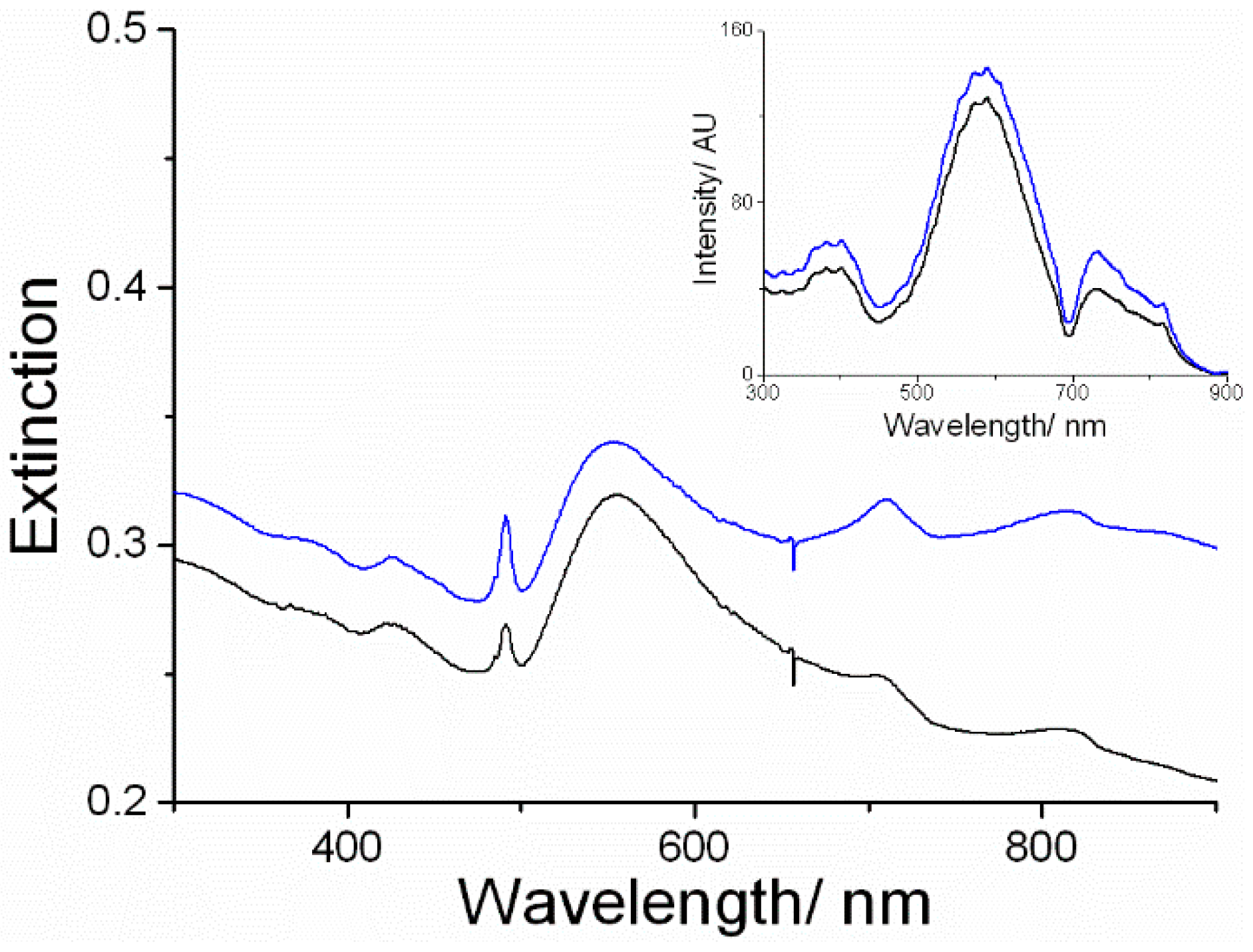
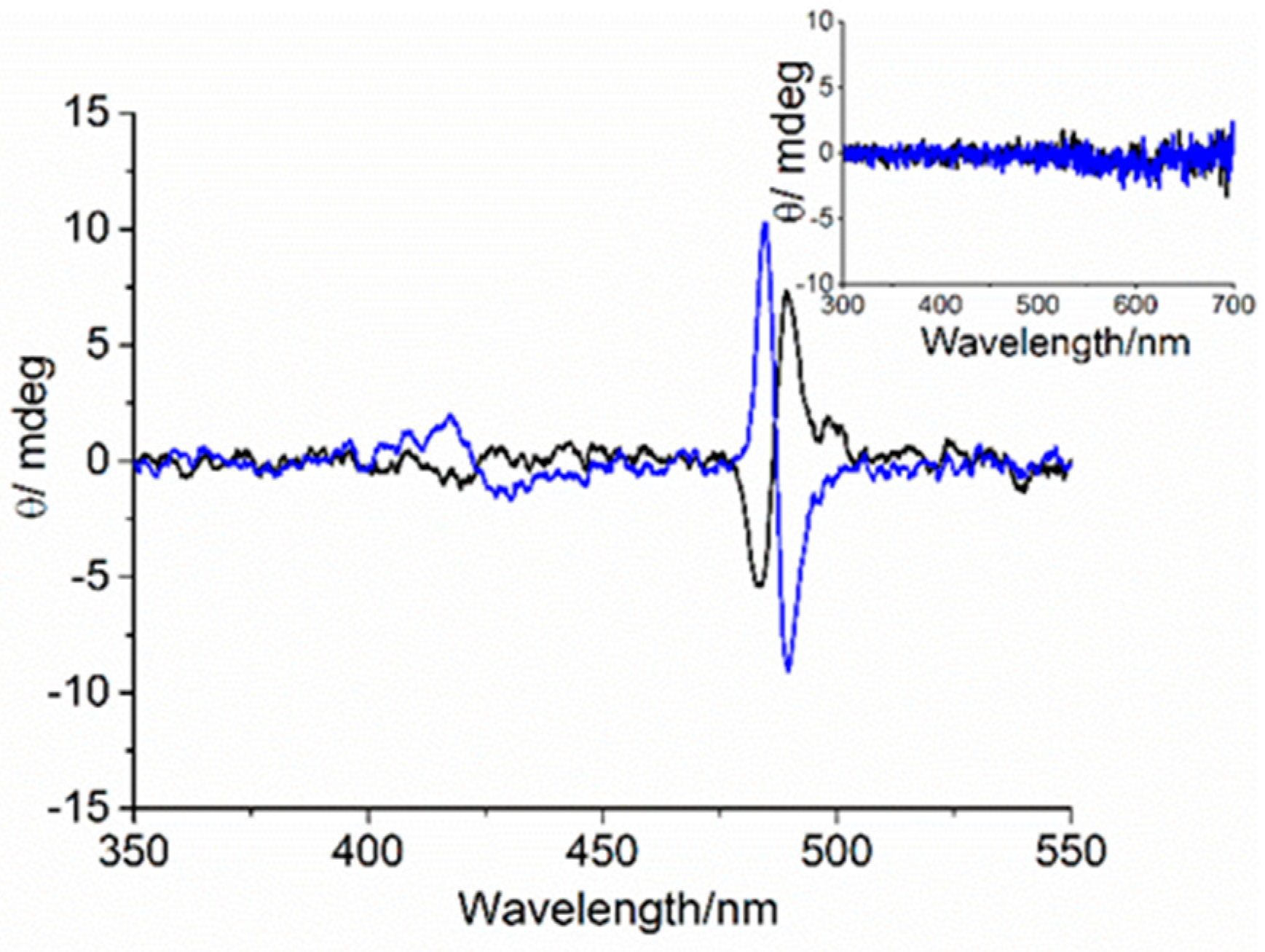
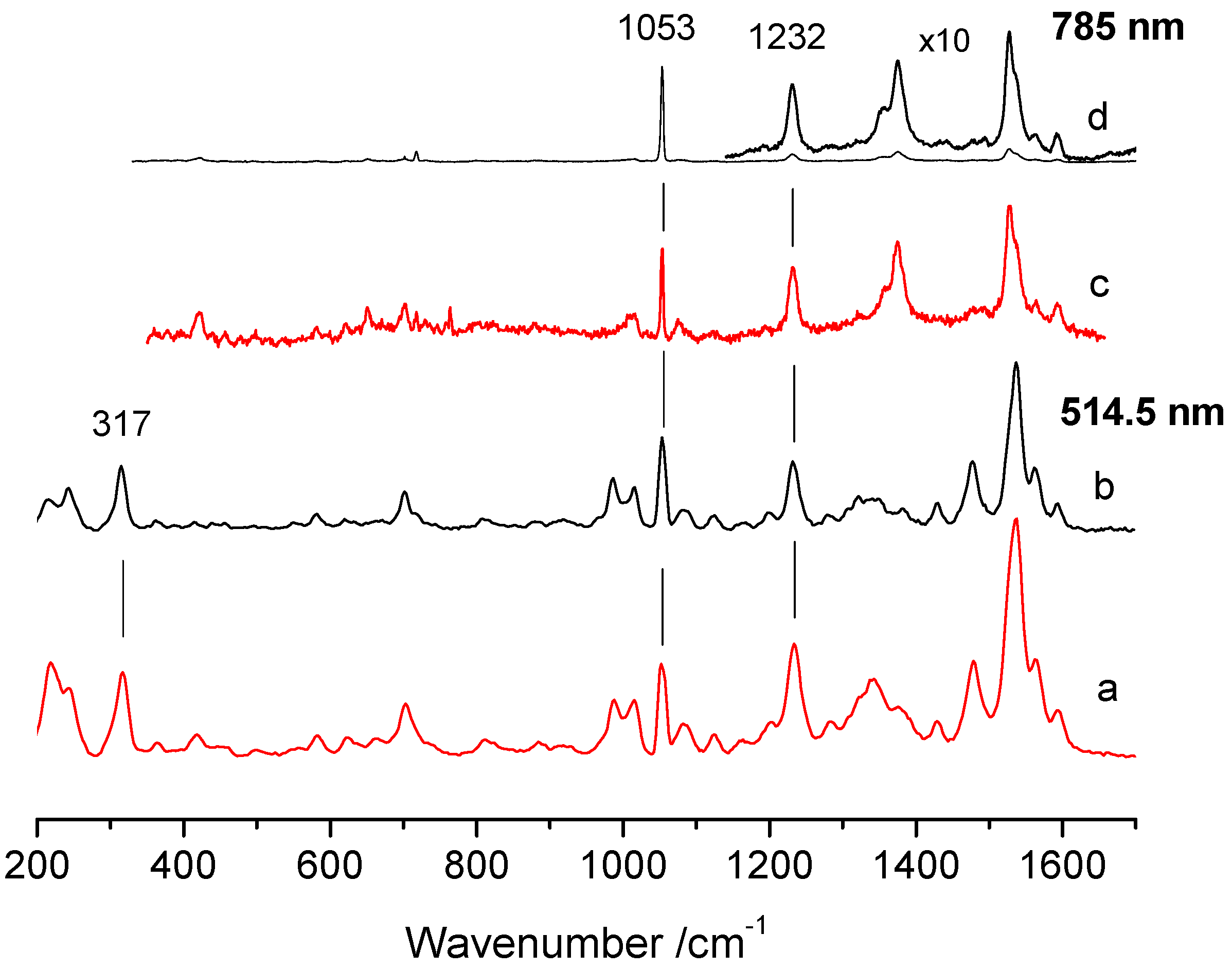
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jin, R.; Zeng, C.; Zhou, M.; Chen, Y. Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities. Chem. Rev. 2016, 116, 10346–10413. [Google Scholar] [CrossRef] [PubMed]
- Schmid, G. The relevance of shape and size of Au55 clusters. Chem. Soc. Rev. 2008, 37, 1909–1930. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, I.; Pradeep, T. Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles. Chem. Rev. 2017, 117, 8208–8271. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, C.; Dickson, R.M. Highly Fluorescent, Water-Soluble, Size-Tunable Gold Quantum Dots. Phys. Rev. Lett. 2004, 93, 077402. [Google Scholar] [CrossRef] [PubMed]
- Van Steerteghem, N.; Van Cleuvenbergen, S.; Deckers, S.; Kumara, C.; Dass, A.; Häkkinen, H.; Clays, K.; Verbiest, T.; Knoppe, S. Symmetry breaking in ligand-protected gold clusters probed by nonlinear optics. Nanoscale 2016, 8, 12123–12127. [Google Scholar] [CrossRef]
- Ramakrishna, G.; Varnavski, O.; Kim, J.; Lee, D.; Goodson, T. Quantum-Sized Gold Clusters as Efficient Two-Photon Absorbers. J. Am. Chem. Soc. 2008, 130, 5032–5033. [Google Scholar] [CrossRef] [PubMed]
- Philip, R.; Chantharasupawong, P.; Qian, H.; Jin, R.; Thomas, J. Evolution of Nonlinear Optical Properties: From Gold Atomic Clusters to Plasmonic Nanocrystals. Nano Lett. 2012, 12, 4661–4667. [Google Scholar] [CrossRef] [PubMed]
- Bertorelle, F.; Russier-Antoine, I.; Calin, N.; Comby-Zerbino, C.; Bensalah-Ledoux, A.; Guy, S.; Dugourd, P.; Brevet, P.-F.; Sanader, Ž.; Krstić, M.; et al. Au10(SG)10: A Chiral Gold Catenane Nanocluster with Zero Confined Electrons. Optical Properties and First-Principles Theoretical Analysis. J. Phys. Chem. Lett. 2017, 8, 1979–1985. [Google Scholar] [CrossRef]
