Facile Strategy for the Synthesis of Gold@Silica Hybrid Nanoparticles with Controlled Porosity and Janus Morphology



Abstract
1. Introduction
2. Materials and Methods
2.1. Characterisation Techniques
2.2. Synthesis
2.2.1. Synthesis of AuNPs
2.2.2. Synthesis of MCM-41 NPs
2.2.3. Synthesis of Au@SiO2 NPs
3. Results and Discussion
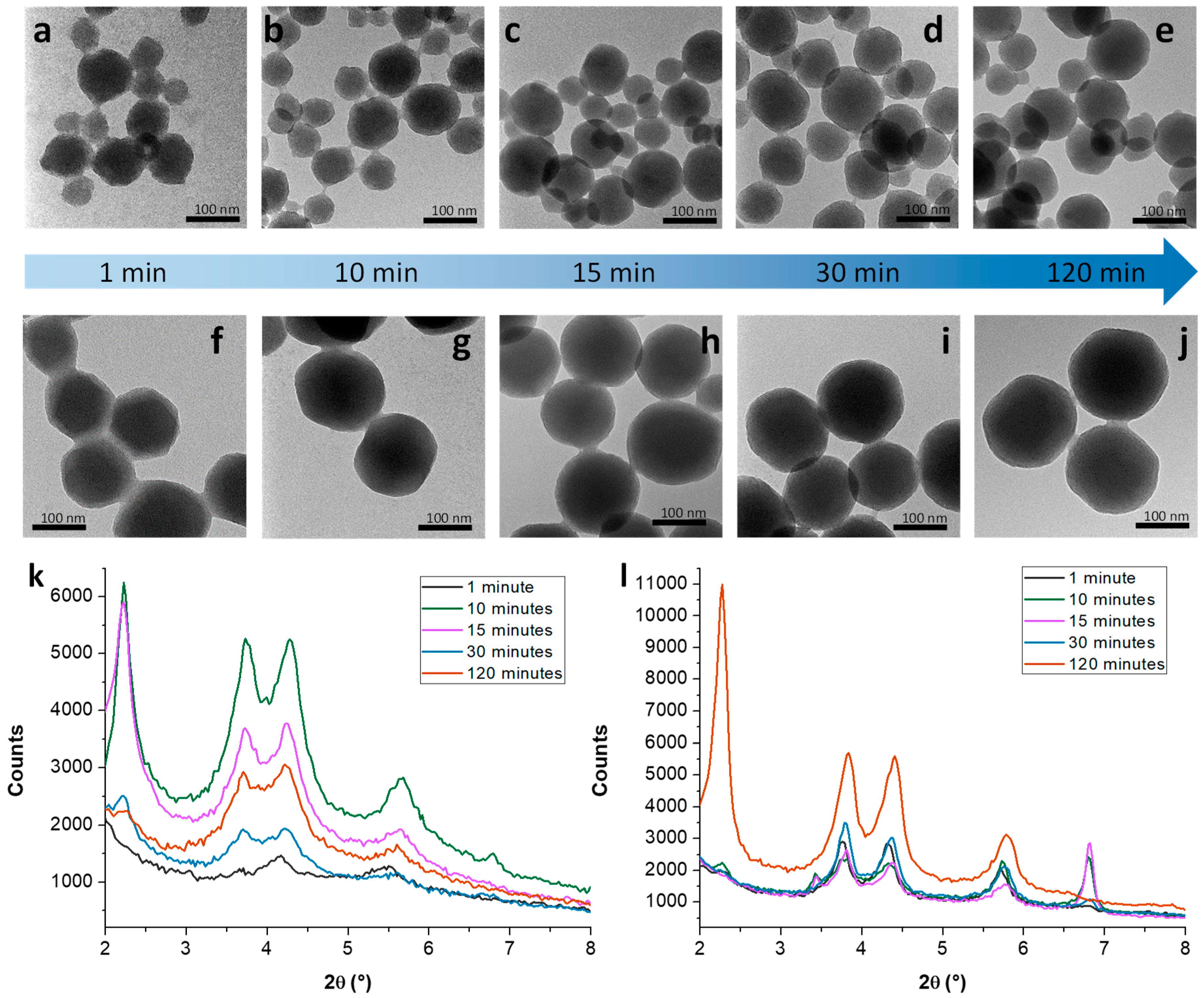
3.1. Mechanistic Studies on MCM-41 NP Growth
3.1.1. Kinetic Study on MCM-41 in the Absence/Presence of Sodium Silicate
3.1.2. Growth Mechanism of MCM-41 NPs in the Absence/Presence of Sodium Silicate
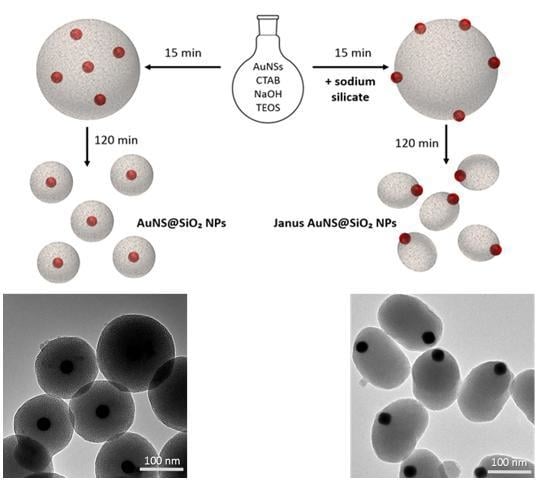
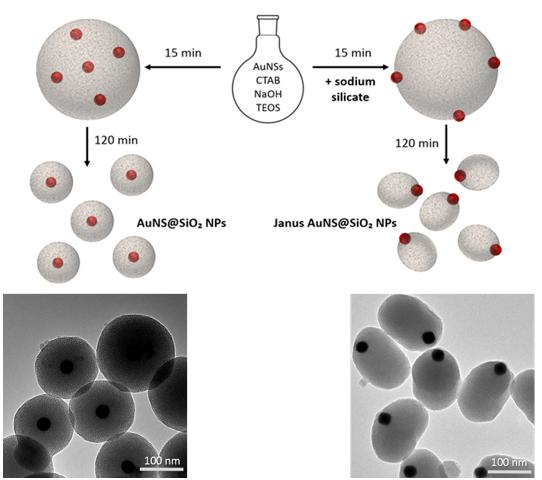
3.2. Incorporation of AuNPs in Mesoporous Silica
3.2.1. Kinetic Study on AuNS@SiO2 NPs in the Absence/Presence of Sodium Silicate
3.2.2. Growth Mechanism of AuNS@SiO2 NPs in the Absence/Presence of Sodium Silicate
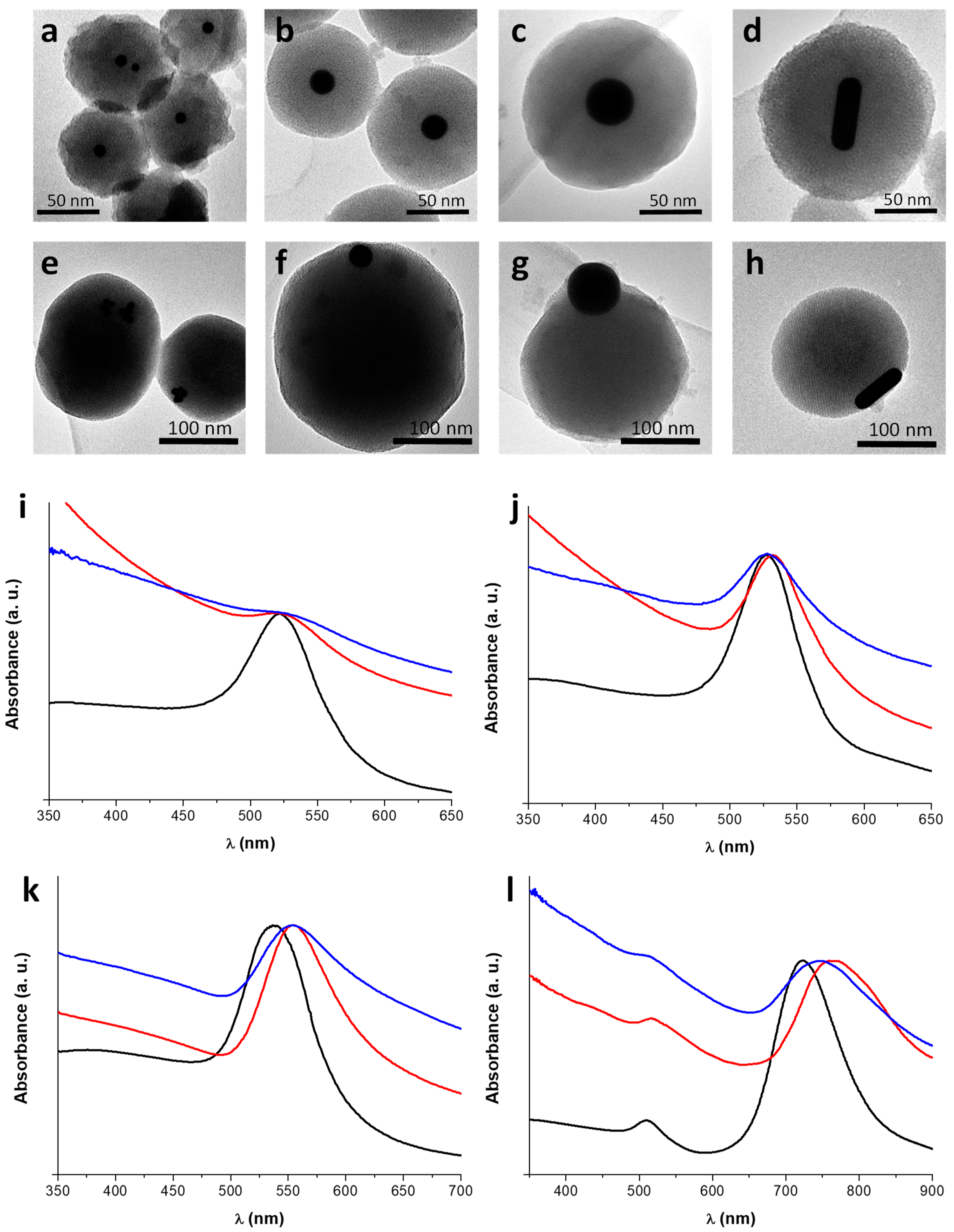
3.3. Incorporation of AuNPs of Different Sizes and Shapes in Mesoporous Silica
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanske, C.; Sanz-Ortiz, M.N.; Liz-Marzán, L.M. Silica-Coated Plasmonic Metal Nanoparticles in Action. Adv. Mater. 2018, 30, 1707003. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Martínez, A.; Pérez-Juste, J.; Liz-Marzán, L.M. Recent Progress on Silica Coating of Nanoparticles and Related Nanomaterials. Adv. Mater. 2010, 30, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Ung, T.; Liz-Marzán, L.M.; Mulvaney, P. Optical Properties of Thin Films of Au@SiO2 Particles. J. Phys. Chem. B 2001, 105, 3441–3452. [Google Scholar] [CrossRef]
- Brandon, M.P.; Ledwith, D.M.; Kelly, J.M. Preparation of saline-stable, silica-coated triangular silver nanoplates of use for optical sensing. J. Colloid Interface Sci. 2014, 415, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Montaño-Priede, J.L.; Coelho, J.P.; Guerrero-Martínez, A.; Peña-Rodríguez, O.; Pal, U. Fabrication of Monodispersed Au@SiO2 Nanoparticles with Highly Stable Silica Layers by Ultrasound-Assisted Stöber Method. J. Phys. Chem. C 2017, 121, 9543–9551. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, R.; Han, L.; Tu, B.; Zhao, D. One-pot synthesis of thermally stable gold@mesoporous silica core-shell nanospheres with catalytic activity. Nano Res. 2013, 6, 871–879. [Google Scholar] [CrossRef]
- Xia, H.X.; Yang, X.Q.; Song, J.T.; Chen, J.; Zhang, M.Z.; Yan, D.M.; Zhang, L.; Qin, M.Y.; Baj, L.Y.; Zhao, Y.D.; et al. Folic acid-conjugated silica-coated gold nanorods and quantum dots for dual-modality CT and fluorescence imaging and photothermal therapy. J. Mater. Chem. B 2014, 2, 1945–1953. [Google Scholar] [CrossRef]
