Development of Magnetically Active Scaffolds for Bone Regeneration

 ,
,
Abstract

1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis of Magnetic Nanoparticles
2.3. Fabrication of PLLA/FeHA Porous Scaffolds
2.4. Cytotoxicity Assay
2.5. In Vitro Degradation
2.6. Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM) Analysis
2.7. X-ray Diffraction Analysis
2.8. Magnetic Analysis
2.9. Differential Scanning Calorimetry (DSC)
2.10. Fourier-Transform Infrared (FTIR)Spectroscopy
3. Results and Discussion
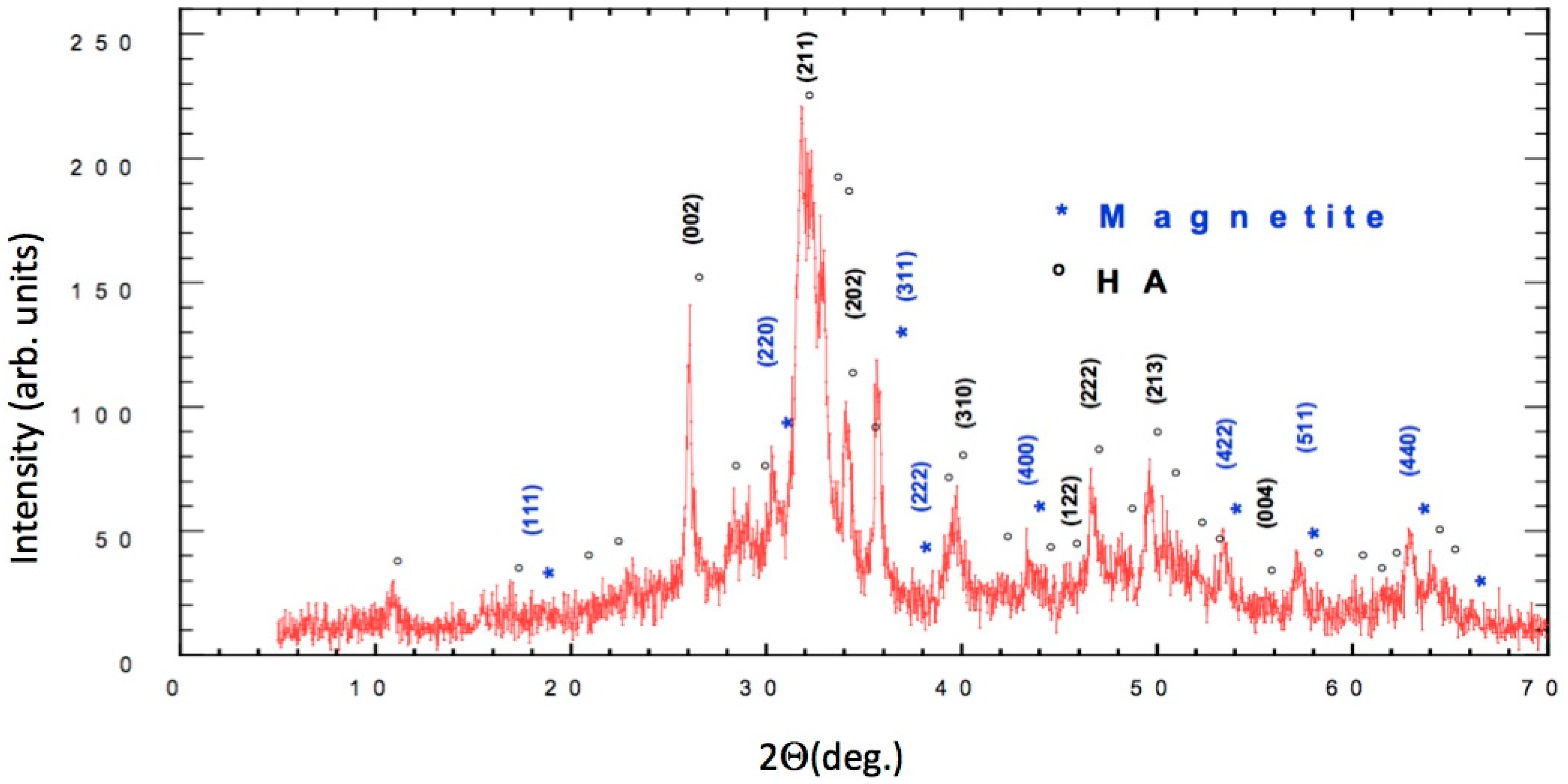
3.1. Characterization of FeHA
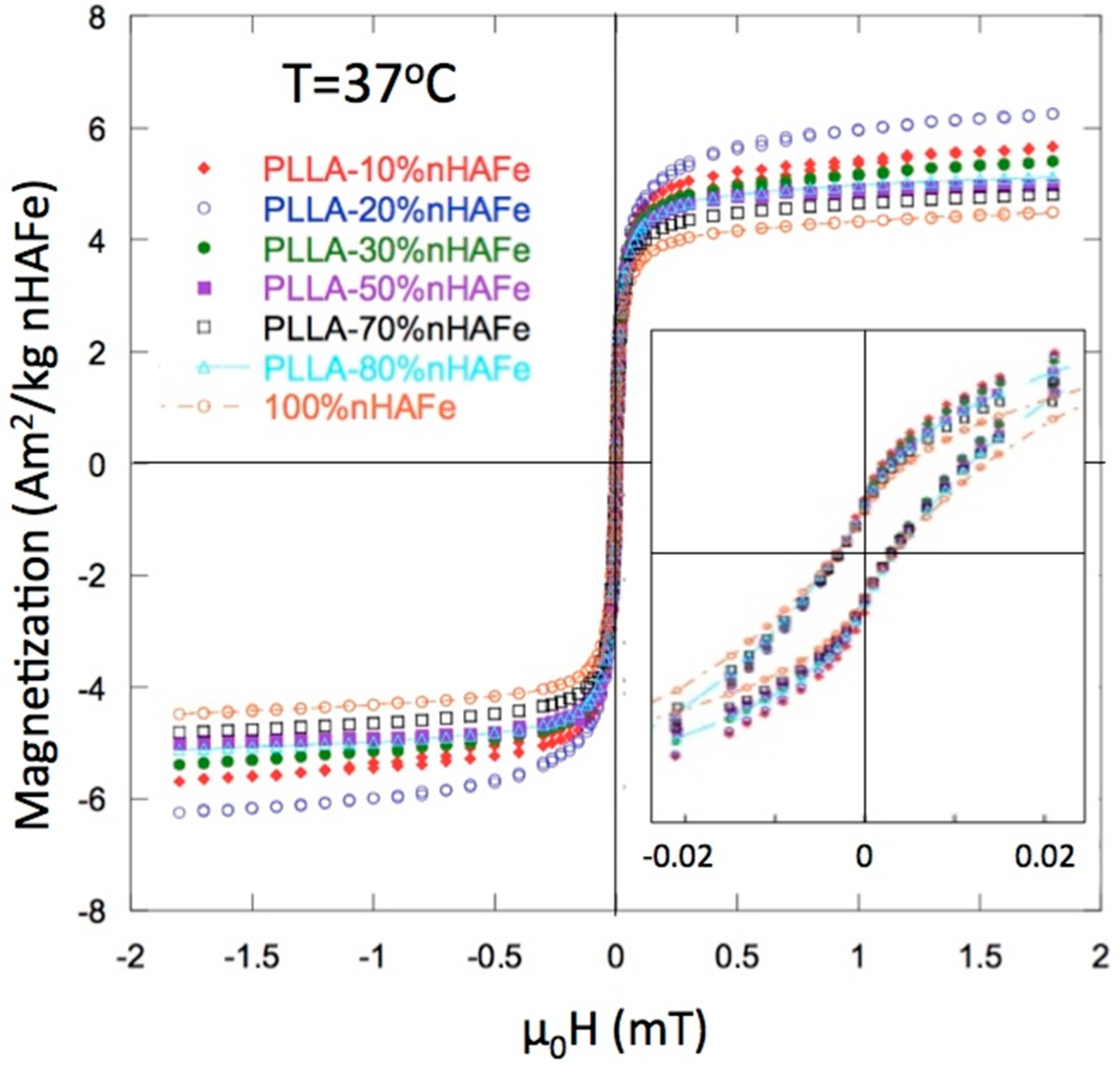
3.2. Magnetic Properties
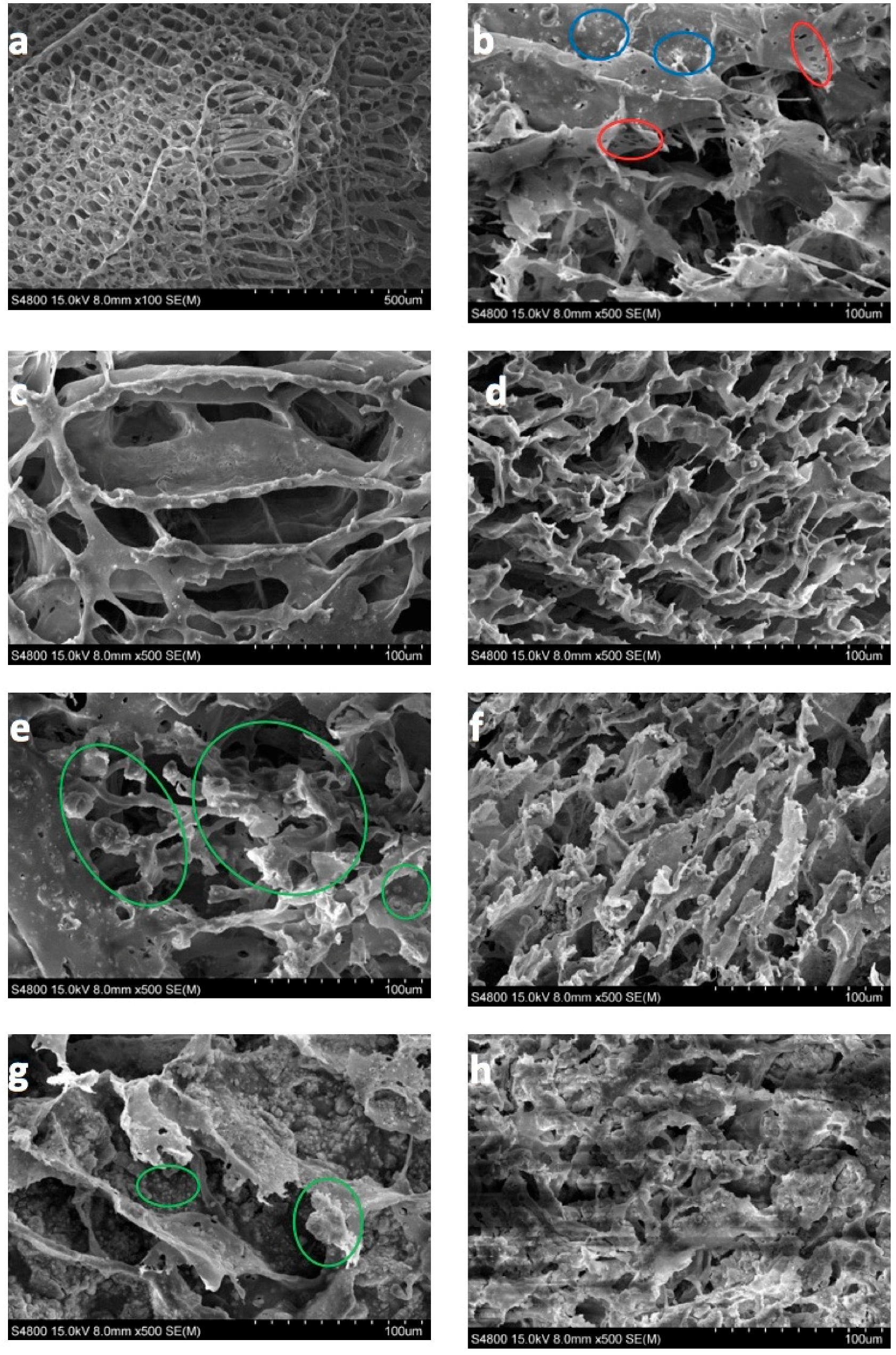
3.3. SEM
3.4. Thermal Analysis
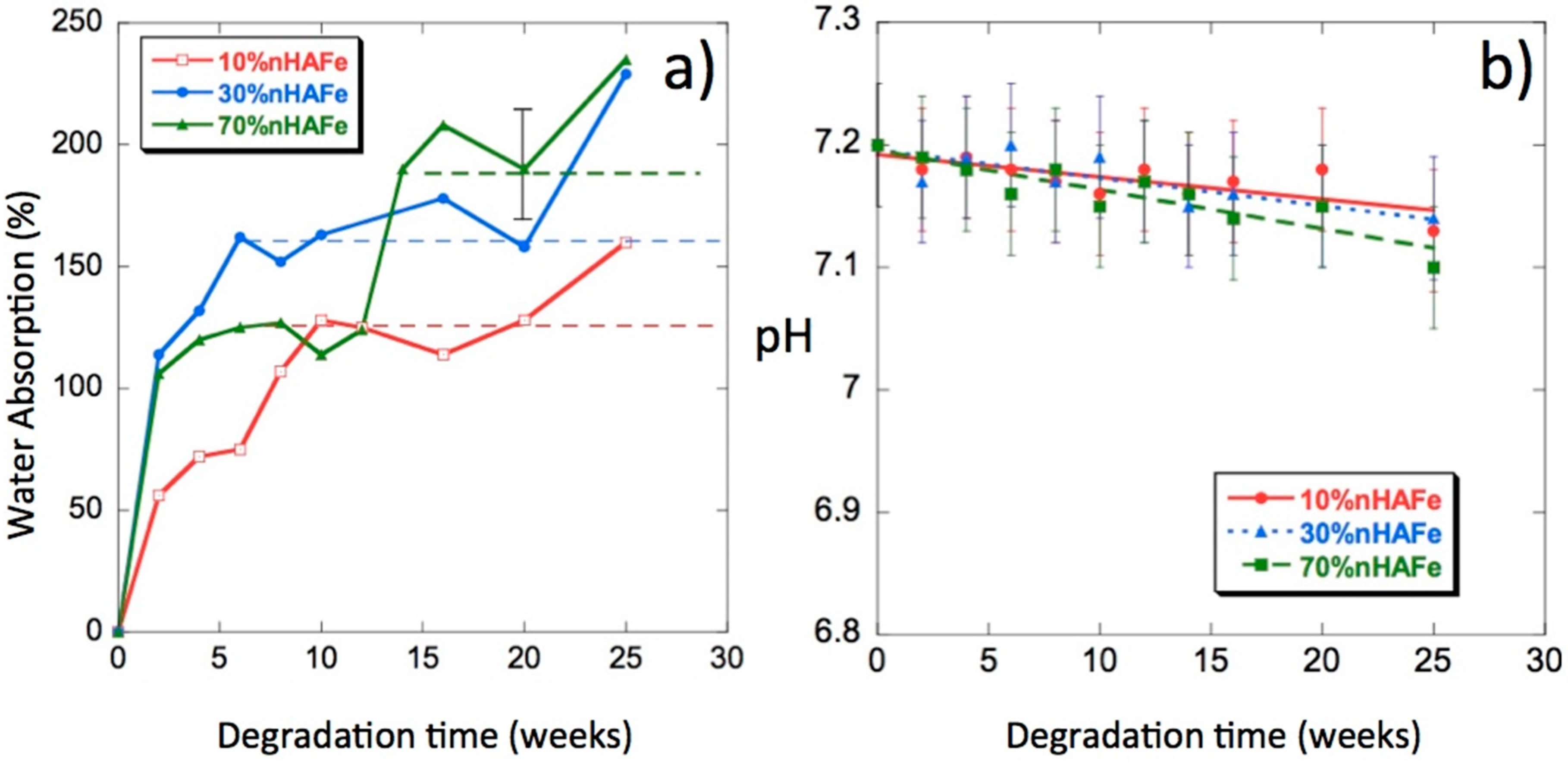
3.5. Water Uptake
3.6. pH
3.7. Mass and Weight Loss
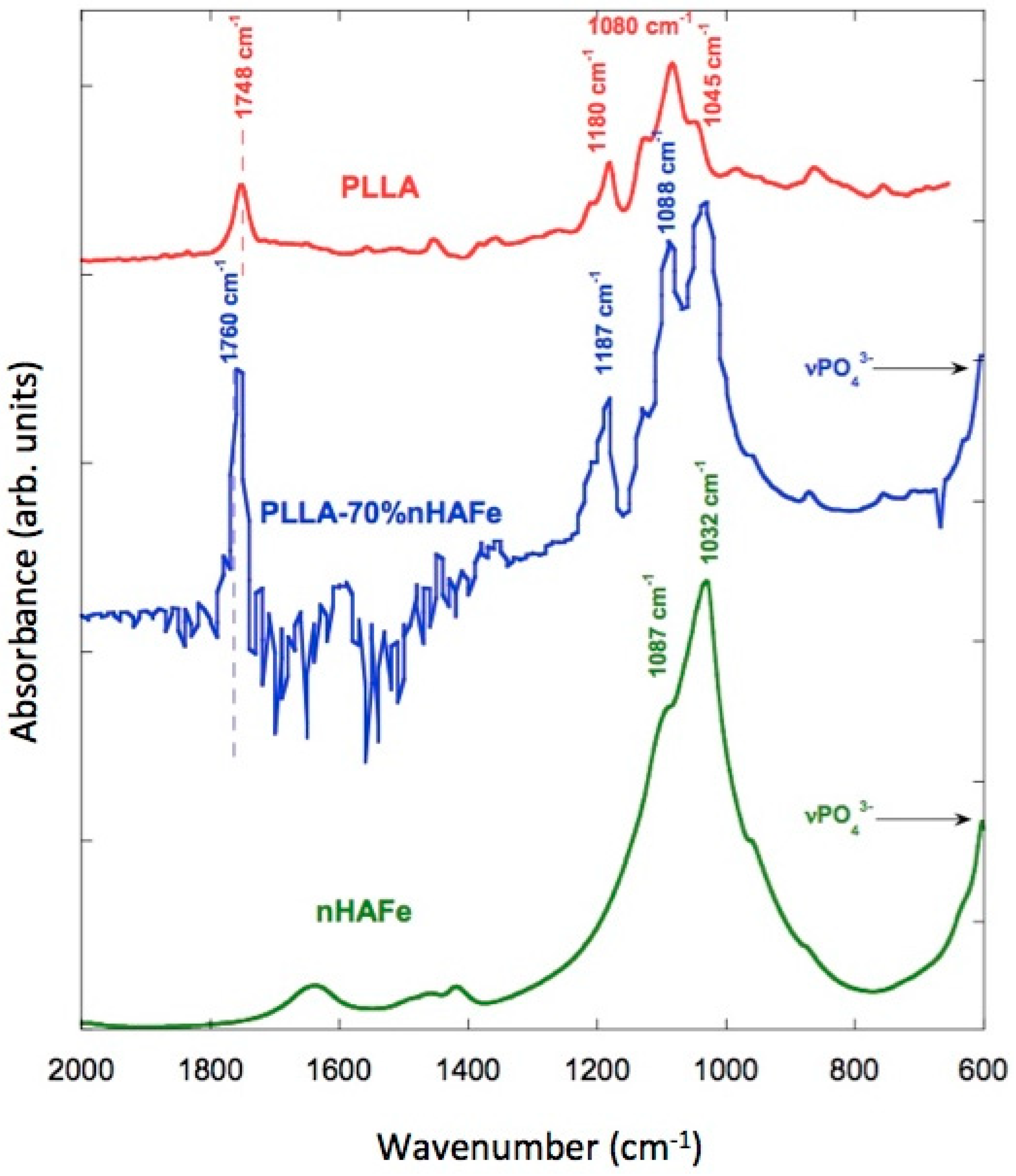
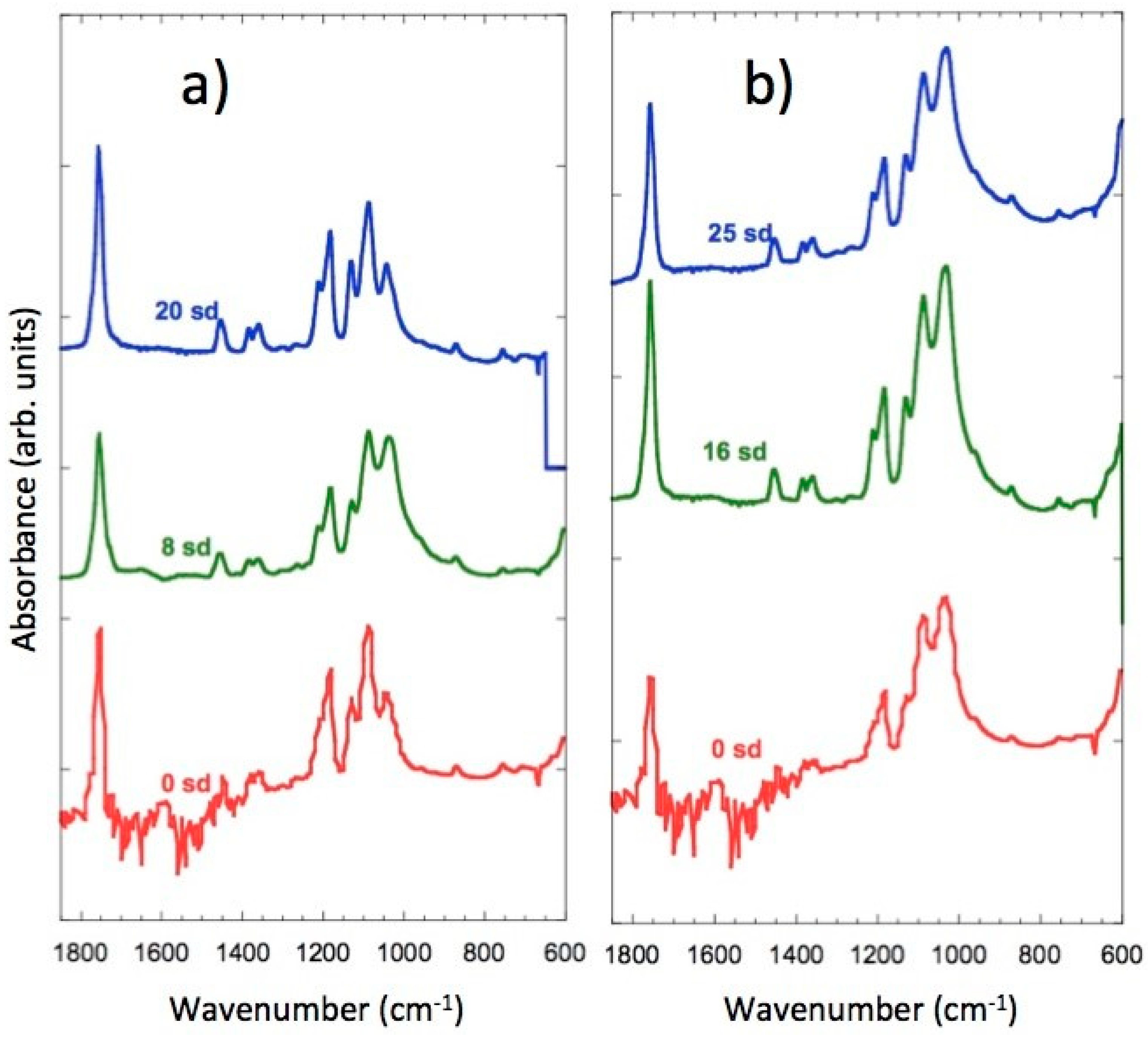
3.8. FTIR
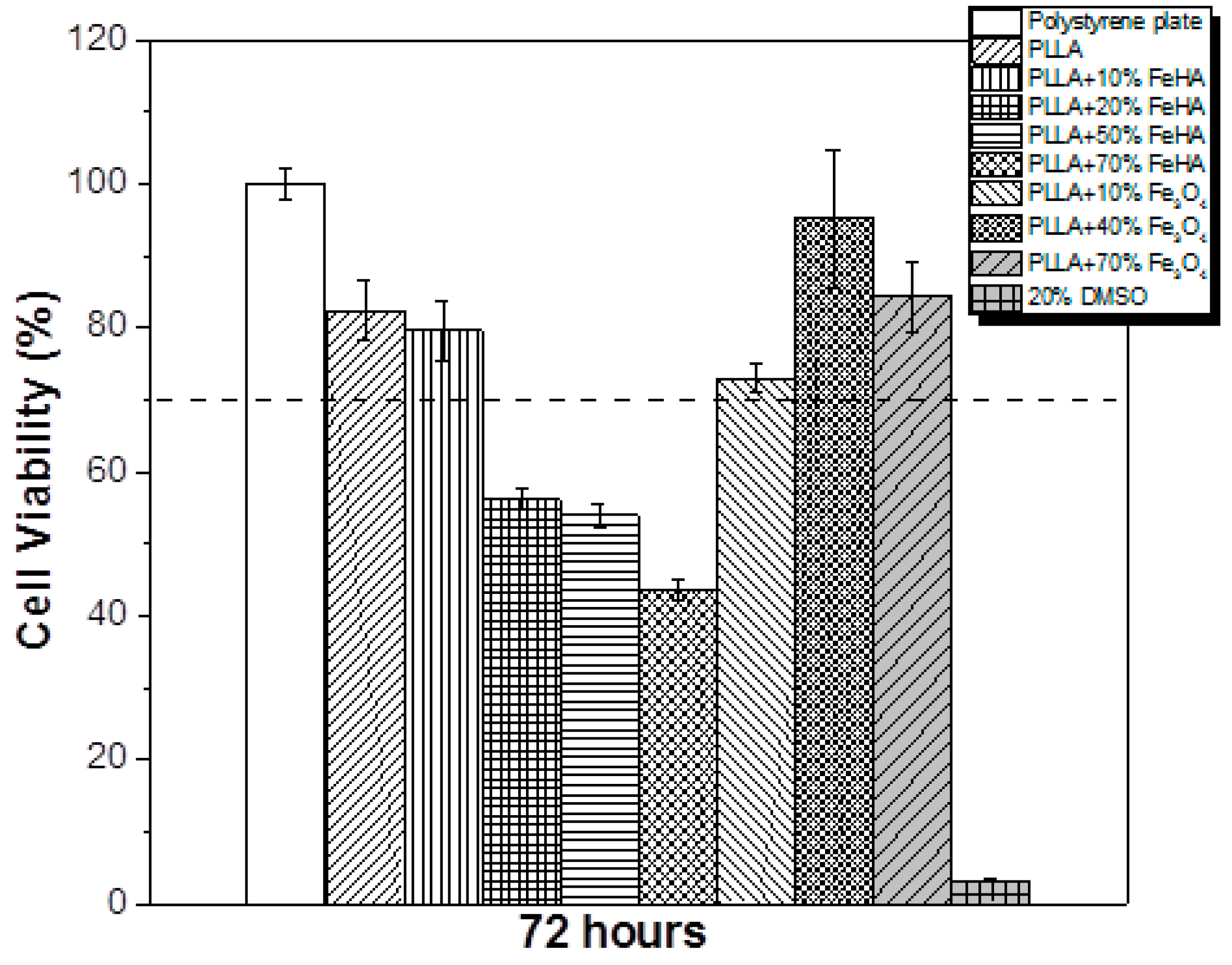
3.9. Cytotoxicity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shalak, R.; Fox, C.F. Preface; Tissue Engineering; Alan R. Liss: New York, NY, USA, 1988; pp. 26–29. [Google Scholar]
- Panseri, S.; Cunha, C.; D’Alessandro, T.; Sandri, M.; Giavaresi, G.; Marcacci, M.; Hung, C.T.; Tampieri, A. Intrinsically superparamagnetic Fe-hydroxyapatite nanoparticles positively influence osteoblast-like cell behavior. J. Nanobiotechnol. 2012, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.F.; Francesko, A.; Ribeiro, C.; Bañobre-López, M.; Martins, P.; Lanceros-Mendez, S. Advances in magnetic nanoparticles for biomedical applications. Adv. Healthc. Mater. 2018, 7, 1700845. [Google Scholar] [CrossRef] [PubMed]