- Russier-Antoine, I.; Bertorelle, F.; Vojkovic, M.; Rayane, D.; Salmon, E.; Jonin, C.; Dugourd, P.; Antoine, R.; Brevet, P.-F. Non-linear optical properties of gold quantum clusters. The smaller the better. Nanoscale 2014, 6, 13572–13578. [Google Scholar] [CrossRef]
- Polavarapu, L.; Manna, M.; Xu, Q.-H. Biocompatible glutathione capped gold clusters as one- and two-photon excitation fluorescence contrast agents for live cells imaging. Nanoscale 2011, 3, 429–434. [Google Scholar] [CrossRef]
- Derfus, A.M.; Chan, W.C.W.; Bhatia, S.N. Probing the Cytotoxicity of Semiconductor Quantum Dots. Nano Lett. 2004, 4, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-D.; Luo, Z.; Chen, J.; Shen, X.; Song, S.; Sun, Y.; Fan, S.; Fan, F.; Leong, D.T.; Xie, J. Ultrasmall Au10−12(SG)10−12 Nanomolecules for High Tumor Specificity and Cancer Radiotherapy. Adv. Mater. 2014, 26, 4565–4568. [Google Scholar] [CrossRef] [PubMed]
- Noguez, C.; Garzón, I.L. Optically active metal nanoparticles. Chem. Soc. Rev. 2009, 38, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Ortega, B.; Burgi, T. Vibrational Properties of Thiolate-Protected Gold Nanoclusters. Acc. Chem. Res. 2018, 51, 2811–2819. [Google Scholar] [CrossRef] [PubMed]
- Knoppe, S.; Buergi, T. Chirality in Thiolate-Protected Gold Clusters. Acc. Chem. Res. 2014, 47, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Govorov, A.O. Plasmonic Circular Dichroism of Chiral Metal Nanoparticle Assemblies. Nano Lett. 2010, 10, 2580–2587. [Google Scholar] [CrossRef] [PubMed]
- Govorov, A.O.; Fan, Z.; Hernandez, P.; Slocik, J.M.; Naik, R.R. Theory of Circular Dichroism of Nanomaterials Comprising Chiral Molecules and Nanocrystals: Plasmon Enhancement, Dipole Interactions, and Dielectric Effects. Nano Lett. 2010, 10, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shi, M.; Zhou, R.; Chen, X.; Chen, H. Blending of HAuCl4 and histidine in aqueous solution: A simple approach to the Au10 cluster. Nanoscale 2011, 3, 2596–2601. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, X.; Long, T.; Lin, M.; Liu, Z.; Huang, C. Histidine-mediated synthesis of chiral fluorescence gold nanoclusters: Insight into the origin of nanoscale chirality. RSC Adv. 2015, 5, 61449–61454. [Google Scholar] [CrossRef]
