Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis




Abstract
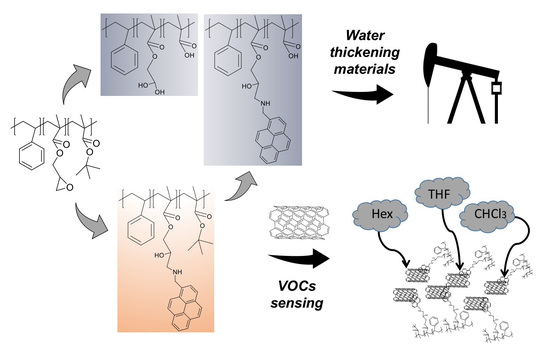
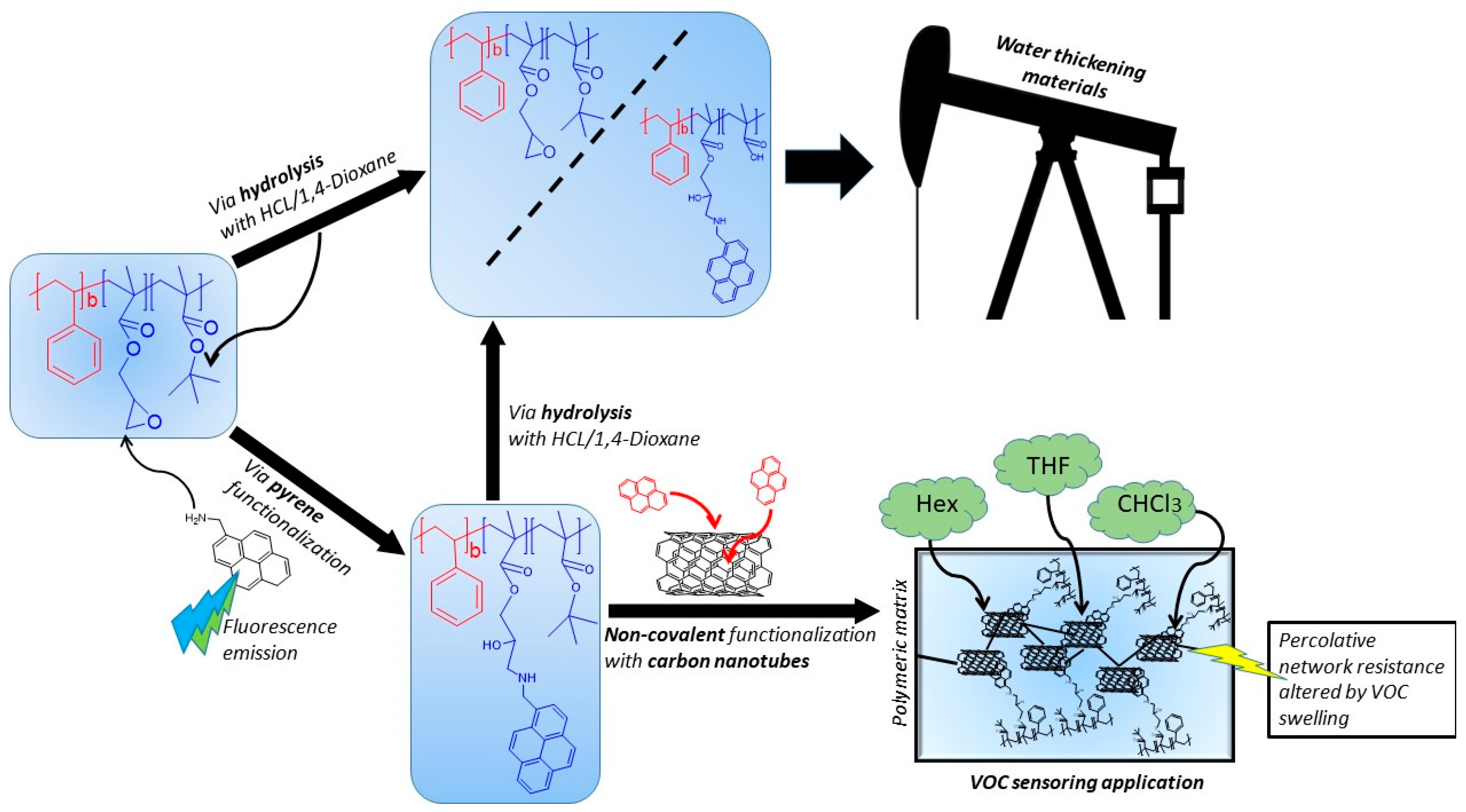
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthetic Procedure, Functionalization, Hydrolysis and Neutralization of Terpolymers
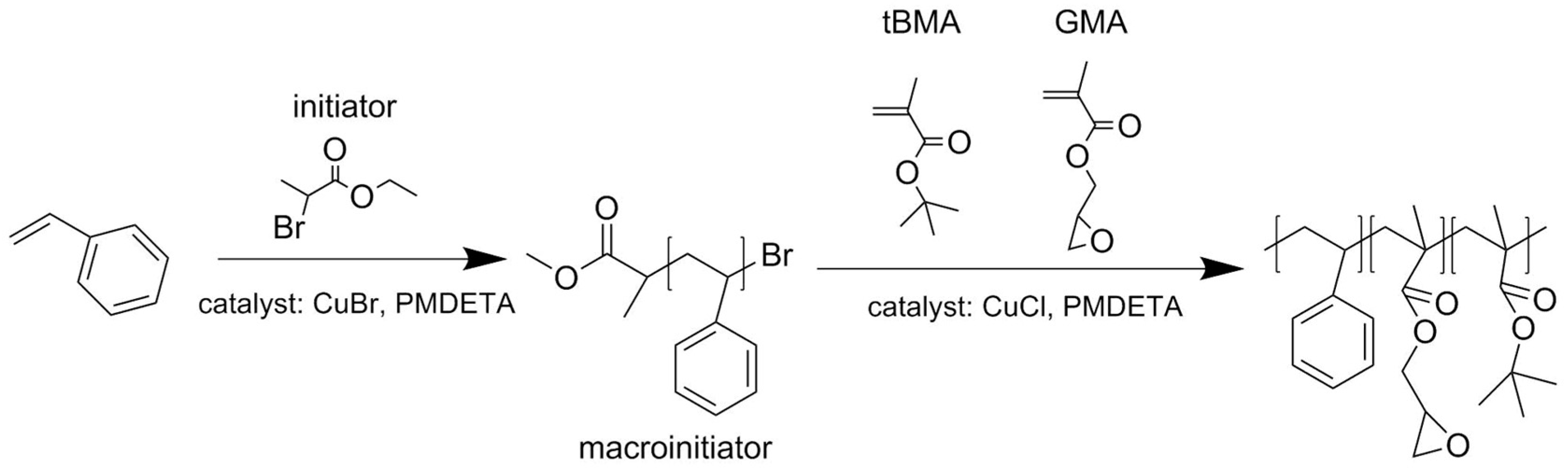
2.2.1. Synthesis of Polystyrene Macroinitiator (PS-Br)
2.2.2. Synthesis of Terpolymer polystyrene-block-(glycidyl methacrylate-co-tert-butyl methacrylate), PS-b-(GMA-r-tBMA)
2.2.3. Kinetic Experiments
2.2.4. Functionalization of PS-b-(GMA-r-tBMA)
2.2.5. Functionalization with 1-AMP
2.2.6. Hydrolysis and Neutralization of PS-b-(GMA-r-tBMA) and TP-AMP
2.3. Nanocomposite and VOC Exposure Setup Preparation
2.4. Characterization and Instruments
3. Results and Discussion
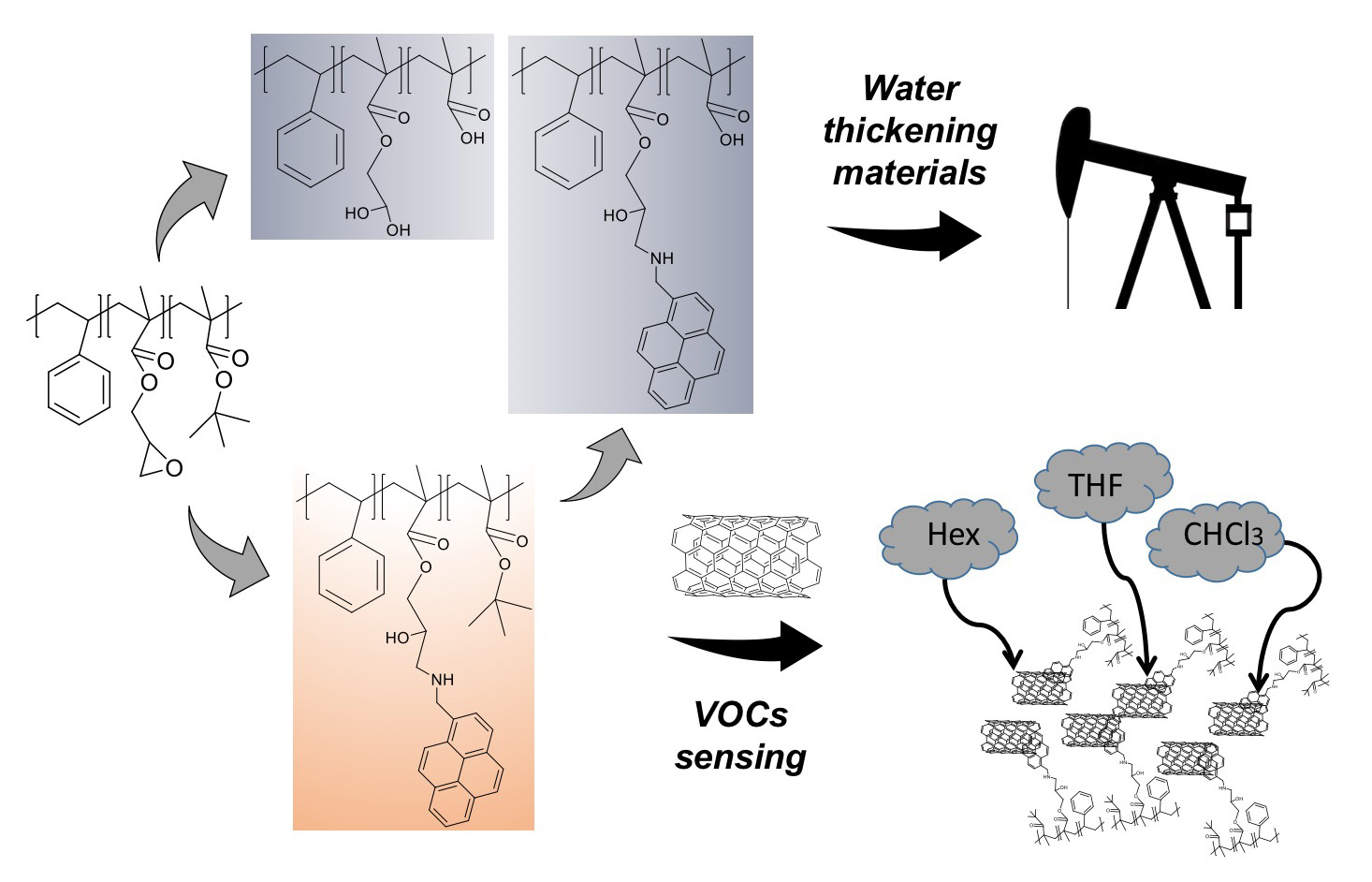
3.1. ATRP Synthesis of PS-b-(tBMA-co-GMA) Terpolymer
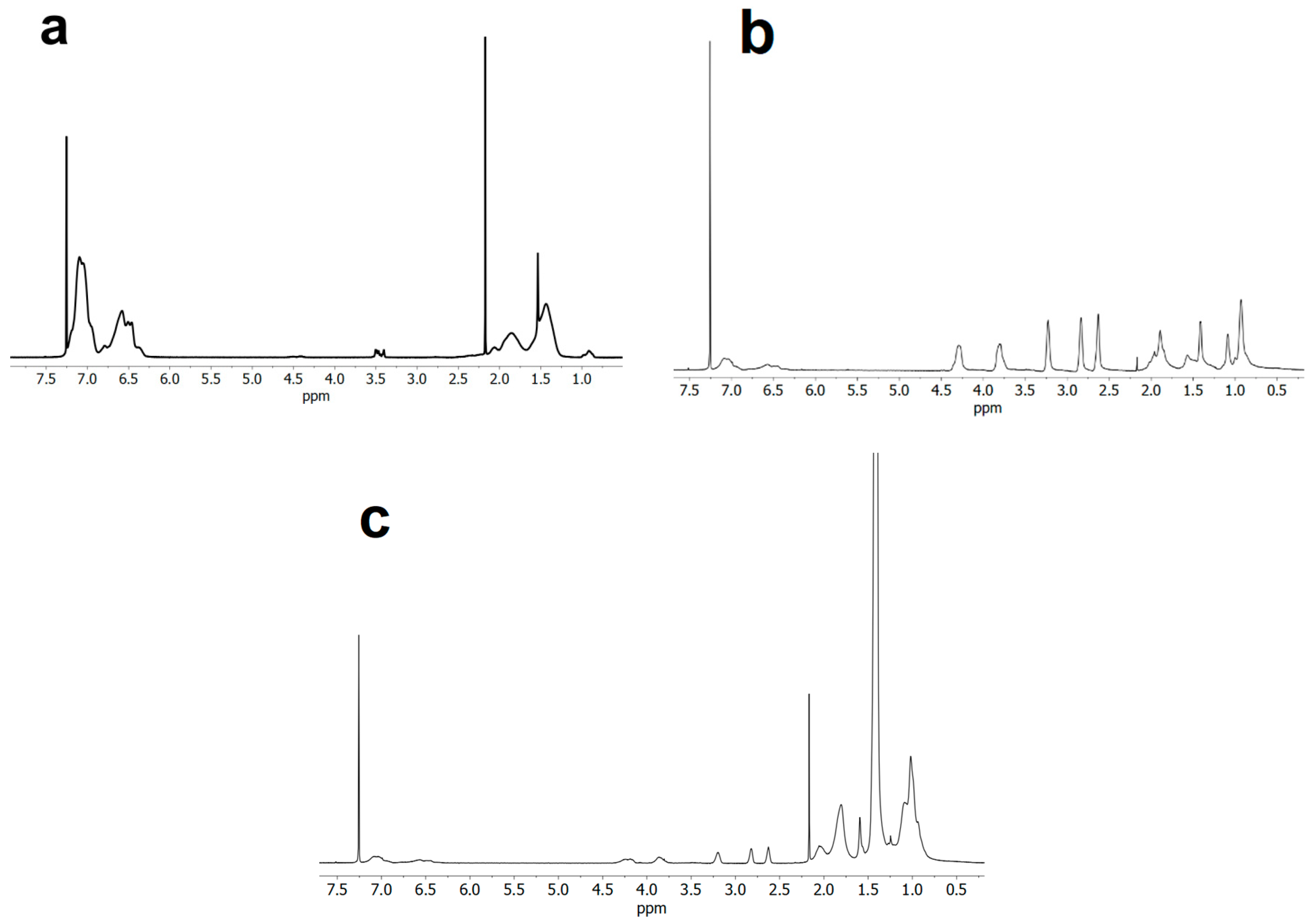
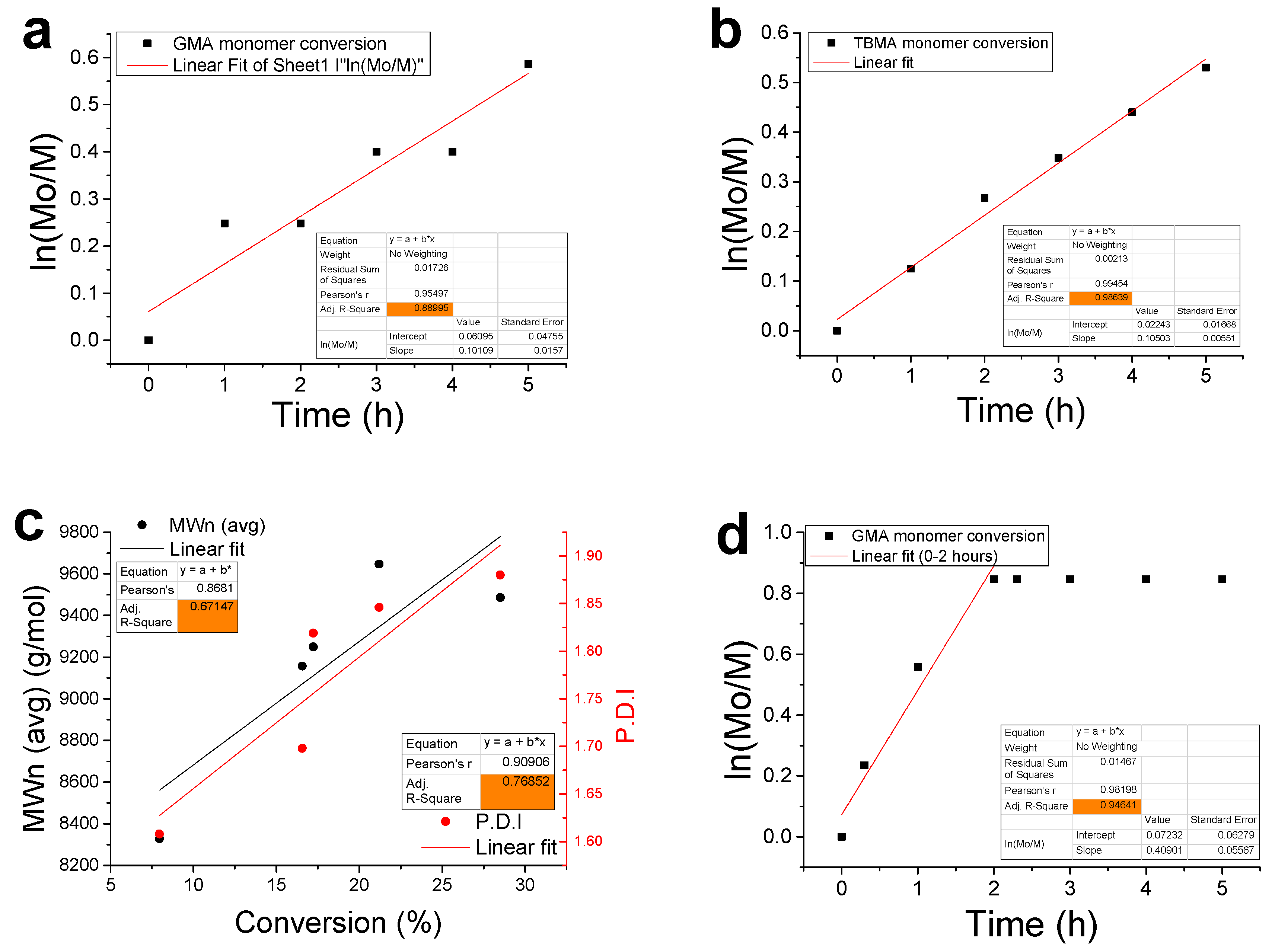
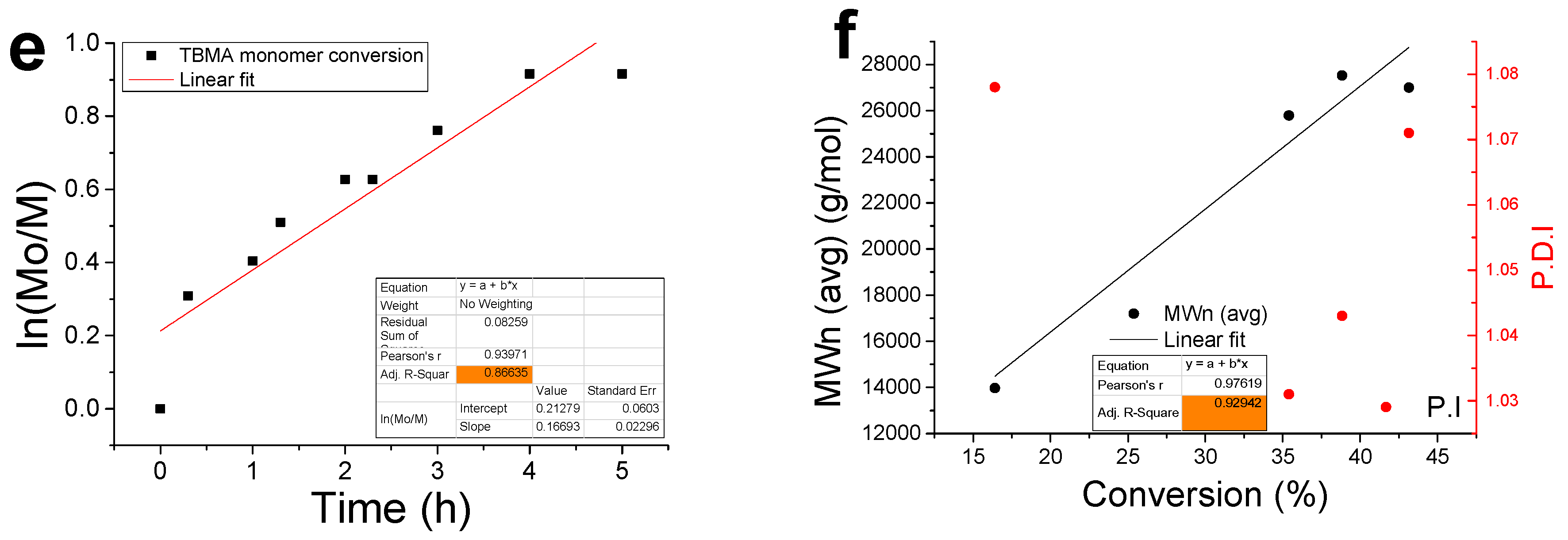
3.2. Kinetic Analysis
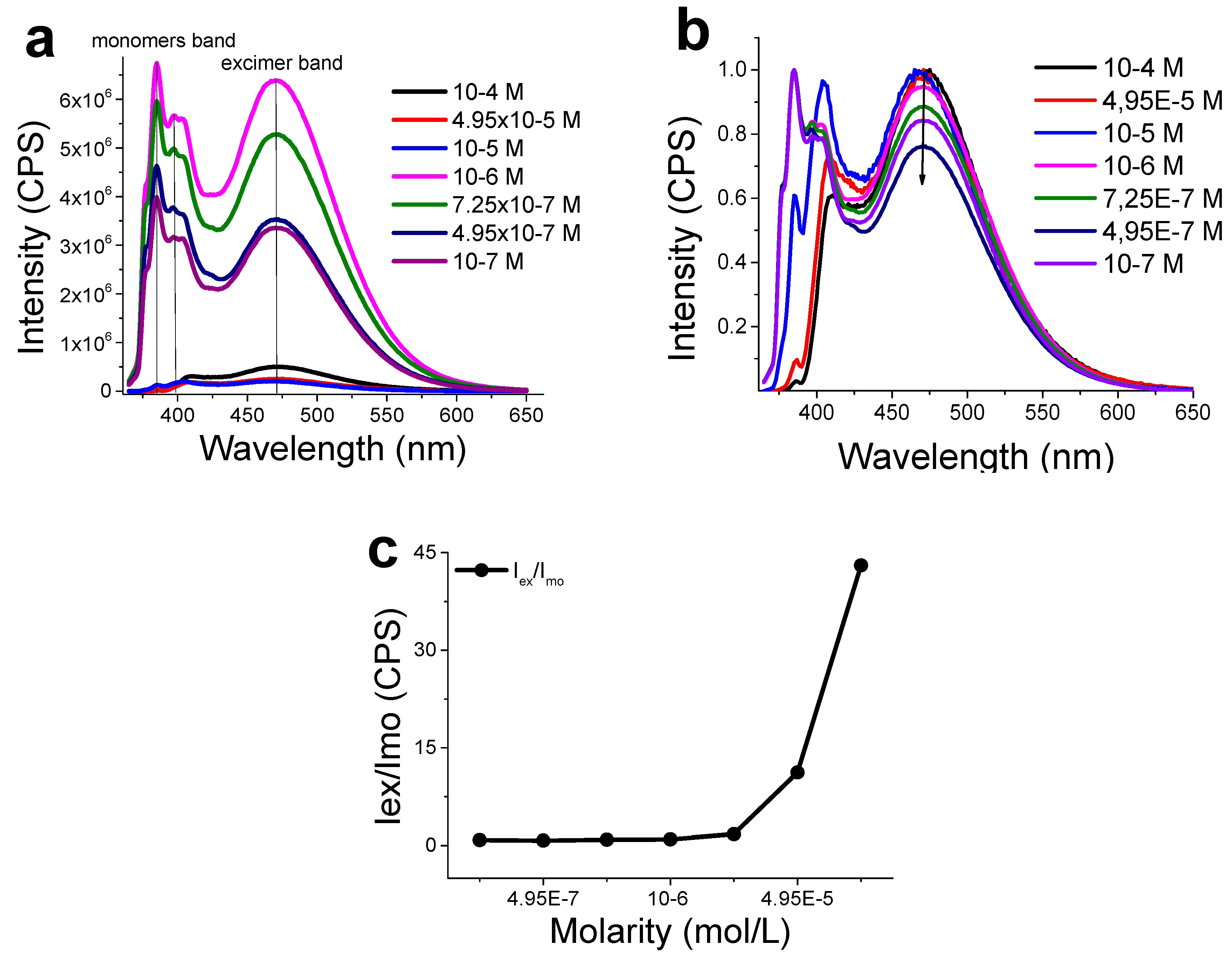
3.3. Functionalization with 1-Pyrenemethylamine (1-AMP)
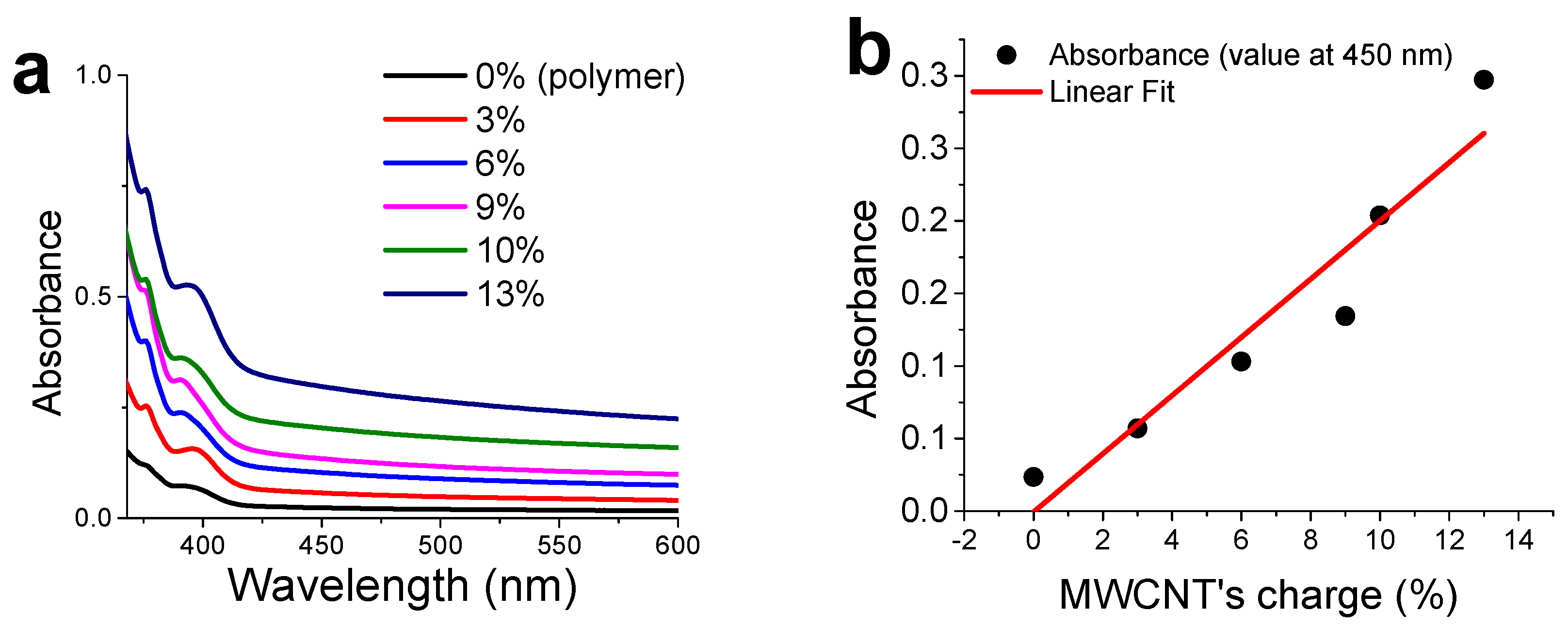
3.4. CNTs Dispersion and Stabilization by AMP-Functionalized Terpolymer
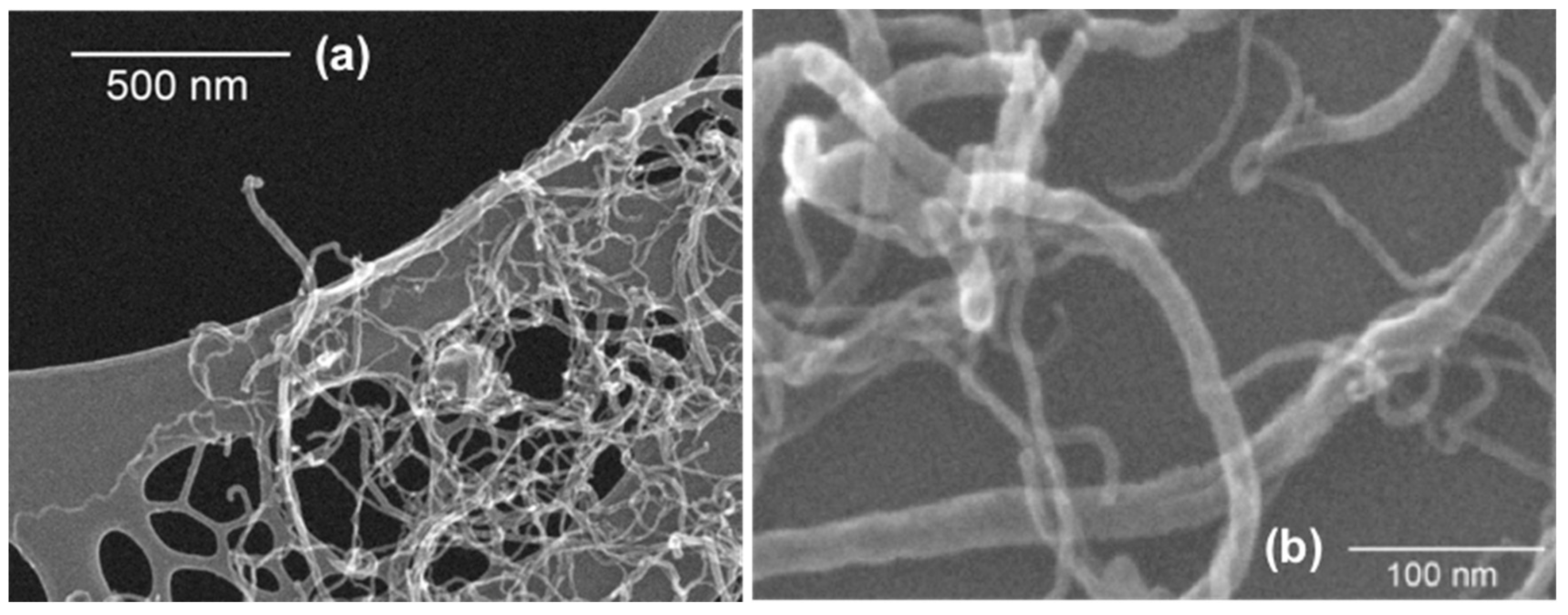
3.5. Scanning Electron Microscopy (SEM) Analysis of CNTs Dispersion
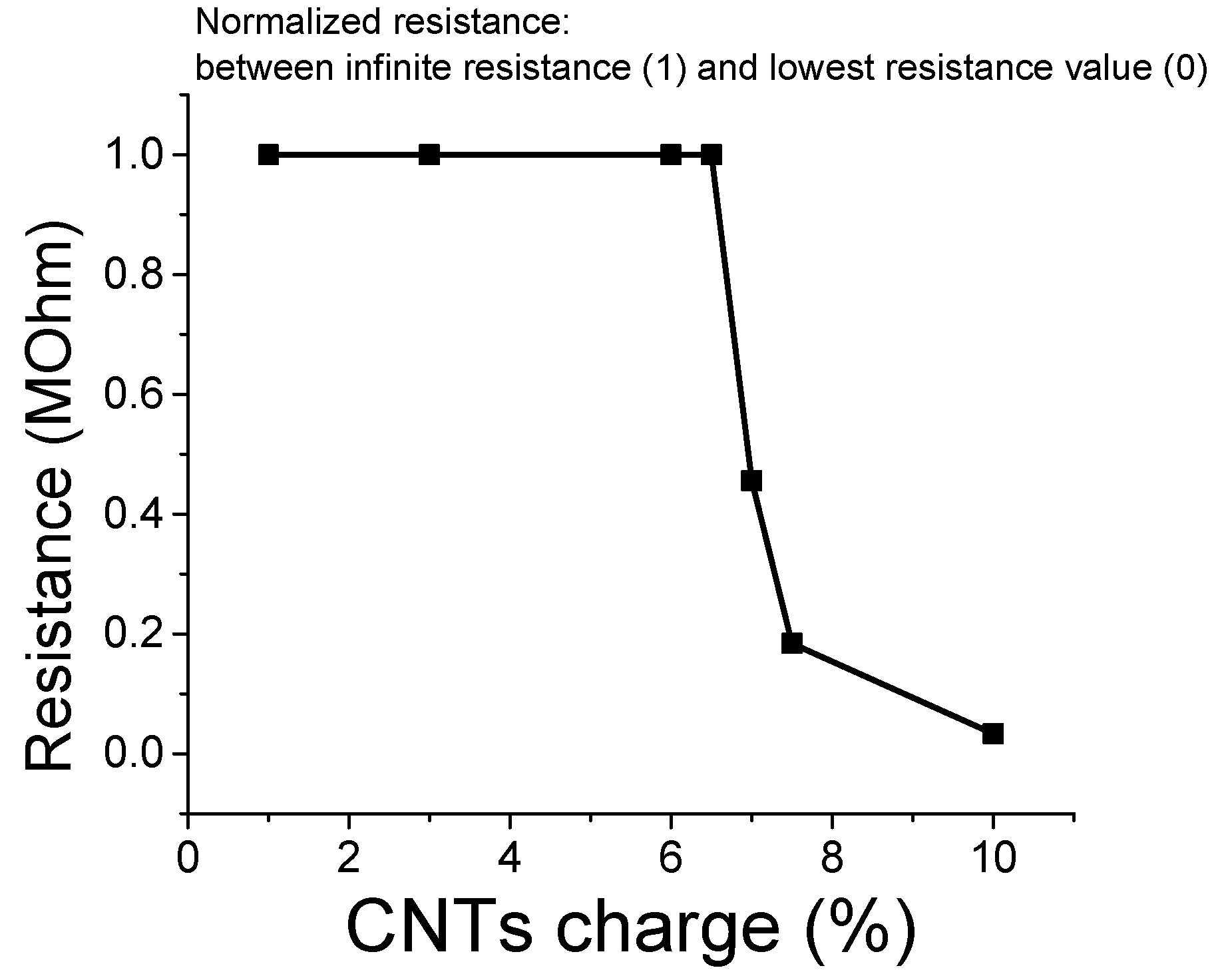
3.6. Percolation Threshold Calculation
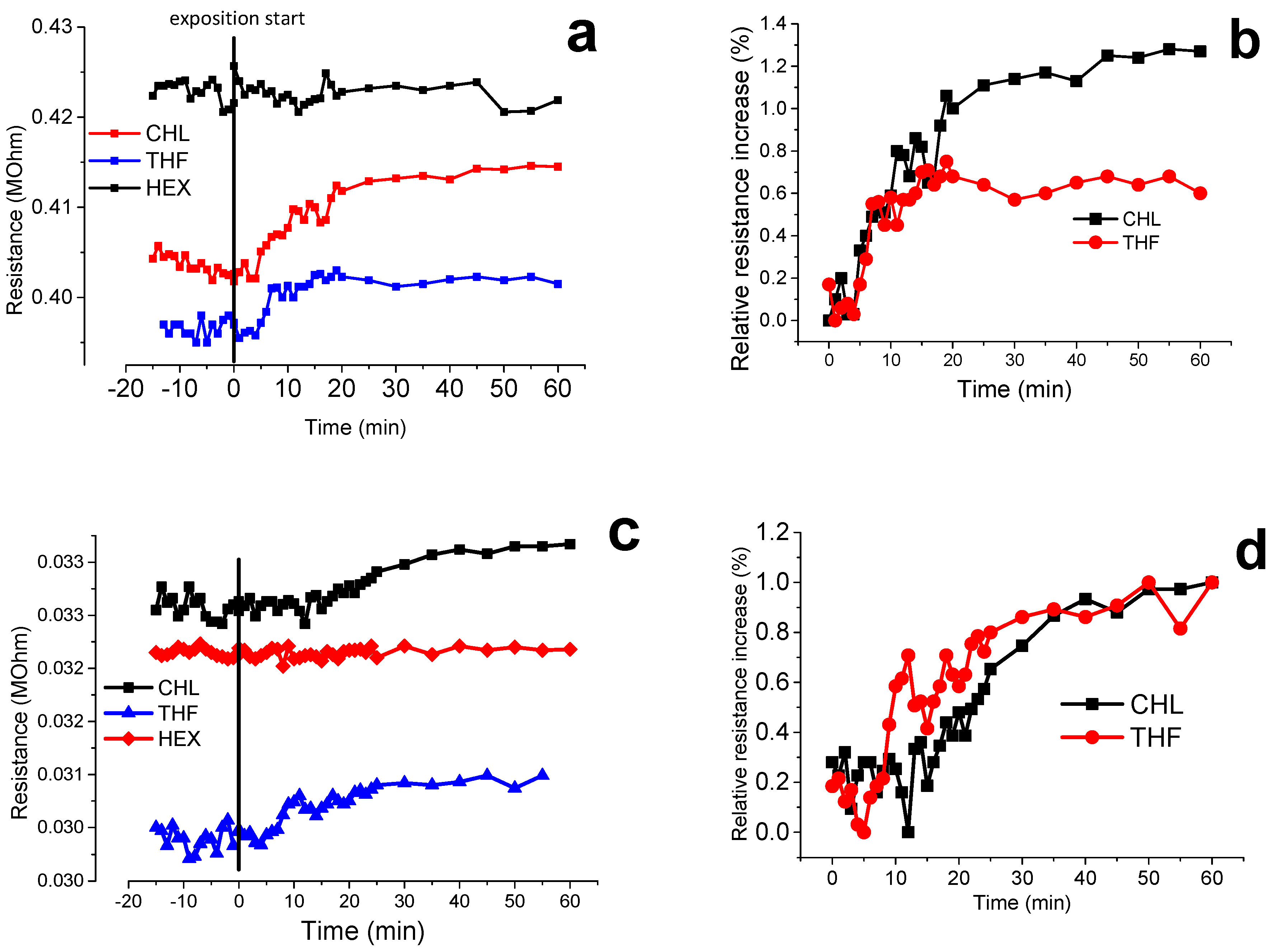
3.7. Volatile Organic Compound (VOCs) Exposure Experiments
3.8. Hydrolysis and Neutralization of TP and AMP-Functionalized Polymers
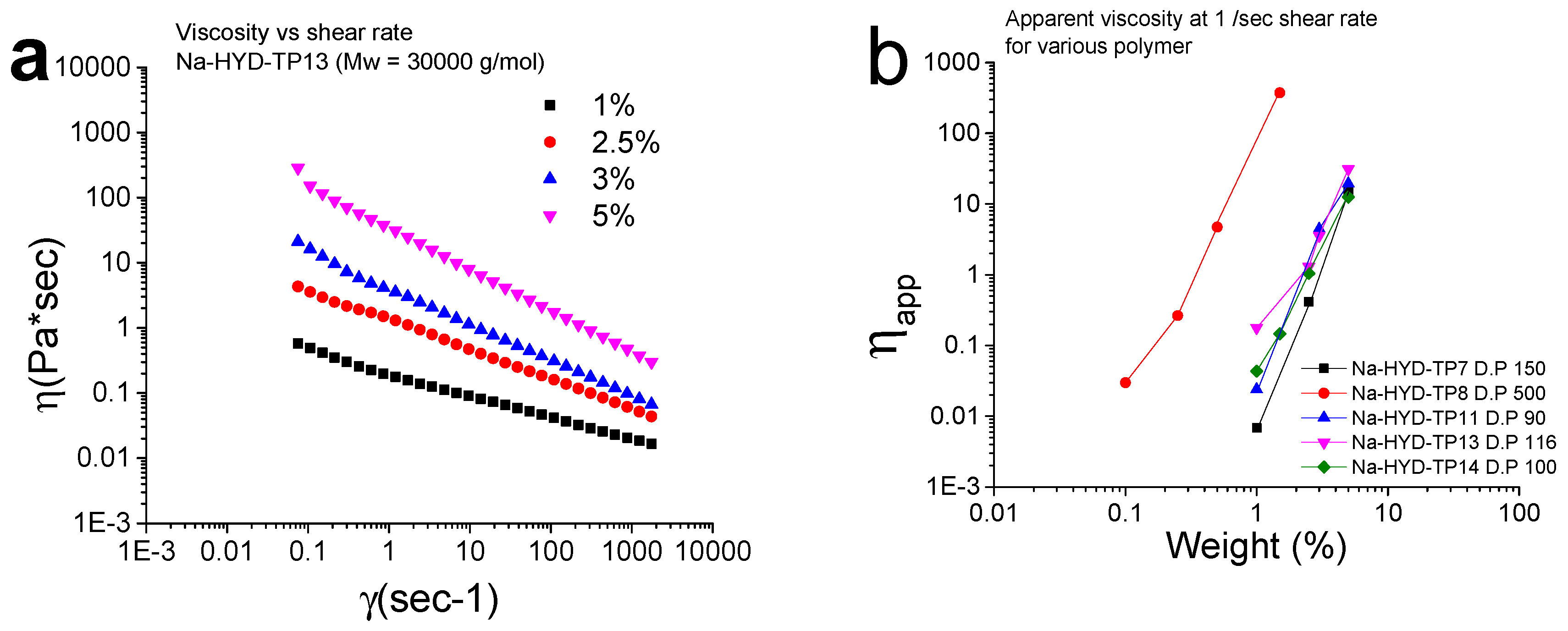
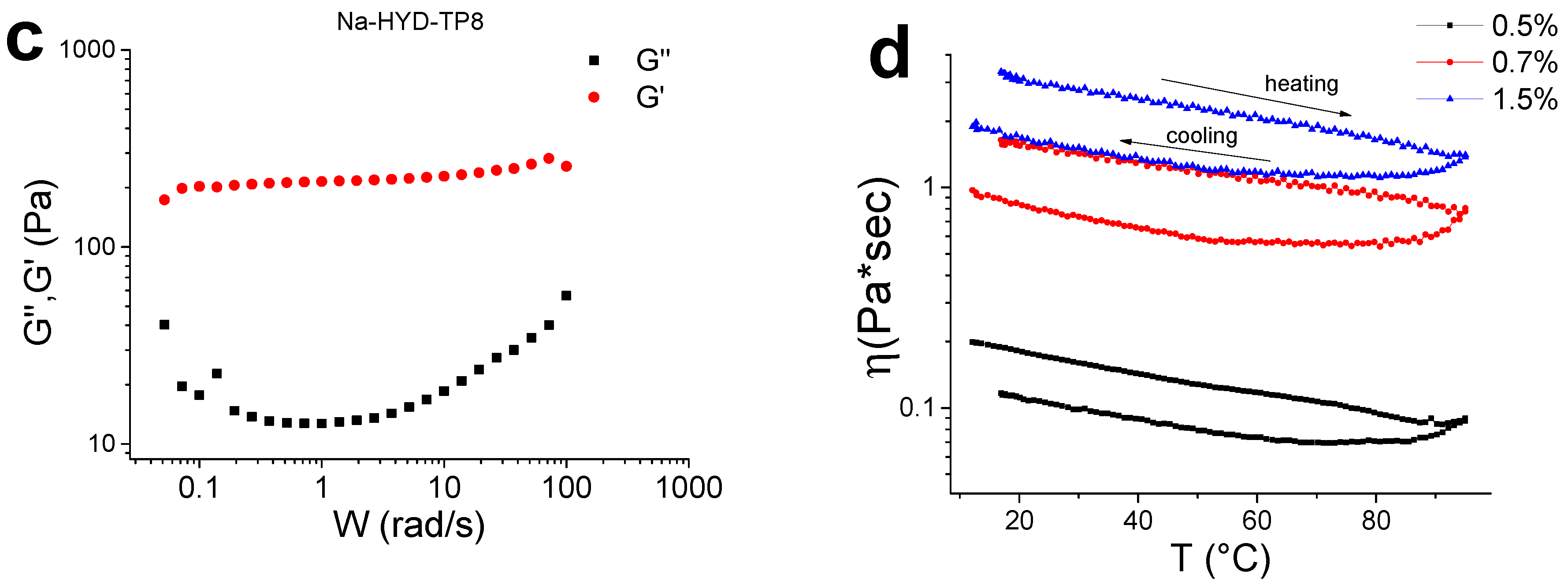
3.9. Rheological Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ganesh, V.A.; Baji, A.; Ramakrishna, S. Smart functional polymers—A new route towards creating a sustainable environment. RSC Adv. 2014, 4, 53352–53364. [Google Scholar] [CrossRef]