- Botella, P.; Ortega, I.; Quesada, M.; Madrigal, R.F.; Muniesa, C.; Fimia, A.; Fernández, E.; Corma, A. Multifunctional hybrid materials for combined photo and chemotherapy of cancer. Dalton Trans. 2012, 12, 9243–9554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, L.; Wang, J.; Juang, X.; Li, X.; Hu, Z.; Ji, Y.; Wu, X.; Chn, C. Mesoporous Silica-Coated Gold Nanorods as a Light-Mediated Multifunctional Theranostic Platform for Cancer Treatment. Adv. Healthc. Mater. 2012, 24, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Dong, B.; Chen, B.; Wang, J.; Xu, L.; Zhang, S.; Song, H. Multifunctional Au@mSiO2/Rhodamine B Isothiocyanate Nanocomposites: Cell Imaging, Photocontrolled Drug Release, and Photothermal Therapy for Cancer Cells. Small 2013, 9, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Ovejero Paredes, K.; Ruiz-Cabello, J.; Martínez-Ruíz, P.; Pingarrón, J.M.; Villalonga, R.; Filice, M. Hybrid Decorated Core@Shell Janus Nanoparticles as a Flexible Platform for Targeted Multimodal Molecular Bioimaging of Cancer. ACS Appl. Mater. Interfaces 2018, 10, 31032–31043. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Sánchez, L.; Gao, Y.; Yu, Y. Janus Nanoparticles for biological imaging and sensing. Analyst 2016, 141, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pauletti, G.; Wang, Y.; Shi, D. Dual Functionalized Janus Nanocomposites of Polystyrene/Fe3O4@SiO2 for Simultaneous Tumor Cell Targeting and Stimulus-Induced Drug Release. Mater. Res. Soc. Symp. Proc. 2014, 25, 3485–3489. [Google Scholar] [CrossRef]
- Aditya, A.; Chattopadhyay, S.; Gupta, N.; Alam, S.; Palillam-Veedu, A.; Pal, M.; Singh, A.; Santhiya, D.; Ansari, K.M.; Ganguli, M. ZnO Nanoparticles Modified with an Amphopathic Peptide Show Improved Photoprotection in Skin. ACS Appl. Meter. Interfaces 2018. [Google Scholar] [CrossRef]
- Reguera, J.; Jiménez de Aberasturi, D.; Henriksen-Lacey, M.; Langer, J.; Espinosa, A.; Szczupak, B.; Wilhelm, C.; Liz-Marzán, L.M. Janus plasmonic-magnetic gold-iron oxide nanoparticles as contrast agents for multimodal imaging. Nanoscale 2017, 9, 9467–9480. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, J.; Xu, X.; Gao, Y.; Li, H.; Liang, F. Preparation of Janus-type catalysts and their catalytic performance at emulsion interface. J. Colloid Interface Sci. 2017, 490, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Faria, J.; Ruiz, M.P.; Resasco, D.E. Phase-Selective Catalysis in Emulsions Stabilized by Janus Silica-Nanoparticles. Adv. Synth. Catal. 