- Wu, Z.; Suhan, J.; Jin, R. One-pot synthesis of atomically monodisperse, thiol-functionalized Au25 nanoclusters. J. Mater. Chem. 2009, 19, 622–626. [Google Scholar] [CrossRef]
- Bain, D.; Maity, S.; Patra, A. Opportunities and challenges in energy and electron transfer of nanocluster based hybrid materials and their sensing applications. Phys. Chem. Chem. Phys. 2019, 21, 5863–5881. [Google Scholar] [CrossRef] [PubMed]
- Mondal, N.; Paul, S.; Samanta, A. Photoinduced 2-way electron transfer in composites of metal nanoclusters and semiconductor quantum dots. Nanoscale 2016, 8, 14250–14256. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.G.; Hawkridge, M.E.; Roper, D.K. Silver disposition and dynamics during electroless metal thin film synthesis. J. Mater. Chem. 2012, 22, 21942–21953. [Google Scholar] [CrossRef]
- Siegel, J.; Lyutakov, O.; Rybka, V.; Kolská, Z.; Švorčík, V. Properties of gold nanostructures sputtered on glass. Nanoscale Res. Lett. 2011, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Claudia Manuela, M.; Flavio Carlo Filippo, M.; Ralph, S. Ordered arrays of faceted gold nanoparticles obtained by dewetting and nanosphere lithography. Nanotechnology 2008, 19, 485306. [Google Scholar]
- Grodzicki, A.; Łakomska, I.; Piszczek, P.; Szymańska, I.; Szłyk, E. Copper(I), silver(I) and gold(I) carboxylate complexes as precursors in chemical vapour deposition of thin metallic films. Coord. Chem. Rev. 2005, 249, 2232–2258. [Google Scholar] [CrossRef]
- Livshits, P.; Inberg, A.; Shacham-Diamand, Y.; Malka, D.; Fleger, Y.; Zalevsky, Z. Precipitation of gold nanoparticles on insulating surfaces for metallic ultra-thin film electroless deposition assistance. Appl. Surf. Sci. 2012, 258, 7503–7506. [Google Scholar] [CrossRef]
