Biofunctional Nanofibrous Substrate for Local TNF-Capturing as a Strategy to Control Inflammation in Arthritic Joints


 , ,
, , 
 and
and 
Abstract
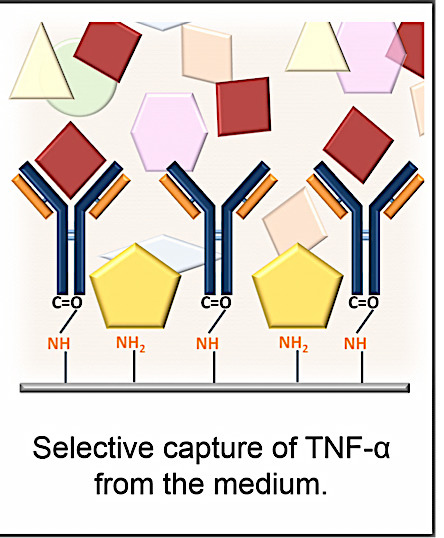
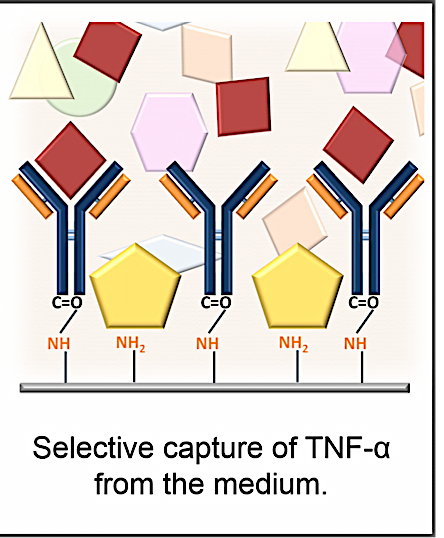
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Production and Functionalization of Nanofiber Meshes
2.2.1. Production of Nanofiber Meshes
2.2.2. Surface Functionalization of Electrospun NFMs
2.2.3. Quantification of Amine Groups Present at the Functionalized NFMs
2.3. Antibody Immobilization
2.4. Characterization Biofunctionalized NFMs
2.4.1. Scanning Electron Microscopy (SEM)
2.4.2. Fluorescence Microscopy
2.5. Capturing of TNF-α Present in Conditioned Medium of Macrophage Culture
Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Biological Assays
2.6.1. Isolation and Cell Culture
2.6.2. Seeding onto NFMs
2.6.3. DNA Quantification
2.6.4. Total Protein Synthesis Quantification
2.6.5. Glycosaminoglycan (GAG) Quantification
2.6.6. Histological Analysis
2.7. Statistical Analysis
3. Results and Discussion
3.1. Antibody Immobilization Efficiency
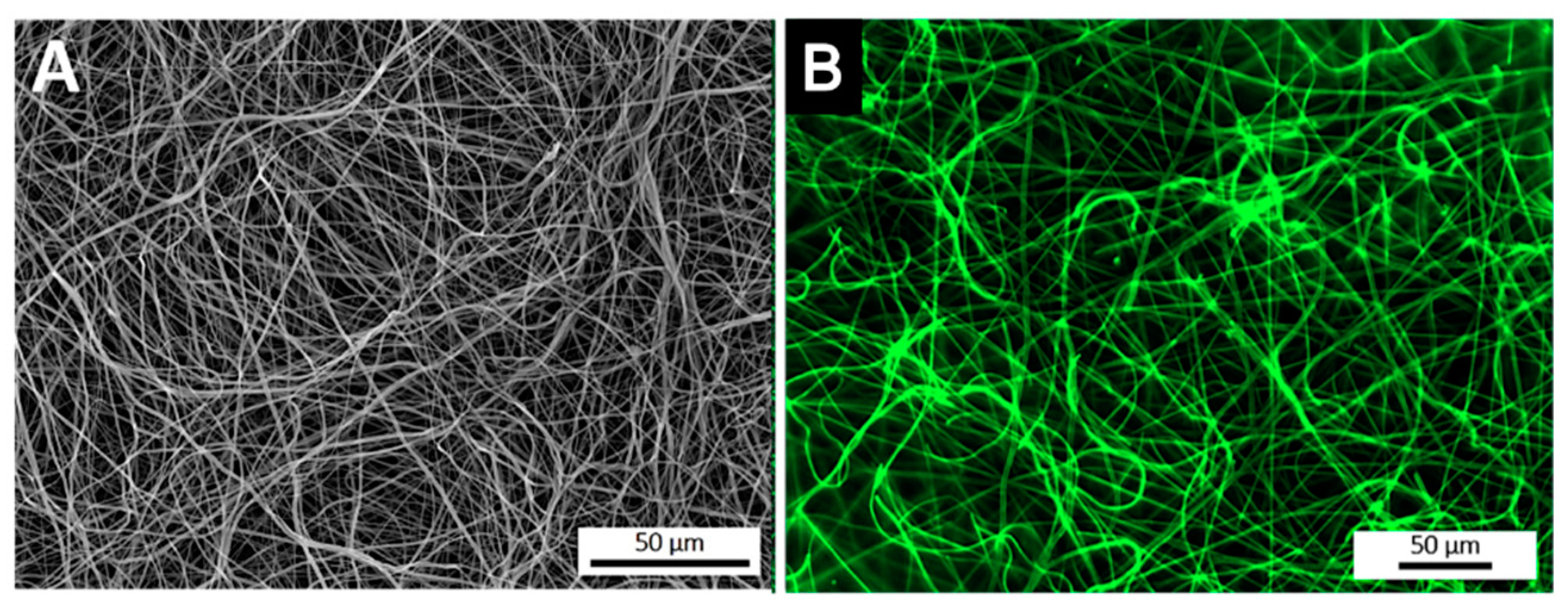
3.2. Spatial Distribution of the Antibody at the Surface of Electrospun Nanofibers
3.3. Quantification of Available Amine Groups
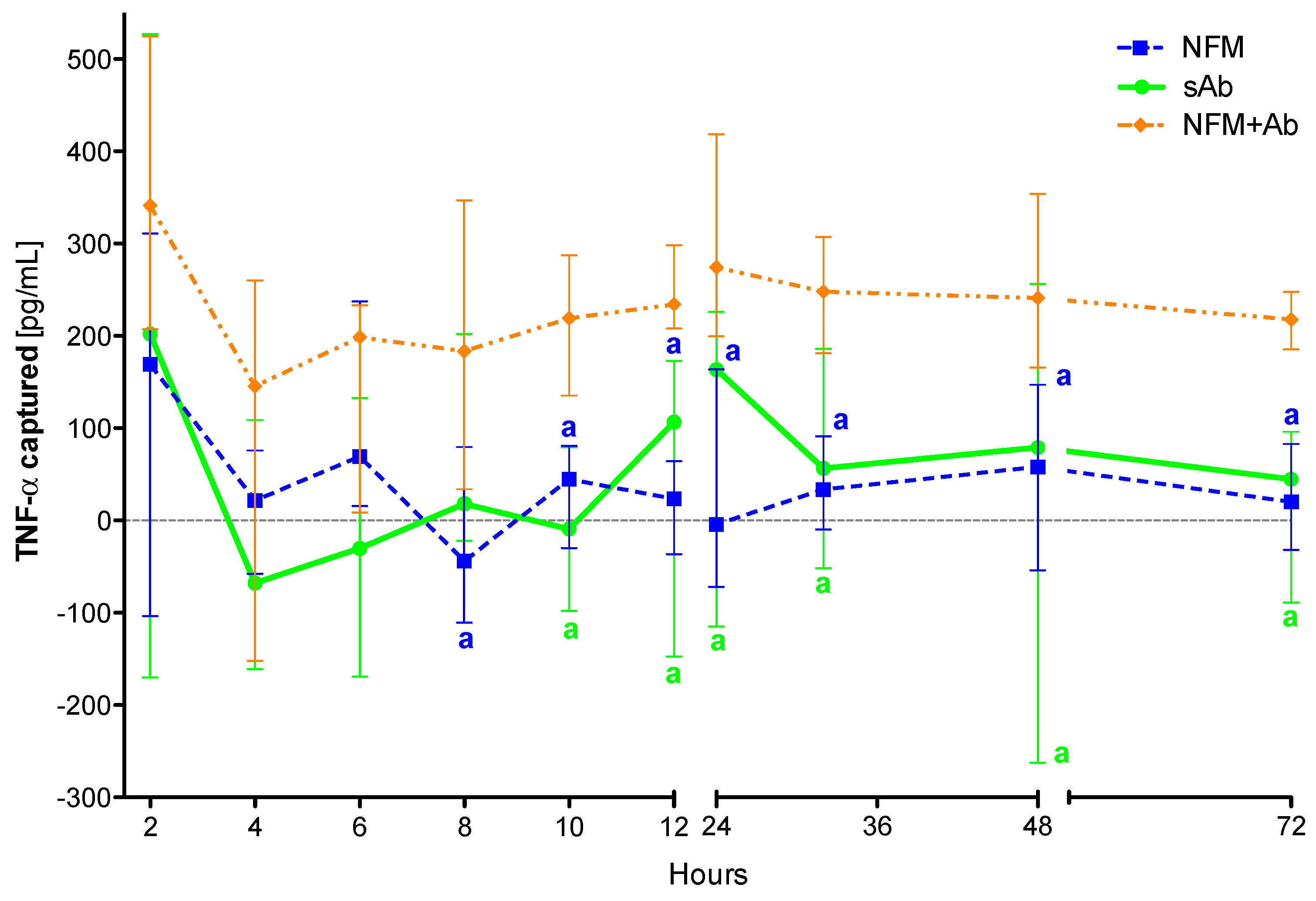
3.4. Quantification of Captured TNF-α
3.4.1. TNF-α Capturing during 3 days
3.4.2. TNF-α Capturing during 15 Days
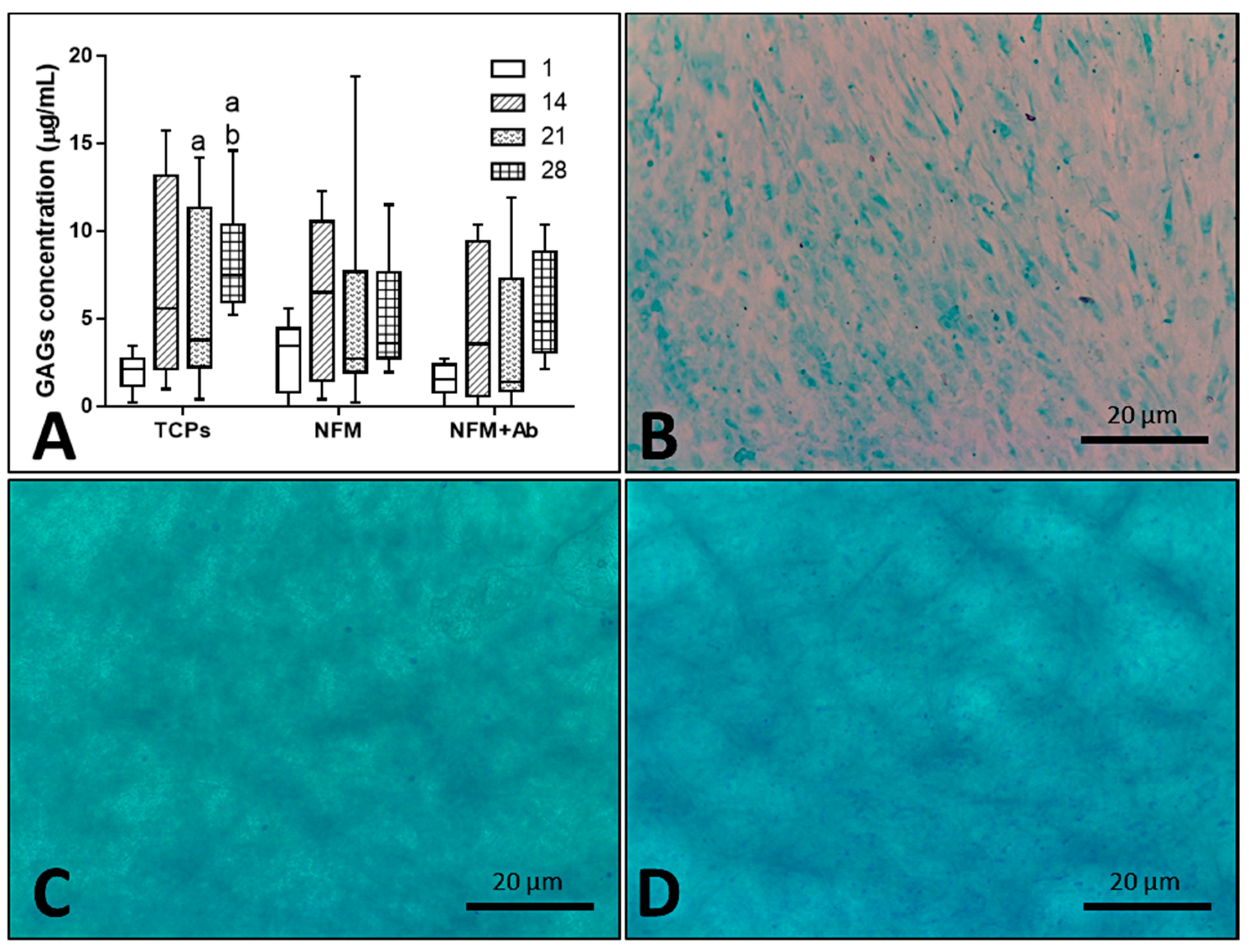
3.5. Biologic Assays
4. Conclusions
5. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Apostolaki, M.; Armaka, M.; Victoratos, P.; Kollias, G. Cellular mechanisms of TNF function in models of inflammation and autoimmunity. Curr. Dir. Autoimmun. 2010, 11, 1–26. [Google Scholar] [CrossRef]