- Mailander, V.; Landfester, K. Interaction of nanoparticles with cells. Biomacromolecules 2009, 10, 2379–2400. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R.; Stark, D.D.; Engelstad, B.L.; Bacon, B.R.; Compton, C.C.; White, D.L.; Jacobs, P.; Lewis, J. Superparamagnetic iron oxide: Pharmacokinetics and toxicity. Am. J. Roentgenol. 1989, 152, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.H.; Hou, S.; Hsueh, Y.; Lin, J.; Wu, H.; Lin, F. The in vivo performance of biomagnetic hydroxyapatite nanoparticles in cancer hyperthermia therapy. Biomaterials 2009, 30, 3956–3960. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Park, S.B.; Yoon, H.; Huh, Y.M.; Haam, S. Preparation of poly E-caprolactone nanoparticles containing magnetite for magnetic drug carrier. Int. J. Pharm. 2006, 324, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Glossop, J.R.; Cartmell, S.H. Tensile strain and magnetic particle force application do not induce MAP3K8 and IL-1Bdifferential gene expression in a similar manner to fluid shear stress in human mesenchymal stem cell. J. Tissue Eng. Regen. Med. 2010, 4, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Panseri, S.; Cunha, C.; D’Alessandro, T.; Sandri, M.; Russo, A.; Giavaresi, G.; Marcacci, M.; Hung, C.T.; Tampieri, A. Magnetic Hydroxyapatite Bone Substitutes to Enhance Tissue Regeneration: Evaluation In Vitro Using Osteoblast-Like Cells and In Vivo in a Bone Defect. PLoS ONE 2012, 7, 38710. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Correia, V.; Martins, P.; Gama, F.M.; Lanceros-Mendez, S. Proving the suitability of magnetoelectric stimuli for tissue engineering applications. Colloids Surf. B Biointerfaces 2016, 140, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Sencadas, V.; Correia, D.M.; Lanceros-Méndez, S. Piezoelectric polymers as biomaterials for tissue engineering applications. Colloids Surf. B Biointerfaces 2015, 136, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Tampieri, A.; Alessandro, T.D.; Sandri, M.; Sprio, S.; Landi, E.; Bertinetti, L.; Panseri, L.; Pepponi, G.; Goettlicher, J.; Bañobre-López, M.; et al. Intrinsic magnetism and hyperthermia in bioactive Fe-doped hydroxyapatite. Acta Biomater. 2012, 8, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Iafisco, M.; Palazzo, B.; Ito, T.; Otsuka, M.; Senna, M.; Delgado-López, J.M.; Gómez-Morales, J.; Tampieri, A.; Prat, M.; Rimondini, L. Preparation of core-shell poly (l-lactic) acidnanocrystalline apatite hollow microspheres for bone repairing applications. J. Mater. Sci. Mater. Med. 2012, 23, 2659–2669. [Google Scholar] [CrossRef] [PubMed]
- Bañobre-López, M.; Piñeiro-redondo, Y.; De Santis, R.; Goria, A.; Ambrosio, L.; Tampieri, A.; Dediu, V.; Rivas, J. Poly (caprolactone) based magnetic scaffolds for bone tissue engineering. J. Appl. Phys. 2011, 109, 07B313. [Google Scholar]
- Díaz, E.; Valle, M.B.; Barandiarán, J.M. Magnetic composite scaffolds of polycaprolactone/nFeHA, for bone-tissue engineering. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 593–600. [Google Scholar] [CrossRef]
- Fisher, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Kolloid-Zeitschrift Zeitschrift Polym. 1973, 251, 980–990. [Google Scholar] [CrossRef]