- De Leo, M.; Pereira, F.C.; Moretto, L.M.; Scopece, P.; Polizzi, S.; Ugo, P. Towards a Better Understanding of Gold Electroless Deposition in Track-Etched Templates. Chem. Mater. 2007, 19, 5955–5964. [Google Scholar] [CrossRef]
- Thorkelsson, K.; Bai, P.; Xu, T. Self-assembly and applications of anisotropic nanomaterials: A review. Nano Today 2015, 10, 48–66. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Regulacio, M.D.; Han, M.-Y. Self-assembly of colloidal one-dimensional nanocrystals. Chem. Soc. Rev. 2014, 43, 2301–2323. [Google Scholar] [CrossRef]
- Freeman, R.G.; Grabar, K.C.; Allison, K.J.; Bright, R.M.; Davis, J.A.; Guthrie, A.P.; Hommer, M.B.; Jackson, M.A.; Smith, P.C.; Walter, D.G.; et al. Self-Assembled Metal Colloid Monolayers: An Approach to SERS Substrates. Science 1995, 267, 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Rich, C.C.; McHale, J.L. Resonance Raman Spectra of Individual Excitonically Coupled Chromophore Aggregates. J. Phys. Chem. C 2013, 117, 10856–10865. [Google Scholar] [CrossRef]
- Hajduková-Šmídová, N.; Procházka, M.; Osada, M. SE(R)RS excitation profile of free-base 5,10,15,20-tetrakis(1-methyl-4-pyridyl) porphyrin on immobilized gold nanoparticles. Vib. Spectrosc. 2012, 62, 115–120. [Google Scholar] [CrossRef]
- Leishman, C.W.; McHale, J.L. Illuminating Excitonic Structure in Ion-Dependent Porphyrin Aggregates with Solution Phase and Single-Particle Resonance Raman Spectroscopy. J. Phys. Chem. C 2016, 120, 12783–12795. [Google Scholar] [CrossRef]
- Friesen, B.A.; Rich, C.C.; Mazur, U.; McHale, J.L. Resonance Raman Spectroscopy of Helical Porphyrin Nanotubes. J. Phys. Chem. C 2010, 114, 16357–16366. [Google Scholar] [CrossRef]