- Mane, S. Functional Polymers: A Review. Can. Chem. Trans. 2016, 4, 316–327. [Google Scholar]
- De las Heras Alarcón, C.; Pennadam, S.; Alexander, C. Stimuli responsive polymers for biomedical applications. Chem. Soc. Rev. 2005, 34, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ko, N.R.; Oh, J.K. Recent advances in stimuli-responsive degradable block copolymer micelles: Synthesis and controlled drug delivery applications. Chem. Commun. 2012, 48, 7542–7552. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.; Huck, W.T.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar] [CrossRef]
- Bae, Y.H.; Okano, T.; Hsu, R.; Kim, S.W. Thermo-sensitive polymers as on-off switches for drug release. Die Makromol. Chemie Rapid Commun. 1987, 8, 481–485. [Google Scholar] [CrossRef]
- Raffa, P.; Wever, D.A.Z.; Picchioni, F.; Broekhuis, A.A. Polymeric surfactants: Synthesis, properties, and links to applications. Chem. Rev. 2015, 115, 8504–8563. [Google Scholar] [CrossRef]
- Grubbs, R.B.; Sun, Z. Shape-changing polymer assemblies. Chem. Soc. Rev. 2013, 42, 7436–7445. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.; Mayadunne, R.T.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition−Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP) Current Status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-polymerization modification reactions of poly(glycidyl methacrylate)s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef]
- Iwakura, Y.; Kurosaki, T.; Ariga, N.; Ito, T. Copolymerization of Methyl Methacrylate with Glycidyl Methacrylate and the Reaction of the Copolymer with Amines. Die Makromol. Chemie 1966, 97, 128–138. [Google Scholar] [CrossRef]
- Höhne, S.; Uhlmann, P. Synthesis of functional block copolymers and terpolymers containing polyglycidyl methacrylate blocks. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 675–684. [Google Scholar] [CrossRef]
