3. Results and Discussion
The particle shape, near-surface configurations, and crystal structure of the experimental NCs were determined using HRTEM analyses. As shown in
Figure 1a, Ni@Pt-1004 NPs (Pt/Ni ratio = 0.4) had ordered atomic arrangements at (111) facets exposing the surface (denoted by red arrows). The presence of a clear twin boundary (denoted by a white line) suggested the formation of semi-coherent interfaces. The interplanar spacing of the (111) facet was determined to be 2.239 Å. This value is about 1.25% smaller than that of the Pt-CNT (2.267 Å) indicating the presence of compressive lattice strain in the shell region. With an average particle size of 2.64 nm in (111) facets (determined by an XRD analysis described in a later section), the surface-to-bulk ratio was estimated to be ~50% In this event, considering the ideal case of conformal deposition of Pt atoms by chemisorption and reduction, formation of an incomplete Pt shell over the Ni-core crystal was expected. In contrast, Ni@Pt-1010 NPs (
Figure 1d) had grown into isotropic spheres, which comprised a complete Pt-shell and Ni core crystal underneath, and this was also confirmed by XPS analyses in a later section. Compared to that of Ni@Pt-1004 NPs, a higher extent of surface defects (denoted by yellow arrows) was observed in Ni@Pt-1010 NPs. The average particle size of Ni@Pt NCs was observed to be around 2~3 nm, which is consistent with XRD findings in a subsequent section.
Shown in
Figure 1b, compared to that of Ni@Pt-1004 NPs, the diameter of NPA-1004006 (i.e., Ni@Pt-1004 NC decorated with 2.0 wt.% of Au atoms) nearly doubled by mixing with Au
3+ ions for 2 min, followed by the addition of a reducing agent (NaBH
4). NPA-1004006 particles grew in a disk-like shape (
Figure 1b) with twin boundaries between the NPs. With a nearly identical coherent length to that of Ni@Pt-1004, the large particle was an agglomeration cluster with structural modulations at semi-coherent interfaces between NPs by the galvanic replacement of Au
3+ to core (Pt/Ni) metals simultaneously with the reduction of residual Pt
4+, Ni
2+, and Au
3+ ions by the reducing agents. Such a hypothesis was confirmed by XRD and XPS analyses in later sections. With an incomplete Pt shell structure, certain parts of the NiO
x core were exposed to the liquid environment. In this event, a high content of Ni atoms participated in the galvanic replacement reaction by interacting with Au
3+ ions. Therefore, Au atoms tended to form metallic clusters between the NPs. As shown in
Figure 1c, with a further increase of Au loading to 9 wt.% (NPA-100402), discrete local domains were formed with lattice fringes pointing in different directions (denoted by green arrows). Such a characteristic further revealed severe galvanic replacement of the NiO
x core followed by agglomeration of Ni@Pt NPs by reduction and deposition of residual metal ions in their interfaces. In this event, because of sufficiently high Au loading, the majority of Au atoms were deposited on the top of and between the NPs. Compared to that of Ni@Pt-1004, restructuring of Ni@Pt-1010 by Au
3+ was insignificant. As shown in
Figure 1e,f, particles tended to grow in a core-shell structure with highly ordered atomic arrangements on the surface with increasing Au contents. Such a characteristic was consistently proven by an XRD analysis, which showed that the coherent length of the Pt crystal (111) facet had increased from 2.72 to 3.34 ± 0.1 nm by adding 2.0 wt.% of Au atoms (namely NPA-1010006). Compared to that of NPA-1010006, further increasing Au atoms to 9 wt.% reduced the coherent length to 3.03 ± 0.1 nm, which could be attributed to restructuring between Pt crystals and Au
3+ ions followed by formation of Au crystals in the shell region (shown by the presence of a shoulder on the left hand side of the Pt (111) peak). EDX results of NPA-1004006 NC has been shown in
Figure S1.
Lattice strain, the coherent length (
Davg), and the crystallinity of pristine NCs were revealed by XRD analyses.