- Trapani, M.; De Luca, G.; Romeo, A.; Castriciano, M.A.; Scolaro, L.M. Spectroscopic investigation on porphyrins nano-assemblies onto gold nanorods. Spectrochim. Acta Mol. Biomol. Spectrosc. 2017, 173, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Romeo, A.; Castriciano, M.A.; Zagami, R.; Pollicino, G.; Monsu Scolaro, L.; Pasternack, R.F. Effect of zinc cations on the kinetics for supramolecular assembling and the chirality of porphyrin J-aggregates. Chem. Sci. 2016, 8, 961–967. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Zagami, R.; Trapani, M.; Bolzonello, L.; Romeo, A.; Castriciano, M.A.; Collini, E.; Monsù Scolaro, L. The role of counter-anions in the kinetics and chirality of porphyrin J-aggregates. Chem. Commun. 2016, 52, 11520–11523. [Google Scholar] [CrossRef]
- Micali, N.; Villari, V.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. From fractal to nanorod porphyrin J-aggregates. Concentration-induced tuning of the aggregate size. J. Phys. Chem. B 2006, 110, 8289–8295. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; Villari, V.; Micali, N.; Scolaro, L.M. Structural rearrangements in 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin J-aggregates under strongly acidic conditions. J. Phys. Chem. B 2003, 107, 8765–8771. [Google Scholar] [CrossRef]
- Zagami, R.; Romeo, A.; Castriciano, M.A.; Monsù Scolaro, L. Inverse kinetic and equilibrium isotopic effect on self-assembly and supramolecular chirality of porphyrin J-aggregates. Chem. Eur.J. 2017, 23, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Zagami, R.; Castriciano, M.A.; Romeo, A.; Trapani, M.; Pedicini, R.; Monsù Scolaro, L. Tuning supramolecular chirality in nano and mesoscopic porphyrin J-aggregates. Dyes Pigments 2017, 142, 255–261. [Google Scholar] [CrossRef]
- Schwab, A.D.; Smith, D.E.; Rich, C.S.; Young, E.R.; Smith, W.F.; de Paula, J.C. Porphyrin nanorods. J. Phys. Chem. B 2003, 107, 11339–11345. [Google Scholar] [CrossRef]