- Korczowska, I. Rheumatoid arthritis susceptibility genes: An overview. World J. Orthop. 2014, 5, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- Quintero, O.L.; Amador-Patarroyo, M.J.; Montoya-Ortiz, G.; Rojas-Villarraga, A.; Anaya, J.-M. Autoimmune disease and gender: Plausible mechanisms for the female predominance of autoimmunity. J. Autoimmun. 2012, 38, J109–J119. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Moroni, L.; Bianchi, I.; Lleo, A. Geoepidemiology, gender and autoimmune disease. Autoimmun. Rev. 2012, 11, A386–A392. [Google Scholar] [CrossRef] [PubMed]
- Amur, S.; Parekh, A.; Mummaneni, P. Sex differences and genomics in autoimmune diseases. J. Autoimmun. 2012, 38, J254–J265. [Google Scholar] [CrossRef] [PubMed]
- Nussinovitch, U.; Shoenfeld, Y. The role of gender and organ specific autoimmunity. Autoimmun. Rev. 2012, 11, A377–A385. [Google Scholar] [CrossRef] [PubMed]
- Pennell, L.M.; Galligan, C.L.; Fish, E.N. Sex affects immunity. J. Autoimmun. 2012, 38, J282–J291. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W.J. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Perricone, C.; Ceccarelli, F.; Valesini, G. An overview on the genetic of rheumatoid arthritis: A never-ending story. Autoimmun. Rev. 2011, 10, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Valesini, G. Tumour necrosis factor α antagonists in the treatment of rheumatoid arthritis: An immunological perspective. BioDrugs 2014, 28 (Suppl. 1), 5–13. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Mcinnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2012, 2205–2219. [Google Scholar] [CrossRef]
- Brennan, F.M.; Foey, A.D. Cytokine regulation in RA synovial tissue: Role of T cell/macrophage contact-dependent interactions. Arthritis Res. 2002, 4 (Suppl. 3), S177–S182. [Google Scholar] [CrossRef]
- Caplan, M.S.; Jilling, T. The pathophysiology of necrotizing enterocolitis. NeoReviews 2001, 2, e103–e109. [Google Scholar] [CrossRef]