Figure 2 compares XRD patterns of the experimental NCs. Values of the
Davg of experimental NCs were calculated from the XRD peak broadening of (111) facets using the Scherrer equation, and their corresponding structural parameters are summarized in
Table 1. As indicated in
Figure 2, peaks
X1 and
X2 centered at 40.2° and 46.4° respectively refer to diffraction signals from (111) and (200) facets of metallic-phase Pt nanocrystals in the NCs. In an XRD pattern, the peak width denotes the relative dimension of long-range ordering in specific facets (i.e., the coherent length,
Davg) and the ratio of peak intensities between the (111) and (200) facets (i.e., h (111)/h (200)), which refers to the extent of preferential crystal growth for samples under investigation. For Ni@Pt NCs (
Figure S2), the
Davg increased by 0.1 nm, which could be attributed to the formation of thin layers of Pt in the shell region over the Ni core crystal with Pt/Ni ratios of 0.4 (Ni@Pt-1004) and 1.0 (Ni@Pt-1010) (
Table S1). A slight shift in diffraction peaks to the low-angle side features lattice expansion by increasing Pt/Ni ratios from 0.4 to 1.0. Meanwhile, the suppression of NiO
x peaks again consistently revealed increased shell coverage with Pt loading. An even closer inspection of
Davg values in (111) and (200) facets reveals the morphology of NCs. The Pt-CNT possessed the highest D (111)/D (200) ratio of 1.33 (higher D (111)) and thus the largest extent of preferential crystal growth along the (111) facet among the experimental NCs. Such a result rationalizes the intrinsic nature of the atomic arrangement in close-packed facets in Pt metal. For Ni@Pt-1004, compared to that of Pt-CNT, the D (111)/D (200) ratio decreased by 0.22, which could be attributed to the competition of crystal growth between galvanic replacement of Pt
4+ to Ni atoms in the open-(High-Miller-index facets) and Pt deposition in close-packed facets (111). In this status, a semi-coherent lattice match was found between facets with truncated surfaces, which can be explained by the formation of twin boundaries as revealed on HRTEM images (
Figure 1a). For Ni@Pt-1010, high contents of Pt
4+ ion reduction and deposition on the NC surface induced severe galvanic replacement to the NiO
x core. Such a phenomenon mostly occurred at interfaceted corners and edges resulting in a high content of surface truncation in NCs. In this event, NCs tended to form a spherical shape (
Figure 1d) due to the strong competition of galvanic replacement to Pt deposition, which was confirmed by a substantially reduced D (111)/D (200) ratio (0.95) compared to that of Pt-CNT.
After adding different contents of Au to Ni@Pt-1004 NCs (
Figure 2a), changes in the lattice strain and crystallinity were obvious due to atomic restructuring. In the case of Ni@Pt-1004 NCs, because of a lower Pt-content (Pt/Ni ratio of 0.4), complete coverage of the Ni-core from Pt atoms was not possible. Thus, with a lesser Au content (NPA-1004006), most of the Au atoms were intercalated with Pt atoms, which aggregated between NCs as revealed by the smeared diffraction peaks across the (111) and (200) facets. Restructuring by formation of Au-rich areas at high-order facets (i.e., (200)) between NCs was significant. These characteristics resulted from the spontaneous trans-metalation between Au
3+ ions and Ni metal atoms (galvanic replacement) accompanied by redeposition of Ni/Au atoms in shell crystals. Such a phenomenon was consistently proven by the suppressed diffraction signals (peaks M1 and M2) of the Au (111) and (200) facets. Further increasing the Au loading to 9 wt.% (NPA-100402) led to obviously increased diffraction signals for the Au (111) and (200) facets, which revealed the presence of discrete Au clusters (~2 nm) on the surface. At the same time, some of the Au atoms tended to deposit on the shell and core regions. Restructuring of Ni@Pt-1010 by interacting with Au
3+ ions showed a completely different behavior compared to that of Ni@Pt-1004. As shown in
Figure 2b, diffraction peaks
X1 and
X2 were not smeared; on the other hand, they were enhanced by the increasing Au contents from 2.0 to 9.0 wt.%. Such characteristics reveal that the long-range ordering of Ni@Pt NCs was improved by Au decoration. In the absence of diffraction peaks from the Au metal phase and the enhanced Au-4f photoemission peaks (
Figure S6, in supplementary information), one can notice that the galvanic replacement of Au
3+ to the NC surface was suppressed, which suggests conformal deposition of Au atoms in the Pt-rich shell.