- Schwab, A.D.; Smith, D.E.; Bond-Watts, B.; Johnston, D.E.; Hone, J.; Johnson, A.T.; de Paula, J.C.; Smith, W.F. Photoconductivity of Self-Assembled Porphyrin Nanorods. Nano Lett. 2004, 4, 1261–1265. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Gentili, D.; Romeo, A.; Cavallini, M.; Scolaro, L.M. Spatial control of chirality in supramolecular aggregates. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Friesen, B.A.; Nishida, K.R.A.; McHale, J.L.; Mazur, U. New Nanoscale Insights into the Internal Structure of Tetrakis(4-sulfonatophenyl) Porphyrin Nanorods. J. Phys. Chem. C 2009, 113, 1709–1718. [Google Scholar] [CrossRef]
- Arai, Y.; Segawa, H. Significantly Enhanced Adsorption of Bulk Self-Assembling Porphyrins at Solid/Liquid Interfaces through the Self-Assembly Process. J. Phys. Chem. B 2012, 116, 13575–13581. [Google Scholar] [CrossRef]
- Villari, V.; Mazzaglia, A.; Trapani, M.; Castriciano, M.A.; De Luca, G.; Romeo, A.; Scolaro, L.M.; Micali, N. Optical enhancement and structural properties of a hybrid organic-inorganic ternary nanocomposite. J. Phys. Chem. C 2011, 115, 5435–5439. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, H.; Wang, J.; Li, Y.F.; Wang, J.; Sang, Y.; Xiao, S.J.; Zhan, L.; Huang, C.Z. Tetrakis(4-sulfonatophenyl)porphyrin-Directed Assembly of Gold Nanocrystals: Tailoring the Plasmon Coupling Through Controllable Gap Distances. Small 2010, 6, 2001–2009. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Collings, P.J. Resonance Light-Scattering—A New Technique for Studying Chromophore Aggregation. Science 1995, 269, 935–939. [Google Scholar] [CrossRef]
- Randazzo, R.; Gaeta, M.; Gangemi, C.M.A.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Chiral Recognition of L- and D- Amino Acid by Porphyrin Supramolecular Aggregates. Molecules 2018, 24, 84. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Donato, M.G.; Villari, V.; Micali, N.; Romeo, A.; Scolaro, L.M. Surfactant-like Behavior of Short-Chain Alcohols in Porphyrin Aggregation. J. Phys. Chem. B 2009, 113, 11173–11178. [Google Scholar] [CrossRef] [PubMed]