- Scherrer, P.; der Grosse, B. Der inneren Struktur von Kolloidteilchen mittels Rontgenstrahlen. Nachr. Ges. Wiss. Gott. 2918, 26, 98–102. [Google Scholar]
- Rodriguez-Carvajal, J. An Introduction to the Program FULLPROF, 2000; Laboratoire León Brillouin (CEA-CNRS): Gif sur Yvette, France, 2001. [Google Scholar]
- Swietek, M.; Tokarz, W.; Tarasiuk, J.; Wronski, S.; Blazewicz, A.M. Magnetic Polymer Nanocomposite for Medical Application. Acta Phys. Pol. A 2014, 4, 891–894. [Google Scholar] [CrossRef]
- Tanaka, H.; Araki, T. Phase inversion during viscoelastic phase separation: Roles of bulk and shear relaxation moduli. Phys. Rev. Lett. 1997, 78, 4966–4969. [Google Scholar] [CrossRef]
- Jackson, C.L.; Shaw, M.T. The phase behaviour and gelation of a rod-like polymer in solution and implications for microcellular foam morphology. Polymer 1990, 31, 1070–1083. [Google Scholar] [CrossRef]
- Schugens, C.; Maquet, V.; Grandfils, C.; Jerome, R.; Teyssie, P. Polylactide macroporous biodegradable implants for cell transplantation. II. Preparation of polylactide foams by liquid-liquid phase separation. J. Biomed. Mater. Res. 1996, 30, 449–461. [Google Scholar] [PubMed]
- Tsuji, H.; Ikada, Y. Properties and morphology of poly (l-lactide): Effects of structural parameters on long-term hydrolysis of poly (l-lactide) I phosphate-bufered solutions. Polym. Degrad. Stabil. 2000, 67, 179–189. [Google Scholar] [CrossRef]
- Zhou, Z.; Yi, Q.; Liu, L.; Liu, X.; Liu, Q. Influence of degradation of poly-L-lactide on mass loss, mechanical properties, and crystallinity in phosphate-buffered solution. J. Macromol. Sci. Part B Phys. 2009, 48, 309–317. [Google Scholar] [CrossRef]