The XPS analysis was performed in order to investigate the surface chemical composition (1~2 nm from the surface) and binding energy (BE) of elements in experimental NCs. The incident X-ray with an excitation energy of 650 eV corresponding to a probing depth of ~2.6 nm was employed to probe the Pt-4f, Ni-2p, and Au-4f orbitals.
Figure 3 reveals the typical fitted XPS spectra in the Pt-4f region of experimental NCs. In the Pt-4f spectrum, doublet peaks at 71 and 74 eV, respectively, emerged as photoelectron emission lines from the Pt-4f
7/2 and Pt-4f
5/2 orbitals. The peaks are further deconvoluted to separate the signals from different oxidation states, and corresponding results are given in
Table 2. Through an analysis of XPS patterns of Ni@Pt-1004 and Ni@Pt-1010 NCs (
Figure 3a,b), it can be seen that most of the Pt was in a zero-valence state (metallic state). In contrast, from the XPS spectra of Ni-2p orbitals (
Figure S3), it is clearly evident that Ni is present in an oxidized (NiO
x) form in both Ni@Pt NCs (i.e., Ni@Pt-1004 and Ni@Pt-1010). An even closer inspection of the intensities of the XPS spectra of Ni-2p (
Figure S3) revealed that NiO
x signals in Ni@Pt-1010 NCs were very much suppressed compared to those of Ni@Pt-1004 NCs. These spectral characteristics confirmed the core-shell structure of Ni@Pt-1010 NCs comprising a Ni core and Pt in the outermost layer. Meanwhile, the profound intensities of Ni-2p emission peaks in Ni@Pt-1004 NC refer to an incomplete Pt-shell over an underlying Ni-crystal. Those results again confirm prior HRTEM and XRD findings.
For Au-cluster-decorated Ni@Pt-1004 NCs (
Figure 3a), significant restructuring of the surface atomic arrangement was observed. NPA-1004006 NCs (2 wt.% Au) exhibited a higher extent of zero-valent Pt (Pt
0) contents on the surface compared to that of NPA-100402 (9 wt.% Au). More evidence of such atomic restructuring came from the XPS analysis of Ni-2p orbitals (
Figure S4). Compared to that of Ni@Pt-1004, we observed that the intensity of the Ni-2p emission peak was gradually suppressed with an increasing amount of Au of 2 to 9 wt.%. Intensities of emission peaks in an XPS spectrum are positively related to the electron density in probing orbitals of target atoms. Therefore, the higher intensity found in the curve of NPA-1004006 revealed that its most abundant 2p electrons were Ni atoms, compared to that of NPA-100402 NCs. Similar spectral changes were observed for Au-cluster-decorated Ni@Pt-1010 NCs (
Figure 3b). Obtained XPS results were very consistent with former structural characterizations. The difference in binding energy of the 4f
7/2 orbital of zero-valent Pt (Pt
0) was not obvious in the experimental NCs, revealing that electron relocation between Pt and neighboring atoms was nearly absent. For comparison XPS spectra of Ni@Pt-1010, NPA-1010006 and NPA-101002 at Ni-2p orbitals are compared in
Figure S5.
XPS spectra of experimental samples of Au-4f orbitals are compared in
Figure S6. Accordingly, intensities of Au-4f
5/2 and Au-4f
7/2 peaks increased with Au contents from 2 (NPA-1004006) to 9 wt.% (NPA-100402). Such a phenomenon shows the increasing exposure of Au with loading, which again consistently proves the formation of Au clusters in Ni@Pt NCs with an incomplete shell structure. In the case of Ni@Pt-1010, when the Au loading was 2.0 wt.%, the doublet peaks in the Au-4f orbital were insignificant. Such a result can be complimentarily explained by the crystal structure parameters. As indicated in
Figure 2b, a significant improvement in Pt crystallinity was found, and diffraction peak shifts were absent when decorated with 2 wt.% of Au atoms on the Ni@Pt surface. These features indicate the formation of atomic Au clusters on the NiO
x@Pt surface. Therefore, the presence of weak emission peaks suggests a discrete and smeared 4f orbital of Au atomic clusters that were finely dispersed in surface defect sites of NiO
x@Pt NCs. By increasing Au to 9 wt.%, pronounced Au-4f peaks rationalized the formation of Au sub-nano- or nanoclusters, as revealed by presence of a diffraction shoulder on the low-angle side of
X1 (111) and a pronounced intensity in
X2 (200) peaks.