2010, 352, 2359–2364. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, M.A.; Rofríguez-Valverde, M.A.; Cabrerizo-Vilchez, M.A.; Hidalgo-Álvarez, R. Surface activity of Janus particles absorbed at fluid-fluid interfaces: Theoretical and experimental aspects. Adv. Colloid Interface Sci. 2016, 233, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yezer, B.A.; Pieve, D.C.; Behrens, S.H. Janus Particles in a Nonpolar Solvent. Langmuir 2016, 32, 3095–3099. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Zhao, Y.; Zhang, D.; Chien, S.; Lo, Y.H. Patterned Fluorescent Particles as Nanoprobes for the Investigation of Molecular Interactions. Nano Lett. 2003, 3, 995–1000. [Google Scholar] [CrossRef]
- Komazaki, Y.; Hirama, H.; Torii, T. Electrically and magnetically dual-driven Janus particles for handwriting-enabled electronic paper. J. Appl. Phys. 2015, 117, 154506. [Google Scholar] [CrossRef]
- Walther, A.; Müller, A.H.E. Janus particles. Soft Matter 2008, 4, 663–668. [Google Scholar] [CrossRef]
- Bellini, S.; Azzato, G.; Grandinetti, M.; Stellato, V.; De Marco, G.; Sun, Y.; Caravella, A. A Novel Connectivity Factor for Morphological Characterization of Membranes and Porous Media: A Simulation Study on Structures of Mono-Sized Spherical Particles. Appl. Sci. 2018, 8, 573. [Google Scholar] [CrossRef]
- Cavallaro, G.; Milioto, S.; Parisi, F.; Lazzara, G. Halloysite Nanotubes Loaded with Calcium Hydroxide: Alkaline Fillers for the Deacidification of Waterlogged Archeological Woods. ACS Appl. Mater. Interfaces 2018, 10, 27355–27364. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Fernández, D.; Langer, J.; Henriksen-Lacey, M.; Liz-Marzán, L.M. Hybrid Au-SiO2 Core-Satellite Colloids as Switchable SERS Tags. Chem. Mater. 2015, 27, 2540–2545. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, D.; Altantzis, T.; Heidari, H.; Bals, S.; Liz-Marzán, L.M. A protecting group approach toward synthesis of Au-silica Janus nanostars. Chem. Commun. 2014, 50, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ghen, G.; Xing, S.; Wu, T.; Chen, H. Scalable Routes to Janus Au-SiO2 and Ternary Ag-Au-SiO2 Nanoparticles. Chem. Mater. 2010, 22, 3826–3828. [Google Scholar] [CrossRef]
- Park, J.H.; Dumani, D.S.; Arsiwala, A.; Emelianov, S.; Kane, R.S. Tunable aggregation of gold-silica Janus nanoparticles to enable contrast-enhanced multiwavelength photoacoustic imaging in vivo. Nanoscale 2018, 10, 15365–15370. [Google Scholar] [CrossRef] [PubMed]