- Li, S.M.; Garreau, H.; Vert, M. Structure–property relationships in the case of the degradation of massive aliphatic poly (α-hydroxyacids) in aqueous media, Part 2: Degradation of lactide–glicolide copolymers. J. Mater. Sci. 1990, 1, 123–130. [Google Scholar]
- Leenslag, J.W.; Pennings, A.J.; Bos, R.R.M.; Rozema, F.R.; Boering, G. Resorbable materials of poly (l-lactide) VIII. In vivo and in vitro degradation. Biomaterials 1987, 8, 311–314. [Google Scholar] [CrossRef]
- Deplaine, H.; Acosta-Santamaría, V.A.; Vidaurre, A.; Gómez Ribelles, J.L.; Doblare, M.; Ochoa, I.; Gallego, G. Evolution of the Properties of a Poly (l-lactic acid) Scaffold with Double Porosity During In Vitro Degradation in a Phosphate-Buffered Saline Solution. J. Appl. Polym. Sci. 2014, 131, 40956–40966. [Google Scholar] [CrossRef]
- Taddei, P.; Di Foggia, M.; Causa, F.; Ambrosio, L.; Fagnano, C. In vitro bioactivity of poly (ε-caprolactone)-apatite (PCL-AP) scaffolds for bone tissue engineering: The influence of the PCL/AP ratio. Int. J. Artif. Organs 2006, 29, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Sandonis, I.; Puerto, I.; Ibañez, I. In Vitro Degradation of PLLA/nHA Composite Scaffolds. Polym. Eng. Sci. 2014, 54, 2571–2578. [Google Scholar] [CrossRef]
- Llop, J.; Jiang, P.; Marradi, M.; Gomez-Vallejo, V.; Echeverria, M.; Yu, S.; Puigivila, M.; Baz, Z.; Szczupak, B.; Perez-Campana, C.; et al. Visualisation of dual radiolabeled poly (lactide-co-glycolide) nanoparticle degradation in vivo using energy-discriminant SPECT. J. Mater. Chem. B 2015, 3, 6293–6300. [Google Scholar] [CrossRef]