- Taylor, P.C. Anti-TNF therapy for rheumatoid arthritis and other inflammatory diseases. Mol. Biotechnol. 2001, 19, 153–168. [Google Scholar] [CrossRef]
- Edrees, A.F.; Misra, S.N.; Abdou, N.I. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: Correlation of TNF-alpha serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin. Exp. Rheumatol. 2005, 23, 469–474. [Google Scholar]
- Rubbert-Roth, A. Assessing the safety of biologic agents in patients with rheumatoid arthritis. Rheumatology 2012, 51, v38–v47. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 1989, 334, 244–247. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-α: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef]
- Jones, E.Y.; Stuart, D.I.; Walker, N.P.C. Structure of tumour necrosis factor. Nature 1989, 338, 225–228. [Google Scholar] [CrossRef]
- Bazzoni, F.; Beutler, B. The Tumor Necrosis Factor Ligand and receptor families. N. Engl. J. Med. 1996, 334, 1717–1725. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Declercq, W.; Beyaert, R.; Fiers, W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 1995, 5, 392–399. [Google Scholar] [CrossRef]
- Willrich, M.A.V.; Murray, D.L.; Snyder, M.R. Tumor necrosis factor inhibitors: Clinical utility in autoimmune diseases. Transl. Res. 2015, 165, 270–282. [Google Scholar] [CrossRef]
- Croft, M.; Duan, W.; Choi, H.; Eun, S.-Y.; Madireddi, S.; Mehta, A. TNF superfamily in inflammatory disease: Translating basic insights. Trends Immunol. 2012, 33, 144–152. [Google Scholar] [CrossRef]
- Hoes, J.N.; Jacobs, J.W.G.; Buttgereit, F.; Bijlsma, J.W.J. Current view of glucocorticoid co-therapy with DMARDs in rheumatoid arthritis. Nat. Rev. Rheumatol. 2010, 6, 693–702. [Google Scholar] [CrossRef]
- Walsh, D.A.; McWilliams, D.F. Mechanisms, impact and management of pain in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 581–592. [Google Scholar] [CrossRef]