- Castro, N.; Constantin, D.; Davidson, P.; Abécassis, B. Solution self-assembly of plasmonic Janus nanoparticles. Soft Matter 2016, 12, 9666–9673. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, Q.; Ni, W.; Zhang, N.; Zheng, X.; Wang, Y.; Shao, D.; Tai, G. Cytotoxicity of various types of gold-mesoporous silica nanoparticles in human breast cancer cells. Int. J. Nanomed. 2015, 10, 6075–6087. [Google Scholar] [CrossRef]
- Fang, L.; Wang, W.; Liu, Y.; Xie, Z.; Chen, L. Janus nanostructures formed by mesoporous silica coating Au nanorods for near-infrared chemo-photothermal therapy. J. Mater. Chem. B 2017, 5, 8833–8838. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Lu, M.; Li, L.; Zhang, Y.; Zheng, X.; Shao, D.; Li, J.; Dong, W. Janus Au-mesoporous silica nanocarriers for chemo-photothermal treatment of liver cancer cells. RSC Adv. 2016, 6, 44498–44505. [Google Scholar] [CrossRef]
- Wang, Y.S.; Shao, D.; Zhang, L.; Zhang, X.L.; Jing, L.; Feng, J.; Xia, H.; Huo, Q.H.; Dong, W.F.; Sun, H.B. Gold nanorods-silica Janus nanoparticles for theranostics. Appl. Phys. Lett. 2015, 106, 173705. [Google Scholar] [CrossRef]
- Llopis-Llorente, A.; Díez, P.; de la Torre, C.; Sánchez, A.; Sancenón, F.; Aznar, E.; Marcos, M.D.; Martínez-Ruíz, P.; Martínez-Máñez, R.; Villalonga, R. Enzyme-Controlled Nanodevice for Acetylcholine-Triggered Cargo Delivery Based on Janus Au-Mesoporous Silica Nanoparticles. Chemistry 2017, 23, 4276–4281. [Google Scholar] [CrossRef] [PubMed]
- Pothorszky, S.; Zámbó, D.; Deák, A. Structural and Optical Properties of Gold/Silica “Mushroom” Particles Prepared by Interfacial Templating. Part. Part. Syst. Charact. 2016, 34, 1600291. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, F.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Kerfe, C.T.; Roth, W.J. The discovery of mesoporous molecular sieves from the twenty year perspective. Chem. Soc. Rev. 2013, 42, 3663–3670. [Google Scholar] [CrossRef]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous Silica Nanoparticles: A comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.; Jarman, B.P.; Caplan, C.; Cooper, S.J.; Riggs, H.J.; Martinelli, J.; Djanashvili, K.; La Mazza, E.; Puntoriero, F. Light-harvesting Antennae using the Host-Chemistry of Mesoporous Organosilica. ChemPhotoChem 2017, 2, 196–206. [Google Scholar] [CrossRef]