- Schmitt, E.A.; Flanagan, D.R.; Linhardt, R.J. Importance of distinct water environments in the hydrolysis of poly (d,l-lactide-co-glycolide). Macromolecules 1994, 27, 743–748. [Google Scholar] [CrossRef]
- Azevedo, M.C.; Reis, R.L.; Claase, M.B.; Grijpma, D.W.; Feijen, J. Development and properties of polycaprolactone/hydroxyapatite composite biomaterials. J. Mater. Sci. Mater. Med. 2003, 14, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, F. Algunas aplicaciones del ácido poli-l-láctico. Revista Académica Colombiana de Ciencias Exactas y Físicas 2013, 37, 125–142. [Google Scholar]
- Zhou, S.; Zheng, X.; Yu, X.; Wang, J.; Weng, J.; Li, X.; Feng, B.; Yin, M. Hydrogen Bonding Interaction of Poly (d,l-Lactide)/hydroxyapatite Nanocomposites. Chem. Mater. 2007, 19, 247–253. [Google Scholar] [CrossRef]
- Partini, M.; Pantani, R. P FTIR analysis of hydrolysis in aliphatic polyesters. Polym. Degrad. Stabil. 2007, 92, 1491–1497. [Google Scholar] [CrossRef]
- Areias, A.C.; Ribeiro, C.; Sencadas, V.; Garcia-Giralt, N.; Diez-Perez, A.; Ribellesde, J.L.G.; Lanceros-Méndezet, S. Influence of crystallinity and fiber orientation on hydrophobicity and biological response of poly (l-lactide) electrospun mats. Soft Matter. 2012, 8, 5818–5825. [Google Scholar] [CrossRef]
- Pan, Y.H.; Wang, H.; Wu, T.; Fan, K.; Huang, H.; Chang, W. Fabrication of Fe3O4/PLLA composites for use in bone tissue engineering. Polym. Compos. 2017, 38, 2881–2888. [Google Scholar] [CrossRef]