- Kalal, J.; Švec, F.; Maroušek, V. Reactions of epoxide groups of glycidyl methacrylate copolymers. J. Polym. Sci. Polym. Symp. 2007, 47, 155–166. [Google Scholar] [CrossRef]
- Durmaz, H.; Dag, A.; Tunca, U.; Hizal, G. Synthesis and characterization of pyrene bearing amphiphilic miktoarm star polymer and its noncovalent interactions with multiwalled carbon nanotubes. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2406–2414. [Google Scholar] [CrossRef]
- Gao, H.; Jones, M.-C.; Tewari, P.; Ranger, M.; Leroux, J.-C. Star-shaped alkylated poly(glycerol methacrylate) reverse micelles: Synthesis and evaluation of their solubilizing properties in dichloromethane. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2425–2435. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Z.; Zhang, J.; Li, Z.; Gao, Y.; Wang, C.; Zhang, H.; Yang, B. Multifunctional nanoparticles/silica microsphere assemblies using polyglycidyl methacrylate shells as supports. J. Colloid Interface Sci. 2009, 339, 83–90. [Google Scholar] [CrossRef]
- Dong, X.; Zheng, Y.; Huang, Y.; Chen, X.; Jing, X. Synthesis and characterization of multifunctional poly(glycidyl methacrylate) microspheres and their use in cell separation. Anal. Biochem. 2010, 405, 207–212. [Google Scholar] [CrossRef]
- Kocak, G.; Solmaz, G.; Dikmen, Z.; Bütün, V. Preparation of Cross-Linked Micelles from Glycidyl Methacrylate Based Block Copolymers and Their Usages as Nanoreactors in the Preparation of Gold Nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 514–526. [Google Scholar] [CrossRef]
- Bains, G.; Patel, A.B.; Narayanaswami, V. Pyrene: A Probe to Study Protein Conformation and Conformational Changes. Molecules 2011, 16, 7909–7935. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, W.; Kovacs, J.Z. A review and analysis of electrical percolation in carbon nanotube polymer composites. Compos. Sci. Technol. 2009, 69, 1486–1498. [Google Scholar] [CrossRef]