- Martín-Aranda, R.M.; Čejka, J. Recent Advances in Catalysis Over Mesoporous Molecular Sieves. Top Catal. 2010, 53, 141. [Google Scholar] [CrossRef]
- Chen, C.Y.; Li, H.X.; Davis, M.E. Studies on mesoporous materials: I. Synthesis and Characterization of MCM-41. Microporous Mater. 1993, 2, 17–26. [Google Scholar] [CrossRef]
- Schmidt, R.; Akporiayea, D.; Stcker, M.; Ellestad, O.H. Synthesis of Al-Containing MCM-41 Materials: Template Interaction and Removal. Stud. Surf. Sci. Catal. 1994, 84, 61–68. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Kennedy, G.J.; Krese, C.T.; Roth, W.J.; Schramm, S.E. Molecular or Supramolecular Templating: Defining the Role of Surfactant Chemistry in the Formation of Microporous and Mesoporous Molecular Sieves. Chem. Mater. 1994, 6, 1816–1821. [Google Scholar] [CrossRef]
- Schmidt, R.; Stöcker, M.; Hansen, E.; Akporiaye, D.; Ellestad, O.H. MCM-41: A model system for adsorption studies on mesoporous materials. Microporous Mater. 1995, 3, 443–448. [Google Scholar] [CrossRef]
- Chenite, A.; Le Page, Y.; Sayari, A. Direct TEM Imaging of Tubules in Calcined MCM-41 Type Mesoporous Materials. Chem. Mater. 1995, 7, 1015–1019. [Google Scholar] [CrossRef]
- Das, D.; Tsai, C.M.; Cheng, S. Improvement of hydrothermal stability of MCM-41 mesoporous molecular sieve. Chem. Commun. 1999, 5, 473–474. [Google Scholar] [CrossRef]
- Vartuli, J.C.; Schmitt, K.D.; Kresge, C.T.; Roth, W.J.; Leonowicz, M.E.; McCullen, S.B.; Hellring, S.D.; Beck, J.S.; Schlenker, J.L. Effect of Surfactant/Silica Molar Ratios on the Formation of Mesoporous Molecular Sieves: Inorganic Mimicry of Surfactant Liquid-Crystal Phases and Mechanistic Implications. Chem. Mater. 1994, 6, 2317–2326. [Google Scholar] [CrossRef]
- Cai, Q.; Li, W.Y.; Xiao, F.S.; Pang, W.Q.; Chen, X.H.; Zou, B.S. The preparation of highly ordered MCM-41 with extremely low surfactant concentration. Microporous Mesoporous Mater. 1999, 32, 1–15. [Google Scholar] [CrossRef]
- Lindén, M.; Schunk, S.A.; Schüth, F. In Situ X-ray Diffraction Study of the Initial Stages of Formation of MCM-41 in a Tubular Reactor. Angew. Chem. Int. Ed. 1998, 37, 821–823. [Google Scholar] [CrossRef]
- Radu, D.R.; Lai, C.Y.; Huang, J.; Shu, X.; Lin, V.S.-Y. Fine-tuning degree of organic functionalization of mesoporous silica nanosphere materials via an interfacially designed co-condensation method. Chem. Commun. 2005, 0, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Grzelczak, M.; Pérez-Juste, J.; Mulvaney, P.; Liz-Marzán, L.M. Shape control in gold nanoparticle synthesis. Chem. Soc. Rev. 2008, 37, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.S.; Glyde, J.C.; Göltner, C.G. Liquid-crystalline phases as templates for the synthesis of mesoporous silica. Nature 1995, 378, 366–368. [Google Scholar] [CrossRef]