- Zagami, R.; Trapani, M.; Castriciano, M.A.; Romeo, A.; Mineo, P.G.; Scolaro, L.M. Synthesis, characterization and aggregation behavior of room temperature ionic liquid based on porphyrin-trihexyl(tetradecyl)phosphonium adduct. J. Mol. Liq. 2017, 229, 51–57. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Leone, N.; Cardiano, P.; Manickam, S.; Scolaro, L.M.; Lo Schiavo, S. A new supramolecular polyhedral oligomeric silsesquioxanes (POSS)-porphyrin nanohybrid: Synthesis and spectroscopic characterization. J. Mater. Chem. C 2013, 1, 4746–4753. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; Villari, V.; Angelini, N.; Micali, N.; Scolaro, L.M. Aggregation behavior of tetrakis(4-sulfonatophenyl)porphyrin in AOT/water/decane microemulsions. J. Phys. Chem. B 2005, 109, 12086–12092. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zu, Y.; Fu, Y.; Meng, R.; Guo, S.; Xing, Z.; Tan, S. Hydrothermal synthesis of histidine-functionalized single-crystalline gold nanoparticles and their pH-dependent UV absorption characteristic. Coll. Surf. B Biointerf. 2010, 76, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Selvan, T.; Spatz, J.P.; Klok, H.-A.; Möller, M. Gold–Polypyrrole Core–Shell Particles in Diblock Copolymer Micelles. Adv. Mater. 1998, 10, 132–134. [Google Scholar] [CrossRef]
- Lakshminarayana, P.; Qing-Hua, X. A single-step synthesis of gold nanochains using an amino acid as a capping agent and characterization of their optical properties. Nanotechnology 2008, 19, 075601. [Google Scholar]
- Akins, D.L.; Zhu, H.R.; Guo, C. Absorption and Raman Scattering by Aggregated meso-Tetrakis(p-sulfonatophenyl)porphine. J. Phys. Chem. 1994, 98, 3612–3618. [Google Scholar] [CrossRef]
- Rotomskis, R.; Augulis, R.; Snitka, V.; Valiokas, R.; Liedberg, B. Hierarchical structure of TPPS4 J-aggregates on substrate revealed by atomic force microscopy. J. Phys. Chem. B 2004, 108, 2833–2838. [Google Scholar] [CrossRef]