- Petrov, P.; Stassin, F.; Pagnoulle, C.; Jérôme, R. Noncovalent functionalization of multi-walled carbon nanotubes by pyrene containing polymers. Chem. Commun. 2003, 0, 2904–2905. [Google Scholar] [CrossRef]
- Meuer, S.; Braun, L.; Schilling, T.; Zentel, R. α-Pyrene polymer functionalized multiwalled carbon nanotubes: Solubility, stability and depletion phenomena. Polymer (Guildf) 2009, 50, 154–160. [Google Scholar] [CrossRef]
- Bahun, G.J.; Wang, C.; Adronov, A. Solubilizing single-walled carbon nanotubes with pyrene-functionalized block copolymers. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 1941–1951. [Google Scholar] [CrossRef]
- Parikh, K.; Cattanach, K.; Rao, R.; Suh, D.-S.; Wu, A.; Manohar, S.K. Flexible vapour sensors using single walled carbon nanotubes. Sensors Actuators B Chem. 2006, 113, 55–63. [Google Scholar] [CrossRef]
- Zhao, J.; Buldum, A.; Han, J.; Lu, J.P. Gas molecule adsorption in carbon nanotubes and nanotube bundles. Nanotechnology 2002, 13, 195. [Google Scholar] [CrossRef]
- Kong, J.; Chapline, M.G.; Dai, H. Functionalized Carbon Nanotubes for Molecular Hydrogen Sensors. Adv. Mater. 2011, 13, 1384–1386. [Google Scholar] [CrossRef]
- Raffa, P.; Brandenburg, P.; Wever, D.A.Z.; Broekhuis, A.A.; Picchioni, F. Polystyrene-poly(sodium methacrylate) amphiphilic block copolymers by ATRP: Effect of structure, pH, and ionic strength on rheology of aqueous solutions. Macromolecules 2013, 46, 7106–7111. [Google Scholar] [CrossRef]