- Ying, J.Y.; Mehnert, C.P.; Wong, M.S. Synthesis and Applications of Supramolecular-Templated Mesoporous Materials. Angew. Chem. Int. Ed. 2004, 38, 56–77. [Google Scholar] [CrossRef]
- Monnier, A.; Schüth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Maxwell, R.S.; Stucky, G.D.; Krishnamurty, M.; Petroff, P.; Firouzi, A.; et al. Cooperative formation of inorganic-organic interfaces in the synthesis of silicate mesostructures. Science 1993, 3, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Michaux, F.; Baccile, N.; Impéror-Clerc, M.; Malfatti, L.; Folliet, N.; Geravis, C.; Manet, S.; Meneau, F.; Pedersen, J.S.; Babonneau, F. In Situ Time-Resolved SAXS Study of the Formation of Mesostructured Organically Modified Silica through Modeling of Micelles Evolution during Surfactant-Templated Self-Assembly. Langmuir 2012, 28, 17477–17493. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Dumée, L.F.; Garvey, C.J.; Feng, C.; She, F.; Rookes, J.E.; Mudie, S.; Cahill, D.; Kong, L.A. New Insight into Growth Mechanism and Kinetics of Mesoporous Silica Nanoparticles by in Situ Small Angle X-ray Scattering. Langmuir 2015, 31, 8478–8487. [Google Scholar] [CrossRef] [PubMed]
- Hanske, C.; González-Rubio, G.; Hamon, C.; Formentín, P.; Modin, E.; Chuvilin, A.; Guerrero-Martínez, A.; Marsal, L.F.; Liz-Marzán, L.M. Large-Scale Plasmonic Pyramidal Supercrystal via Templated Sef-Assembly of Monodisperse Gold Nanospheres. J. Phys. Chem. C 2017, 121, 10899–10906. [Google Scholar] [CrossRef]
- Scarabelli, L.; Grzelczak, M.; Liz-Marzán, L.M. Tuning Gold Nanorod Synthesis through Prereduction with Salicylic Acid. Chem. Mater. 2013, 25, 4232–4238. [Google Scholar] [CrossRef]
- Holt, S.A.; Foran, G.J.; White, J.W. Observation of Hexagonal Crystalline Diffraction from Growing Silicate Films. Langmuir 1999, 15, 2540–2542. [Google Scholar] [CrossRef]
- Zhao, Y.; Trewying, B.G.; Slowing, I.I.; Lin, V.S.-Y. Mesoporous Silica Nanoparticle-Based Double Drug Delivery System for Glucose-Responsive Controlled Release of Insulin and Cyclic AMP. J. Am. Chem. Soc. 2009, 131, 8398–8400. [Google Scholar] [CrossRef] [PubMed]