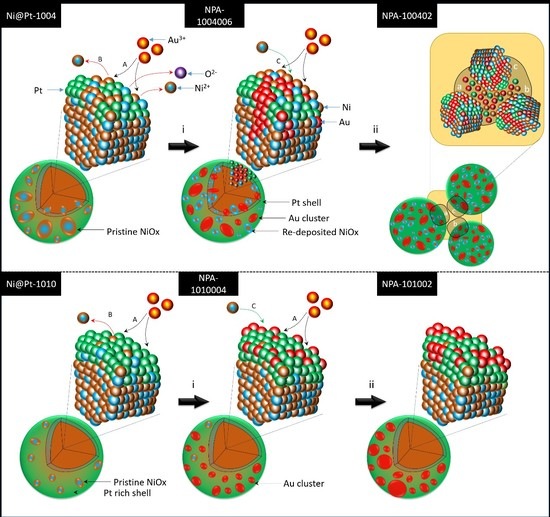
By cross-referencing results of the physical characterization, the effects of Au
3+ loading and Pt contents on the evolution of atomic structures of Ni@Pt NCs was systematically determined, and corresponding structural models are given in
Scheme 1—where the upper and lower layers respectively present changes in the atomic structure with increasing Au
3+ loading for Ni@Pt-1004 and Ni@Pt-1010. Accordingly, a significant galvanic replacement on oxidation followed by dissolution of Ni
0 to Ni
2+ appeared by interacting Ni@Pt-1004 with 2 wt.% of Au
3+ (step i in the upper layer of
Scheme 1) with the reaction of Au
3+ + Ni
0 (or Pt
0) Au
0 + 3/2Ni
2+ (or 4/3Pt
4+), where Au
3+ has a higher selectivity for Ni
0 than Pt
0 due to the larger electronegativity difference. In this event, Au atoms tended to penetrate the core region, thus resulting in the coexistence of nanosized Au and Pt clusters in NiO
x@Pt NPs. By increasing the loading to 9 wt.%, galvanic replacement between Au
3+ ion and core crystals was further enhanced, which caused the severe interparticle agglomeration by dissolution of core metal atoms accompanied by the rapid redeposition of residual metal ions between interfaces (i.e., regions a, b, and c in step ii of the upper layer of
Scheme 1) of the NPs. Compared to those of Ni@Pt-1004, the effects of Au
3+ loading on the structural evolution were suppressed by the high contents of the Pt shell structure in Ni@Pt-1010. As shown in the bottom layer of
Scheme 1, Au
3+ tended to be adsorbed and was reduced by NaBH
4 to form atomic clusters on the NP surface with a loading of 2.0 wt.% (NPA-1010006). By increasing the loading to 9.0 wt.%, the Au
3+ ions tended to form homoatomic bonds and thus grew into sub-nanometer crystals on the NPA-101002 surface (step ii in the bottom layer of
Scheme 1). These atomic structural arrangements provide direct information explaining ORR activities of the experimental NCs.
In heterogeneous catalysts, dissociation of chemisorbed oxygen molecules (i.e., the oxygen adsorption strength) is a cardinal performance-determining factor in ORRs. Lowering the oxygen adsorption energy reduces the applied energy for initiating ORRs at reaction sites and relocating them to neighboring atoms. In this way, the reaction kinetics and ORR activities of NCs can be substantially improved. In this study, the surface composition design of catalysts within the sub-nano scale played a key role in ORR performances of the NCs. By cross-referencing physical inspection results (the upper layer of
Scheme 1), the surface chemical configuration of NiO
x@Pt comprised mixtures of sub-nano Au and Pt clusters in the shell region and Au clusters intercalated with NiO
x in the core when the Pt/Ni ratio was 0.4 and Au was 2.0 wt.% With an Au content of 9.0 wt.%, severe interparticle agglomeration due to galvanic replacement (Au
3+ + Pt/Ni → Au + Pt
4+/Ni
2+) accompanied by Au crystal growth and redeposition of residual metal ions between NCs occurred. All three pathways dramatically reduced the degree of heteroatomic intermixing on the surface among reaction sites; therefore, ORR activities of those NCs were substantially suppressed.
Results of the electrochemical analyses consistently elucidated the above scenarios.