- Ðorđević, L.; Arcudi, F.; D’Urso, A.; Cacioppo, M.; Micali, N.; Bürgi, T.; Purrello, R.; Prato, M. Design principles of chiral carbon nanodots help convey chirality from molecular to nanoscale level. Nat. Commun. 2018, 9, 3442. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, M.; Raciti, D.; Randazzo, R.; Gangemi, C.M.A.; Raudino, A.; D’Urso, A.; Fragalà, M.E.; Purrello, R. Chirality Enhancement of Porphyrin Supramolecular Assembly Driven by a Template Preorganization Effect. Angew. Chem. Int. Ed. 2018, 57, 10656–10660. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Point | R1053/1232 | R1053/1232 | R1053/317 |
|---|---|---|---|
| 785 nm | 514.5 nm | ||
| Without Au | |||
| 9 | 5.9 | 0.7 | 0.9 |
| 8 | 8.9 | 1.2 | 1.3 |
| 7 | 34.5 | 2.4 | 2.7 |
| 6 | 13.3 | 1.4 | 1.4 |
| 5 | 24.3 | 2.2 | 2.7 |
| 4 | 16.2 | 1.4 | 1.5 |
| With Au | |||
| 12 | 1.0 | 0.9 | 1.0 |
| 11 | 1.3 | 0.9 | 1.0 |
| 10 | 1.5 | 0.5 | 0.7 |
| 3 | 1.3 | 0.8 | 1.0 |
| 2 | 2.0 | 0.5 | 0.6 |
| 1 | 3.5 | 2.8 | 3.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trapani, M.; Castriciano, M.A.; Romeo, A.; De Luca, G.; Machado, N.; Howes, B.D.; Smulevich, G.; Scolaro, L.M. Nanohybrid Assemblies of Porphyrin and Au10 Cluster Nanoparticles. Nanomaterials 2019, 9, 1026. https://doi.org/10.3390/nano9071026
Trapani M, Castriciano MA, Romeo A, De Luca G, Machado N, Howes BD, Smulevich G, Scolaro LM. Nanohybrid Assemblies of Porphyrin and Au10 Cluster Nanoparticles. Nanomaterials. 2019; 9(7):1026. https://doi.org/10.3390/nano9071026
Chicago/Turabian StyleTrapani, Mariachiara, Maria Angela Castriciano, Andrea Romeo, Giovanna De Luca, Nelson Machado, Barry D. Howes, Giulietta Smulevich, and Luigi Monsù Scolaro. 2019. "Nanohybrid Assemblies of Porphyrin and Au10 Cluster Nanoparticles" Nanomaterials 9, no. 7: 1026. https://doi.org/10.3390/nano9071026
APA StyleTrapani, M., Castriciano, M. A., Romeo, A., De Luca, G., Machado, N., Howes, B. D., Smulevich, G., & Scolaro, L. M. (2019). Nanohybrid Assemblies of Porphyrin and Au10 Cluster Nanoparticles. Nanomaterials, 9(7), 1026. https://doi.org/10.3390/nano9071026