- Raffa, P.; Stuart, M.C.A.; Broekhuis, A.A.; Picchioni, F. The effect of hydrophilic and hydrophobic block length on the rheology of amphiphilic diblock Polystyrene-b-Poly(sodium methacrylate) copolymers prepared by ATRP. J. Colloid Interface Sci. 2014, 428, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Meijerink, M.; van Mastrigt, F.; Franken, L.E.; Stuart, M.C.A.; Picchioni, F.; Raffa, P. Triblock copolymers of styrene and sodium methacrylate as smart materials: Synthesis and rheological characterization. Pure Appl. Chem. 2017, 89, 1641–1658. [Google Scholar] [CrossRef]
- Raffa, P.; Broekhuis, A.A.; Picchioni, F. Polymeric surfactants for enhanced oil recovery: A review. J. Pet. Sci. Eng. 2016, 145, 723–733. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Jakubowski, W. Atom transfer radical polymerization of functional monomers employing Cu-based catalysts at low concentration: Polymerization of glycidyl methacrylate. J. Polym. Sci. Pol. Chem. 2011, 49, 918–925. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer molecular weight analysis by1H NMR spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Wang, T.-L.; Liu, Y.-Z.; Jeng, B.-C.; Cai, Y.-C. The Effect of Initiators and Reaction Conditions on the Polymer Syntheses by Atom Transfer Radical Polymerization. J. Polym. Res. 2005, 12, 67–75. [Google Scholar] [CrossRef]
- Chakraborti, A.K.; Rudrawar, S.; Kondaskar, A. An efficient synthesis of 2-amino alcohols by silica gel catalysed opening of epoxide rings by amines. Org. Biomol. Chem. 2004, 2, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Haldar, S.; Chattopadhyay, A. Organization and dynamics in micellar structural transition monitored by pyrene fluorescence. Biochem. Biophys. Res. Commun. 2009, 390, 728–732. [Google Scholar] [CrossRef]
- Numata, Y.; Nirasawa, T.; Suzuka, I. Excited states of pyrene excimer observed by photodissociation spectroscopy in a supersonic jet. J. Photochem. Photobiol. A Chem. 2010, 209, 27–31. [Google Scholar] [CrossRef]