Figure 4a compares CV curves of the commercial Johnson Matthey-Pt/C catalyst (Johnson Matthey-Pt/C) with the experimental NCs (i.e., Ni@Pt-1004, NPA-1004006, and NPA-100402). Electrochemical active surface areas (ECSAs) are calculated based on corresponding CV curves using the oxygen desorption peak in the backward potential sweeping curve (detailed ECSA data of various ORR catalysts are listed in
electronic supplementary information in Table S2). Three distinctive potential regions are found in a CV curve, including an under-potential deposition of hydrogen (UPD-H) region at 0 < E < 0.4 V, a double-layer region between 0.4 and 0.6 V, and chemisorption of oxygen species at >0.6 V vs. the RHE because of hydrogen adsorption/desorption, OH- ligand chemisorption, and the formation of alpha Pt oxide (E
Oads; forward scan) as well as a reduction in Pt oxides (E
Odes; backward scan). In this way, the position and width of each peak are susceptible to the chemical composition and structure of the NC surface. For the Johnson Matthey-Pt/C, positions of two characteristic peaks (H
1 and H
2) in the forward scan denoted the potential to be applied for dissociation of H
+ from close-packed (111) and opened (200) facets and the corresponding current, respectively. In contrast, peaks H
1* and H
2* respectively refer to current responses of H
+ adsorption in the (111) and (200) facets. For Ni@Pt-1004, compared to the CV profile of the Johnson Matthey-Pt/C, a downshift of peaks H
1 and H
2 in the forward scan and an upshift of peaks H
1* and H
2* in the backward scan refer to a decreased energy barrier for redox desorption/adsorption of H
+. As consistently shown by XRD observations, a substantially higher intensity of peak H
1 than that of peak H
2 (i.e., weakened H
+ interactions on (200) facets) revealed preferential crystal growth at (111) facets in all experimental NCs.
Compared to that of the Johnson Matthey-Pt/C, Ni@Pt-1004 showed a higher surface area for H
2 evolution as revealed by the larger area of the UPD_H region. Meanwhile, the broadened and smeared UPD_H peaks revealed a high density of surface defects in Ni@Pt-1004. This statement is consistently illustrated by the pronounced oxygen adsorption peak (E
Oads) with a potential shift by ca. ~0.17 V (i.e., easy oxidation of the Ni@Pt-1004 surface) compared to that of the Johnson Matthey-Pt/C. Compared to the CV profile of NCs without Au decoration, a slight amount of Au decoration reduced the surface defect sites of Ni@Pt-1004, as consistently revealed by the significant suppression of the E
Oads and E
Odes peaks in NPA-1004006. Moreover, the position of the oxide reduction peak (E
Odes) was upshifted to high-potential sites at the same time. These two observations integrally bring out the fact that atomic decoration by Au clusters can fix the defect and simultaneously suppress the surface oxidation on the Ni@Pt-1004 surface. In the presence of discrete 4f orbitals in atomic clusters, a strong repulsive force to the chemisorbed O (O
ads) was formed at Au atoms. This relocated the O
ads to neighboring atoms around the Pt and Au interfaces and thus substantially boosted the ORR kinetic current (
Jk) of NPA-1004006 to ∼75 mA cm
−2 (details discussed below in the LSV analysis,
Figure 4c). For NPA-100402, the H
+ adsorption peak, “H
1*”, in the backward scan together with a smeared CV profile in forward scan was observed in the UPD_H region. Such a feature can be rationalized by its nanostructure, where the surface of the NC consists of nanosized Au/Pt clusters. Meanwhile, a severe interparticle agglomeration by the strong galvanic replacement accompanied by rapid reduction of residual metal ions was found (
Figure 1c,
Scheme 1); therefore, the heteroatomic intermix and the amount of reaction sites dramatically decreased. Formation of cluster-in-cluster structures turned 4f orbitals from a discrete state into a band structure. In this state, both the Au and Pt atoms possessed bulk properties, and consequently the
Jk of NPA-100402 was dramatically reduced by 87% (65.62 mA cm
−2) to 9.4 mA cm
−2 (
Table S2), compared to that of NPA-1004006. This value was even lower than that of Ni@Pt-1004, indicating that the impact of the heterogeneous interface between nanoclusters on ORR activity was limited.