- Hong, Z.; Zhan, P.; He, C.; Qiu, X.; Liu, A.; Chen, L.; Chen, X.; Jing, X. Nano-composite of poly (l-lactide) and surface grafted hydroxyapatite: Mechanical properties and biocompatibility. Biomaterials 2005, 26, 6296–6304. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % FeHA Nominal | Moment (Am2/kg FeHA) | µ0 Hc (mT) | % FeHA Recalculated |
|---|---|---|---|
| 100 (1) | 4.45 | 3.18 | - |
| 10 | 5.58 | 2.77 | 12.5 |
| 20 | 6.16 | 2.73 | 27.6 |
| 30 | 5.33 | 2.64 | 36 |
| 50 | 4.86 | 2.65 | 54.5 |
| 70 | 4.77 | 2.50 | 74.9 |
| 80 | 4.86 | 2.27 | 87.2 |
| PLLA/FeHA% dt (Week) | First Run | Second Run | Xc% | CF% | |||||
|---|---|---|---|---|---|---|---|---|---|
| Tm (°C) | ΔHm (J/g) | Tcc (°C) | ΔHcc (J/g) | Tg (°C) | Tc (°C) | ΔHc (J/g) | |||
| 0% | 184 | 41.1 | 76 | 4.7 | 56 | 96 | 2.3 | 39 | 6 |
| 10% | 183 | 44.1 | 75.5 | 3.4 | 58.5 | 98 | 9.7 | 44 | 22 |
| 4 dt | 183 | 41.8 | 76.5 | 2.3 | 58 | 97 | 8.8 | 42 | 21 |
| 8 dt | 182 | 43.2 | 75 | 1.9 | 59 | 97 | 6.9 | 44 | 16 |
| 12 dt | 182 | 44 | - | - | 58 | 98 | 6.8 | 47 | 15 |
| 16 dt | 182 | 40.7 | - | - | 57 | 99 | 6.4 | 44 | 16 |
| 20 dt | 182 | 40.1 | 77.5 | 1.5 | 58 | 97 | 5.7 | 42 | 14 |
| 25 dt | 181 | 38 | 77 | 2.7 | 56 | 98 | 5.2 | 38 | 14 |
| 20% | 184 | 36.1 | 76.1 | 4.9 | 60 | 101 | 18.2 | 33.5 | 50 |
| 30% | 183 | 31.7 | - | - | 61 | 103 | 20.4 | 34 | 64 |
| 4 dt | 183 | 24.2 | 77 | 2.5 | 62 | 105 | 19 | 23 | 78 |
| 8 dt | 182 | 28.8 | 77 | 1.43 | 61 | 102 | 17 | 29 | 59 |
| 12 dt | 182 | 29 | 86 | 1.3 | 61 | 103 | 15 | 30 | 52 |
| 16 dt | 181 | 30.3 | - | - | 58 | 100 | 12 | 33 | 40 |
| 20 dt | 181 | 28.6 | 74 | 2.3 | 57 | 98.5 | 10 | 28 | 35 |