- Kathiravan, A.; Sundaravel, K.; Jaccob, M.; Dhinagaran, G.; Rameshkumar, A.; Arul Ananth, D.; Sivasudha, T. Pyrene Schiff Base: Aggregation Induced Emission, and Antimicrobial Properties. J. Phys. Chem. B 2014, 118, 13573–13581. [Google Scholar] [CrossRef]
- De Halleux, V.; Mamdouh, W.; De Feyter, S.; De Schryver, F.; Levin, J.; Geerts, Y.H. Emission properties of a highly fluorescent pyrene dye in solution and in the liquid state. J. Photochem. Photobiol. A Chem. 2006, 178, 251–257. [Google Scholar] [CrossRef]
- Bains, G.K.; Kim, S.H.; Sorin, E.J.; Narayanaswami, V. The extent of pyrene excimer fluorescence emission is a reflector of distance and flexibility: Analysis of the segment linking the LDL receptor-binding and tetramerization domains of apolipoprotein E3. Biochemistry 2012, 51, 6207–6219. [Google Scholar] [CrossRef]
- Saltiel, C.; Manickavasagam, S.; Mengüc, M.P.; Andrews, R. Light-scattering and dispersion behavior of multiwalled carbon nanotubes. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2005, 22, 1546–1554. [Google Scholar] [CrossRef]
- Malhofer, A.; Rother, M.; Zakharko, Y.; Graf, A.; Schießl, S.P.; Zaumseil, J. Direct visualization of percolation paths in carbon nanotube/polymer composites. Org. Electron. Phys. Mater. Appl. 2017, 45, 151–158. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Winey, K.I. Polymer nanocomposites containing carbon nanotubes. Macromolecules 2006, 39, 5194–5205. [Google Scholar] [CrossRef]
- Hsu, J.C.; Cao, W.; Yang, F.; Yang, T.J.; Lee, S. Absorption behavior of poly(methyl methacrylate)-multiwalled carbon nanotube composites: Effects of UV irradiation. Phys. Chem. Chem. Phys. 2017, 19, 7359–7369. [Google Scholar] [CrossRef]
- Vayer, M.; Vital, A.; Sinturel, C. New insights into polymer-solvent affinity in thin films. Eur. Polym. J. 2017, 93, 132–139. [Google Scholar] [CrossRef]