On the other hand, as shown in
Figure 4b, changes in profiles in CV curves were insignificant by decoration with Au atoms on Ni@Pt-1010. In the case of 2.0 wt.% Au decoration, a slight suppression of both E
Oads and E
Odes was found for NPA-1010006 NCs compared to that of Ni@Pt-1010. These characteristics resemble the same redox response to O adsorption and desorption when decorating Au atomic clusters on the Ni@Pt-1004 surface. Increasing the Au content to 9.0 wt.% did not further suppress the redox peaks of O evolutions (E
Oads/E
Odes); however, they moved in an opposite direction. Due to the strong preference for homoatomic bonding, Au atoms tended to form sub-nano clusters instead of a conformal coating or defect sites on NC Pt surfaces. Those characteristics reduced the heteroatomic intermix and amount of O
2-splitting sites in NCs. The former suppresses intrinsic activity, and the latter reduces the number of active sites; therefore, the
Jk of NPA-101002 was substantially reduced by 70% (to ∼22 mA cm
−2), compared to that of NPA1010006. Again, given that most of the Au atoms were deposited as sub-nanometer clusters or atomic clusters (shown by absence of an Au diffraction peak in the XRD pattern of
Figure 2b), a high
Jk (∼22 mA cm
−2) was expected.
To further rationalize the impacts of Au decoration and Pt contents on ORR activities of Ni@Pt NPs, an LSV analysis was employed.
Figure 4c,d demonstrates LSV curves of NPA compared to control samples (Ni@Pt-1004 and Ni@Pt-1010) and the Johnson Matthey-Pt/C, where corresponding electrochemical parameters are summarized in
Figure 5, and
Tables S2 and S3. As shown in
Figure 4c and
Table S2, the onset potential (Voc vs. the RHE) of experimental samples followed the trend of the Johnson Matthey-Pt/C (0.910 V) < NPA-1004006 (0.964 V) < NPA-100402 (0.969 V) < Ni@Pt-1004 (0.990 V). Among them, the highest Voc indicates the lowest activation energy for initiating ORRs on Ni@Pt-1004 surfaces and can be rationalized by a large extent of heteroatomic intermix between Pt and Ni sub-nanoclusters. On such a surface, Pt clusters are in charge of splitting (dissociating) the oxygen molecule (O
2). After O
2 splitting, the two chemisorbed oxygen atoms are relocated to the high-oxygen-affinity Ni atoms for the subsequent reduction reaction. Since the dimensions of Pt clusters are quite small (i.e., the interface ratio between Ni domain is high), a rapid relocation of chemisorbed oxygen atoms to Ni sites is expected; therefore, the
Jk of Ni@Pt-1004 doubled compared to that of the Johnson Matthey-Pt/C. By decorating with 2.0 wt.% Au atoms on Ni@Pt-1004, the atomic Au clusters reduced surface defects of the Pt shell and covered the Ni core crystal. In this event, activation energy for O
2 splitting increased, which reduced the intrinsic activities of surface sites and thus the Voc by 0.026 V. Such a phenomenon would seemingly suppress the activity of surface sites on the NC surface; however, it actually went the opposite way. As revealed by results of physical structural inspections, decoration with a slight amount of Au atoms (2.0 wt.%) resulted in a surface structure comprising combinations of Au, Pt, and Ni sub-nanoclusters on the NC surface. The presence of sub-nano or atomic Au clusters in defect sites protected the NC surface from oxidation (shown by the suppressed E
Oads peak in the CV curve). In this event, relocation of the O
ads was dramatically facilitated, resulting in a quantum leap of
Jk by 6.7-fold compared to that of NCs without Au decoration (i.e., Ni@Pt-1004). By increasing Au to 9.0 wt.%, compared to that of NPA-1004006, the
Jk of NPA-100402 substantially decreased by ~87% to 9.4 mAcm
−2. This value was almost the same as that of Ni@Pt-1004, indicating that all reaction sites (Pt, Au, and Ni) possessed metallic properties of NPs (bulk) without facilitation of heteroatomic intermixing or ligand effects in ORRs. Meanwhile, as shown in
Figure 4c, similar slopes of diffusion (V < 0.8 V vs. the RHE) and kinetic limit (V > 0.8 V vs. the RHE) current regions indicated that the redox responses of Ni@Pt-1004 were not greatly influenced by a high Au loading. Such electrochemical properties are understandable due to the fact that Au atoms tend to form large nanocrystals by homogeneous crystal growth on the surface and galvanic replacement in the core region of Ni@Pt NCs. Compared to those of Ni@Pt-1004, Au decoration showed similar effects on ORR activity of Ni@Pt NCs with a conformal Pt shell (Ni@Pt-1010).