| Sample | Degradation Time (Weeks) | Mw | Mn | I |
|---|---|---|---|---|
| PLLA | 0 | 144,221 | 104,042 | 1.386 |
| PLLA/FeHA 10 wt % | 0 | 98,633 | 55,206 | 1.787 |
| 16 | 94,867 | 53,228 | 1.782 | |
| 20 | 92,094 | 53,123 | 1.734 | |
| 25 | 52,704 | 32,946 | 1.600 | |
| PLLA/FeHA 30 wt % | 0 | 66,625 | 47,961 | 1.389 |
| 5 | 55,750 | 30,984 | 1.799 | |
| 16 | 49,382 | 20,093 | 2.458 | |
| 20 | 89,927 | 45,269 | 1.986 | |
| 25 | 49,972 | 36,484 | 1.370 | |
| (*) | 10,668 | 9,873 | 1.081 | |
| PLLA/FeHA 70 wt % | 0 | 81,855 | 44,573 | 1.836 |
| 15 | 98,065 | 45,623 | 2.149 | |
| 25 | 86,760 | 53,289 | 1.628 | |
| (*) | 10,982 | 10,511 | 1.045 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, E.; Valle, M.B.; Ribeiro, S.; Lanceros-Mendez, S.; Barandiarán, J.M. Development of Magnetically Active Scaffolds for Bone Regeneration. Nanomaterials 2018, 8, 678. https://doi.org/10.3390/nano8090678
Díaz E, Valle MB, Ribeiro S, Lanceros-Mendez S, Barandiarán JM. Development of Magnetically Active Scaffolds for Bone Regeneration. Nanomaterials. 2018; 8(9):678. https://doi.org/10.3390/nano8090678
Chicago/Turabian StyleDíaz, Esperanza, Mᵃ Blanca Valle, Sylvie Ribeiro, Senentxu Lanceros-Mendez, and José Manuel Barandiarán. 2018. "Development of Magnetically Active Scaffolds for Bone Regeneration" Nanomaterials 8, no. 9: 678. https://doi.org/10.3390/nano8090678
APA StyleDíaz, E., Valle, M. B., Ribeiro, S., Lanceros-Mendez, S., & Barandiarán, J. M. (2018). Development of Magnetically Active Scaffolds for Bone Regeneration. Nanomaterials, 8(9), 678. https://doi.org/10.3390/nano8090678