- Paoletti, C.; He, M.; Salvo, P.; Melai, B.; Calisi, N.; Mannini, M.; Cortigiani, B.; Bellagambi, F.G.; Swager, T.M.; Di Francesco, F.; et al. Room temperature amine sensors enabled by sidewall functionalization of single-walled carbon nanotubes. RSC Adv. 2018, 8, 5578–5585. [Google Scholar] [CrossRef]
- Grabowska, B.; Holtzer, M. Structural Examination of The Cross-Linking Reaction Mechanism of Polyacrylate Binding Agents. Arch. Metall. Mater. 2009, 54, 427–437. [Google Scholar]
- Kimerling, A.S.; Rochefort, W.E.; Bhatia, S.R. Rheology of Block Polyelectrolyte Solutions and Gels: A Review. Ind. Eng. Chem. Res. 2006, 45, 6885–6889. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [Sty]:[I]:[C]:[L] | Sty (mL) | Solvent (mL) | Mn 1 (g/mol) | Time (h) | Yield (%) | Sty Unit | PDI |
|---|---|---|---|---|---|---|---|---|
| PS1 | 30:1:1:1 | 20 | Bulk | 3700 | 1.5 | 55 | 33 | 1.1 |
| PS2 | 30:1:1:1 | 40 | 20 (toluene) | 2500 | 3 | 45 | 21 | 1.1 |
| Polymer | Molar Ratio 1 | Solvent (Anisole) Volume % | Time (h) | T (°C) | Mn (GPC) g/mol | Mn (NMR) g/mol | PDI 2 | Sty-GMA-TBMA Units |
|---|---|---|---|---|---|---|---|---|
| TP1 | 1:1:1:270:30 | 25 | 18 | 90 | 26,400 | 23,000 | 1.2 | 33-27-239 |
| TP2 | 1:1:1:270:30 | 25 | 4 | 90 | 18,700 | 41,300 | 1.6 | 33-23-155 |
| TP3 | 1:1:1:210:90 | 25 | 2 | 90 | 11,800 | 45,500 | 1.85 | 33-99-197 |
| TP4 | 1:1:1:210:90 | 25 | 1 | 90 | 14,700 | 17,200 | 1.59 | 33-52-45 |
| TP5 | 1:1:1:210:90 | 25 | 0.5 | 90 | 11,300 | 19,650 | 1.47 | 33-21-39 |
| TP6 | 1:1:1:270:30 | 50 | 48 | 30 | 12,900 | 20,200 | 1.36 | 33-109-9 |
| TP7 | 1:1:1:270:30 | 50 | 5 | 60 | 35,100 | 29,700 | 1.09 | 33-33-152 |
| TP8 | 1:1:1:270:30 | 50 | 15 | 60 | 31,900 | 118,000 | 1.13 | 33-57-531 |
| TP9 | 1:1:1:210:90 | 50 | 5 | 60 | 29,600 | 12,400 | 1.25 | 33-37-26 |
| TP10 | 1:1:1:255:45 | 50 | 5 | 60 | 24,400 | 27,150 | 1.1 | 33-23-144 |
| TP11 | 1:1:1:105:45 | 50 | 10 | 60 | 15,400 | 18,700 | 1.67 | 33-31-85 |
| TP12 | 1:1:1:210:90 | 50 | 8 | 60 | 30,700 | 48,900 | 1.15 | 33-133-196 |