- Rossi, D.; Modena, V.; Sciascia, S.; Roccatello, D. Rheumatoid arthritis: Biological therapy other than anti-TNF. Int. Immunopharmacol. 2015, 27, 185–188. [Google Scholar] [CrossRef]
- Alves da Silva, M.; Martins, A.; Teixeira, A.A.; Reis, R.L.; Neves, N.M. Impact of biological agents and tissue engineering approaches on the treatment of rheumatic diseases. Tissue Eng. Part B Rev. 2010, 16, 331–339. [Google Scholar] [CrossRef]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Katsikis, P.; Brennan, F.M.; Walker, J.; Bijl, H.; Ghrayeb, J.; et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993, 36, 1681–1690. [Google Scholar] [CrossRef]
- Buchan, G.; Barrett, K.; Turner, M.; Chantry, D.; Maini, R.N.; Feldmann, M. Interleukin-1 and tumour necrosis factor mRNA expression in rheumatoid arthritis: Prolonged production of IL-1 alpha. Clin. Exp. Immunol. 1988, 73, 449–455. [Google Scholar]
- Keffer, J.; Probert, L.; Caziaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Haworth, C.; Brennan, F.M.; Chantry, D.; Turner, M.; Maini, R.N.; Feldmann, M. Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: Regulation by tumor necrosis factor-alpha. Eur. J. Immunol. 1991, 21, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.O.; Feldmann, M.; Maini, R.N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 1992, 89, 9784–9788. [Google Scholar] [CrossRef]
- Dimitroulas, T.; Nikas, S.N.; Trontzas, P.; Kitas, G.D. Biologic therapies and systemic bone loss in rheumatoid arthritis. Autoimmun. Rev. 2013, 12, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Astrakhantseva, I.V.; Efimov, G.A.; Drutskaya, M.S.; Kruglov, A.A.; Nedospasov, S.A. Modern anti-cytokine therapy of autoimmune diseases. Biochemistry 2014, 79, 1308–1321. [Google Scholar] [CrossRef]