Figure 4d compares LSV curves of experimental NCs (NPA-1010006 and NPA-101002) with those of the control sample (Ni@Pt-1010) and the Johnson Matthey-Pt/C; corresponding electrochemical properties are summarized in
Table S3. Accordingly, VOC and E
1/2 followed the same trend as that of Ni@Pt-1004 with increasing Au contents from 2.0 to 9.0 wt.%. The same scenario to NPA NCs with low Pt contents held for NPA-1010006, except that Au atoms did not penetrate into the core region to form nanosized clusters, therefore NPA-101002 retained a high
Jk value (21.99 mA cm
−2) in ORRs.
Surface activities (SAs) of electrocatalysts in ORRs are calculated by normalizing the
Jk to the ECSA of the oxygen desorption region in the CV curve. These values are an important index for the average intrinsic activity of reaction sites on NC surfaces. As shown in
Table S2, the ECSA was 81.2 cm
2 mg
−1 for Ni@Pt-1004 and decreased to 50.0 cm
2 mg
−1 by decorating with 2.0 wt.% Au atoms, again proving that Au atoms tended to reside in defect sites of the Pt shell. Further increasing the Au content to 9.0 wt.% did not affect the ECSA value. This result, consistent with that proven by the XRD analysis, suggests that Au atoms tended to grow in homoatomic nanocrystals instead of capping on the Pt shell surface. Accordingly, SAs were 13.89 mA cm
−2 for NPA-1004006 and 1.83 mA cm
−2 for NPA-100402. With similar ESCAs, the significantly enhanced SA elucidates conformation of the substantially improved intrinsic ORR activity by the presence of atomic/sub-nano Au clusters simultaneously with Pt and Ni nanoclusters on the NPA-1004006 surface. For Ni@Pt with a conformal Pt shell, Au decoration mainly occurred on the NPA-1010006 surface, as indicated by a reduction in the ECSA (64.4 cm
2 mg
−1) by 11.9% compared to that of Ni@Pt-1010 (73.1 cm
2 mg
−1). Compared to that of NPA-1010006, the ESCA increased by 11.3% when decorated with 9.0 wt.% of Au on the Ni@Pt-1010 surface, which can be attributed to the formation of sub-nano Au clusters on the NC surface. In this event, SAs were 5.64 mA cm
−2 for NPA-1010006 and 1.61 mA cm
−2 for NPA-101002. Compared to those of NPA with Pt/Ni = 0.4, the substantially suppressed SA reveals the truth that the combination of atomic Au/Pt clusters with Ni atoms in neighboring sites can support exceptional reaction kinetics of ORRs (i.e.,
Jk and SA).
Mass activity (MA) refers to the current density per unit weight of active sites and is calculated by normalizing the residual current at 0.85 V vs. the RHE with respect to the loading amount of metal Pt in NCs. As illustrated in
Figure 5a, the MA of Ni@Pt was slightly improved by 26% compared that of the Johnson Matthey-Pt/C. By adding 2.0 wt.% Au atoms on the surface, the MA of NPA-1004006 substantially improved by 7.1-fold compared to that of Ni@Pt-1004. With a slight increment of noble metal loading, such a dramatic improvement in the MA depicts the truth for boosting the activity of NCs by syngeneic collaboration between sub-nano Au, Pt, and Ni domains in the reaction pathways in ORRs. Such a scenario was further confirmed by the MA of NPA-100402. In this NC, the MA was significantly reduced by 87% compared to that of NPA-1004006. Given that the difference in noble metal loading was small (7.0 wt.%), the dramatic difference in the MA again proves changes in intrinsic activities instead of mass differences. The same phenomenon exists in changes of the MA with respect to Au loading in Ni@Pt-1010 again complementarily proves the synergetic effects on ORR activities of NCs. Electrochemical results of control samples with commercial catalyst has been compared in
Figure S7 and corresponding parameters has been summarized in
Table S4.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}