- Gayam, S.R.; Wu, S.-P. Redox responsive Pd(II) template rotaxane nanovalve capped mesoporous silica nanoparticles: A folic acid mediated biocompatible cancer-targeted drug delivery system. J. Mater. Chem. B 2014, 2, 7009–7016. [Google Scholar] [CrossRef]
- Du, X.; He, J. Elaborate control over the morphology and structure of mercapto-functionalized mesoporous silicas as multipurpose carriers. Dalton Trans. 2010, 39, 9063–9072. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hu, T.; Zhang, X.; Huo, K.; Chu, P.K.; He, J. One-Step Synthesis of Monodisperse and Hierarchically Mesostructured Silica Particles with a Thin Shell. Langmuir 2010, 26, 13556–13563. [Google Scholar] [CrossRef] [PubMed]
- Szajna-Fuller, E.; Huang, Y.; Rapp, J.L.; Chaka, G.; Lin, V.S.-Y.; Pruski, M.; Bakac, A. Kinetics of oxidation of an organic amine with a Cr(V) salen complex in homogeneous aqueous solution and on the surface of mesoporous silica. Dalton Trans. 2009, 0, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.F. Purification of Caustic Soda and Production of Caustic Compound. United. States Patent 2418372, 1 April 1947. [Google Scholar]
- Gianotti, E.; Bertolino, C.A.; Benzi, C.; Nicotra, G.; Caputo, G.; Castino, R.; Isidoro, C.; Coluccia, S. Photoactive Hybrid Nanomaterials: Indocyanine Immobilized in Mesoporous MCM-41 for “In-Cell” Bioimaging. Appl. Mater. Interfaces 2009, 1, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, A.; Hijazi, A.K.; Al Hmaideen, A.I.; Kühn, F.E. Grafting of [Cu(NCCH3)6][B{C6H3(m-CF3)2}4]2 on the surface of aminosilane modified SBA-14. Microporous Mesoporous Mater. 2006, 96, 293–300. [Google Scholar] [CrossRef]
- Ogawa, M. Formation of Novel Oriented Transparent Films of Layered Silica-Surfactant Nanocomposites. J. Am. Chem. Soc. 1994, 116, 7941–7942. [Google Scholar] [CrossRef]
- Lofgren, J.E.; Ozin, G.A. Controlling morphology and porosity to improve performance of molecularly imprinted sol-gel silica. Chem. Soc. Rev. 2014, 43, 911–933. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Fukushima, Y. Particle size control of mono-dispersed super-microporous silica spheres. J. Mater. Chem. 2003, 13, 2577–2581. [Google Scholar] [CrossRef]
- Meena, S.K.; Sulpizi, M. From Gold Nanoseed to Nanorods: The microscopic Origin of the Anisotropic Growth. Angew. Chem. Int. Ed. 2016, 55, 11960–11964. [Google Scholar] [CrossRef] [PubMed]
- González-Rubio, G.; Díaz-Núñez, P.; Rivera, A.; Prada, A.; Tardajos, G.; González-Izquierdo, J.; Bañares, L.; Llombart, P.; Macdowell, L.G.; Alcolea-Palafox, M.; et al. Femtosecond laser reshaping yields gold nanorods with ultranarrow surface plasmon resonances. Science 2017, 358, 640–644. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sodium Silicate | [Au0], mol L−1/10−4 (AuNP Size) |