- Quan, L.-D.; Thiele, G.M.; Tian, J.; Wang, D. The development of novel therapies for rheumatoid arthritis. Expert Opin. Ther. Pat. 2008, 18, 723–738. [Google Scholar] [CrossRef]
- Tarner, I.H.; Müller-Ladner, U. Drug delivery systems for the treatment of rheumatoid arthritis. Expert Opin. Drug Deliv. 2008, 5, 1027–1037. [Google Scholar] [CrossRef]
- Gokhale, K.S.; Jonnalagadda, S. Preparation and evaluation of sustained release infliximab microspheres. PDA J. Pharm. Sci. Technol. 2013, 67, 255–266. [Google Scholar] [CrossRef]
- McInnes, S.J.P.; Turner, C.T.; Al-Bataineh, S.A.; Airaghi Leccardi, M.J.I.; Irani, Y.; Williams, K.A.; Cowin, A.J.; Voelcker, N.H. Surface engineering of porous silicon to optimise therapeutic antibody loading and release. J. Mater. Chem. B 2015, 3, 4123–4133. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, W.; Na, K. Temperature-modulated noncovalent interaction controllable complex for the long-term delivery of etanercept to treat rheumatoid arthritis. J. Control. Release 2013, 171, 143–151. [Google Scholar] [CrossRef]
- Alves da Silva, M.L.; Martins, A.; Costa-Pinto, A.R.; Costa, P.; Faria, S.; Gomes, M.; Reis, R.L.; Neves, N.M. Cartilage Tissue Engineering Using Electrospun PCL Nanofiber Meshes and MSCs. Biomacromolecules 2010, 11, 3228–3236. [Google Scholar] [CrossRef]
- Alves da Silva, M.L.; Costa-Pinto, A.R.; Martins, A.; Correlo, V.M.; Sol, P.; Bhattacharya, M.; Faria, S.; Reis, R.L.; Neves, N.M. Conditioned medium as a strategy for human stem cells chondrogenic differentiation. J. Tissue Eng. Regen. Med. 2015, 9, 714–723. [Google Scholar] [CrossRef]
- Piai, J.F.; Alves da Silva, M.L.; Martins, A.; Torres, A.; Faria, S.; Reis, R.L.; Muniz, E.C.; Neves, N.M. Chondroitin sulfate immobilization at the surface of electrospun nanofiber meshes for cartilage tissue regeneration approaches. Appl. Surf. Sci. 2017, 403, 112–125. [Google Scholar] [CrossRef]