| TP13 | 1:1:1:210:90 | 50 | 8 | 60 | 32,500 | 27,450 | 1.12 | 33-63-116 |
| TP14 | 1:1:1:210:90 | 50 | 5 | 60 | 30,900 | 24,500 | 1.14 | 33-49-106 |
| Sample | GMA Molar Amount in the Polymer | AMP: Polymer (Molar Ratio) | SiO2 w/w on Polymer | Reaction Time (h) |
|---|---|---|---|---|
| TP9-PYR(0) | 36% | 1.0 | 0% | 48 1 |
| TP9-PYR (1) | 36% | 2.5 | 0% | 48 1 |
| TP7-PYR (2) | 16% | 1.5 | 10% | 48 |
| TP9-PYR(3) | 39% | 1.5 | 5% | 24 |
| TP11-PYR(4) | 24% | 1.0 | 10% | 24 |
| TP11-PYR(5) | 24% | 1.0 | 7.5% | 48 |
| TP9-PYR(6) | 39% | 1.0 | 7.5% | 48 |
| TP13-PYR(7) | 31% | 1.0 | 7.5% | 48 |
| TP13-PYR(8) | 31% | 1.0 | 7.5% | 48 |
| TP14-PYR(9) | 28% | 1.0 | 5% | 48 |
| Sample | CNTs Feed (%) | Average Residue (%) | CNTs Average Effective Charge (%) |
|---|---|---|---|
| TP9-PYR(6) | 0% | 7.3 | 0 |
| D3 | 3% | 9.8 | 2.4 |
| D6 | 6% | 13.0 | 5.6 |
| D75 | 7.5% | 13.9 | 6.5 |
| D8 | 8% | 14.9 | 7.5 |
| D9 | 9% | 16.0 | 8.5 |
| D10 | 10% | 15.9 | 8.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Sacco, F.; Pucci, A.; Raffa, P. Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials 2019, 9, 458. https://doi.org/10.3390/nano9030458
Di Sacco F, Pucci A, Raffa P. Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials. 2019; 9(3):458. https://doi.org/10.3390/nano9030458
Chicago/Turabian StyleDi Sacco, Federico, Andrea Pucci, and Patrizio Raffa. 2019. "Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis" Nanomaterials 9, no. 3: 458. https://doi.org/10.3390/nano9030458
APA StyleDi Sacco, F., Pucci, A., & Raffa, P. (2019). Versatile Multi-Functional Block Copolymers Made by Atom Transfer Radical Polymerization and Post-Synthetic Modification: Switching from Volatile Organic Compound Sensors to Polymeric Surfactants for Water Rheology Control via Hydrolysis. Nanomaterials, 9(3), 458. https://doi.org/10.3390/nano9030458