|---|---|---|
| System A | NO | - |
| System B | YES | - |
| System C | NO | 7.6 (30 nm) |
| System D | YES | 7.6 (30 nm) |
| AuNS@SiO2 NPs (10 nm) | NO | 1.6 (10 nm) |
| Janus AuNS@SiO2 NPs (10 nm) | YES | 1.6 (10 nm) |
| AuNS@SiO2 NPs (30 nm) | NO | 7.6 (30 nm) |
| Janus AuNS@SiO2 NPs (30 nm) | YES | 7.6 (30 nm) |
| AuNS@SiO2 NPs (68 nm) | NO | 55.2 (68 nm) |
| Janus AuNS@SiO2 NPs (68 nm) | YES | 55.2 (68 nm) |
| AuNR@SiO2 NPs | NO | 7.9 (55 × 19 nm) |
| Janus AuNR@SiO2 NPs | YES | 7.9 (55 × 19 nm) |
| 1 min | 10 min | 15 min | 30 min | 120 min | |
|---|---|---|---|---|---|
| System A | 73 ± 20 | 76 ± 22 | 84 ± 25 | 81 ± 20 | 81 ± 21 |
| System B | 127 ± 31 | 148 ± 26 | 145 ± 28 | 159 ± 22 | 157 ± 17 |
| Sample | Diameter (nm) | LSPR Wavelength (nm) | AuNPs LSPR Wavelength (nm) | LSPR Shift (nm) |
|---|---|---|---|---|
| AuNS@SiO2 NPs (10 nm) | 99 ± 11 | 523 | 522 | 1 |
| Janus AuNS@SiO2 NPs (10 nm) | 203 ± 51 | 523 | 522 | 1 |
| AuNS@SiO2 NPs (30 nm) | 153 ± 15 | 531 | 527 | 4 |
| Janus AuNS@SiO2 NPs (30 nm) | 215 ± 34 | 528 | 527 | 1 |
| AuNS@SiO2 NPs (68 nm) | 207 ± 15 | 554 | 537 | 17 |
| Janus AuNS@SiO2 NPs (68 nm) | 221 ± 39 | 554 | 537 | 17 |
| AuNR@SiO2 NPs | 145 ± 10 | 745 | (509), 722 | 23 |
| Janus AuNR@SiO2 NPs | 231 ± 35 | 765 | (509), 722 | 43 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana Vega, M.; Guerrero Martínez, A.; Cucinotta, F. Facile Strategy for the Synthesis of Gold@Silica Hybrid Nanoparticles with Controlled Porosity and Janus Morphology. Nanomaterials 2019, 9, 348. https://doi.org/10.3390/nano9030348
Santana Vega M, Guerrero Martínez A, Cucinotta F. Facile Strategy for the Synthesis of Gold@Silica Hybrid Nanoparticles with Controlled Porosity and Janus Morphology. Nanomaterials. 2019; 9(3):348. https://doi.org/10.3390/nano9030348
Chicago/Turabian StyleSantana Vega, Marina, Andrés Guerrero Martínez, and Fabio Cucinotta. 2019. "Facile Strategy for the Synthesis of Gold@Silica Hybrid Nanoparticles with Controlled Porosity and Janus Morphology" Nanomaterials 9, no. 3: 348. https://doi.org/10.3390/nano9030348
APA StyleSantana Vega, M., Guerrero Martínez, A., & Cucinotta, F. (2019). Facile Strategy for the Synthesis of Gold@Silica Hybrid Nanoparticles with Controlled Porosity and Janus Morphology. Nanomaterials, 9(3), 348. https://doi.org/10.3390/nano9030348