- Oliveira, C.; Costa-Pinto, A.R.; Reis, R.L.; Martins, A.; Neves, N.M. Biofunctional nanofibrous substrate comprising immobilized antibodies and selective binding of autologous growth factors. Biomacromolecules 2014, 15, 2196–2205. [Google Scholar] [CrossRef]
- Monteiro, N.; Martins, A.; Pires, R.; Faria, S.; Fonseca, N.A.; Moreira, J.N.; Reis, R.L.; Neves, N.M. Immobilization of bioactive factor-loaded liposomes on the surface of electrospun nanofibers targeting tissue engineering. Biomater. Sci. 2014, 2, 1195–1209. [Google Scholar] [CrossRef]
- Kakabakos, S.E.; Tyllianakis, P.E.; Evangelatos, G.P.; Ithakissios, D.S. Colorimetric determination of reactive solid-supported primary and secondary amino groups. Biomaterials 1994, 15, 289–297. [Google Scholar] [CrossRef]
- Riddles, P.W.; Blakeley, R.L.; Zerner, B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid)—A reexamination. Anal. Biochem. 1979, 94, 75–81. [Google Scholar] [CrossRef]
- Caporali, R.; Pallavicini, F.B.; Filippini, M.; Gorla, R.; Marchesoni, A.; Favalli, F.B.; Sarzi-Puttini, P.; Atzeni, F.; Montecucco, C. Treatment of rheumatoid arthritis with anti-TNF-alpha agents: A reappraisal. Autoimmun. Rev. 2009, 8, 274–280. [Google Scholar] [CrossRef]
- Turková, J. Oriented immobilization of biologically active proteins as a tool for revealing protein interactions and function. J. Chromatogr. B Biomed. Sci. Appl. 1999, 722, 11–31. [Google Scholar] [CrossRef]
- Joos, H.; Wildner, A.; Hogrefe, C.; Reichel, H.; Brenner, R.E. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res. Ther. 2013, 15, R119. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, A.I.; Beekhuizen, M.; ‘t Hart, M.C.; Radstake, T.; Dhert, W.; Saris, D.; van Osch, G.; Creemers, L.B. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 2014, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Timmen, M.; Hidding, H.; Wieskötter, B.; Baum, W.; Pap, T.; Raschke, M.J.; Schett, G.; Zwerina, J.; Stange, R. Influence of antiTNF-alpha antibody treatment on fracture healing under chronic inflammation. BMC Musculoskelet. Disord. 2014, 15, 184. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | SH/NH2 Groups [mol/cm] |
|---|---|
| Untreated | (3.54 ± 5.61) × 10−9 |
| UV-O activated | (5.86 ± 1.12) × 10−9 |
| Aminolysis-treatment | (17.9 ± 6.06) × 10−9 |
| Immobilized Antibody | (8.65 ± 3.41) × 10−9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacelo, E.; Alves da Silva, M.; Cunha, C.; Faria, S.; Carvalho, A.; Reis, R.L.; Martins, A.; Neves, N.M. Biofunctional Nanofibrous Substrate for Local TNF-Capturing as a Strategy to Control Inflammation in Arthritic Joints. Nanomaterials 2019, 9, 567. https://doi.org/10.3390/nano9040567
Bacelo E, Alves da Silva M, Cunha C, Faria S, Carvalho A, Reis RL, Martins A, Neves NM. Biofunctional Nanofibrous Substrate for Local TNF-Capturing as a Strategy to Control Inflammation in Arthritic Joints. Nanomaterials. 2019; 9(4):567. https://doi.org/10.3390/nano9040567
Chicago/Turabian StyleBacelo, Elisa, Marta Alves da Silva, Cristina Cunha, Susana Faria, Agostinho Carvalho, Rui L. Reis, Albino Martins, and Nuno M. Neves. 2019. "Biofunctional Nanofibrous Substrate for Local TNF-Capturing as a Strategy to Control Inflammation in Arthritic Joints" Nanomaterials 9, no. 4: 567. https://doi.org/10.3390/nano9040567
APA StyleBacelo, E., Alves da Silva, M., Cunha, C., Faria, S., Carvalho, A., Reis, R. L., Martins, A., & Neves, N. M. (2019). Biofunctional Nanofibrous Substrate for Local TNF-Capturing as a Strategy to Control Inflammation in Arthritic Joints. Nanomaterials, 9(4), 567. https://doi.org/10.3390/nano9040567