Polarization- and Angular-Resolved Optical Response of Molecules on Anisotropic Plasmonic Nanostructures


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Origin of Polarization Characteristics in Spectroscopy
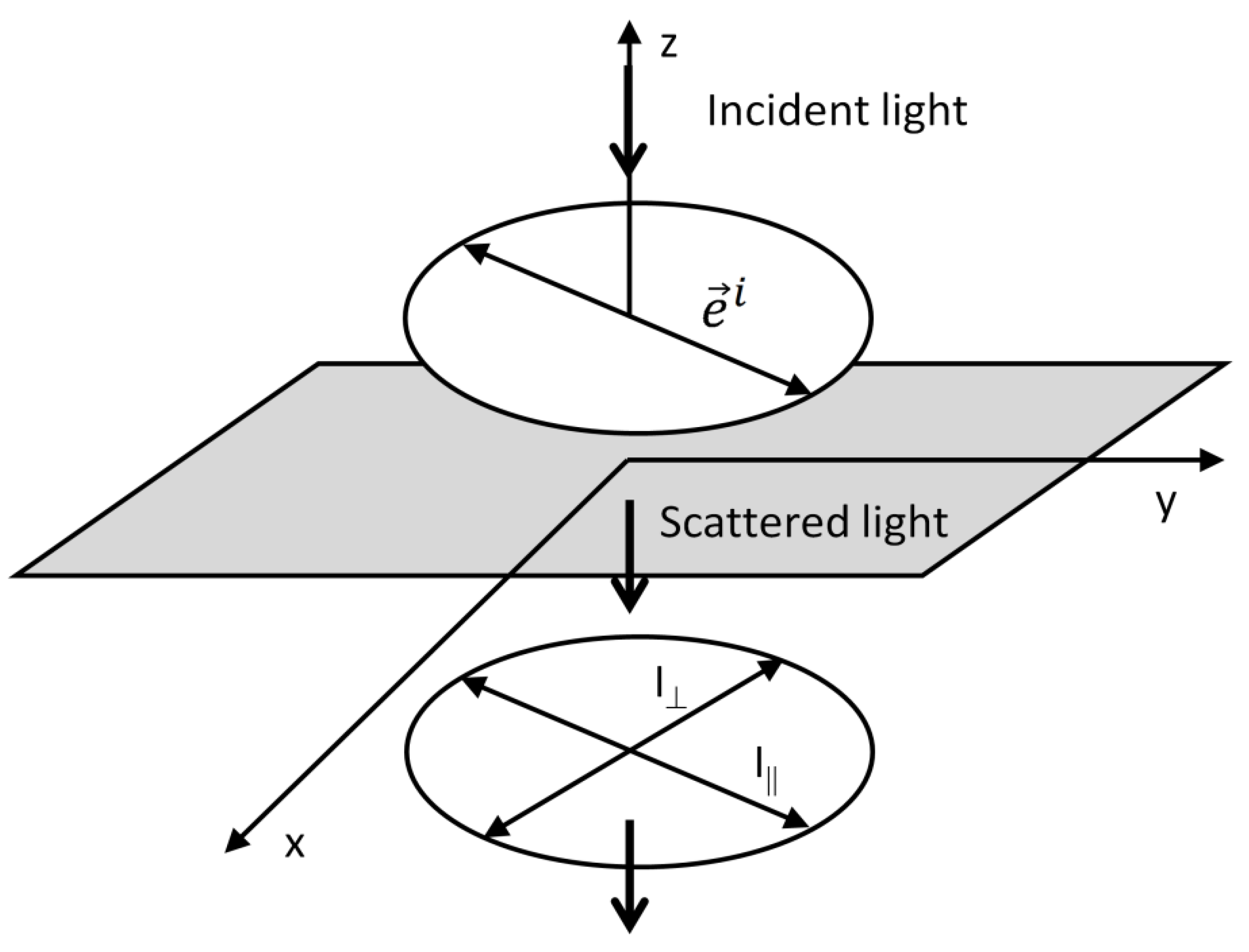
3. Polarization and Angular Dependences in Raman Spectroscopy
3.1. Derivation of Raman Intensities as a Function of the Polarization and Angular Arrangement
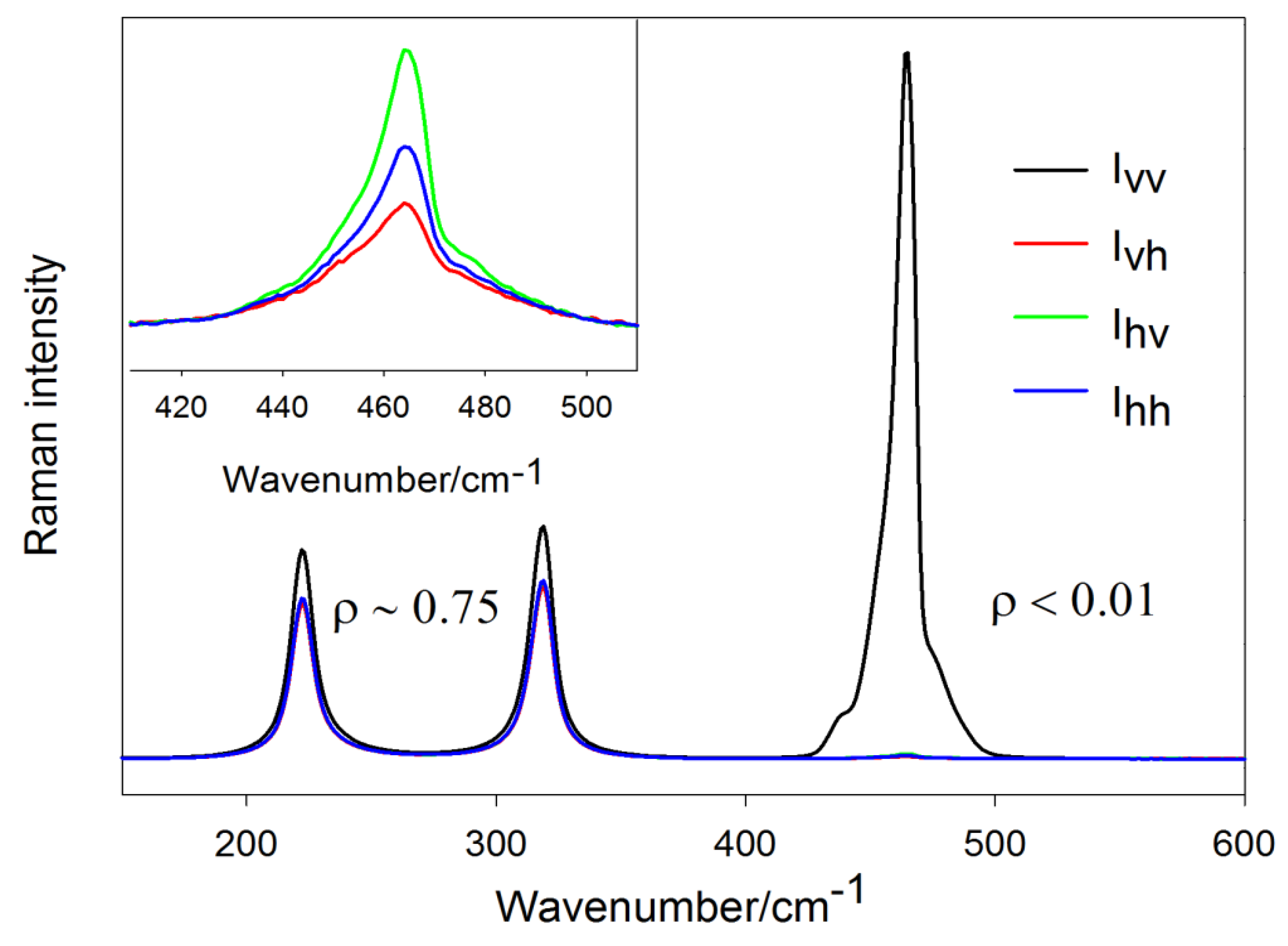
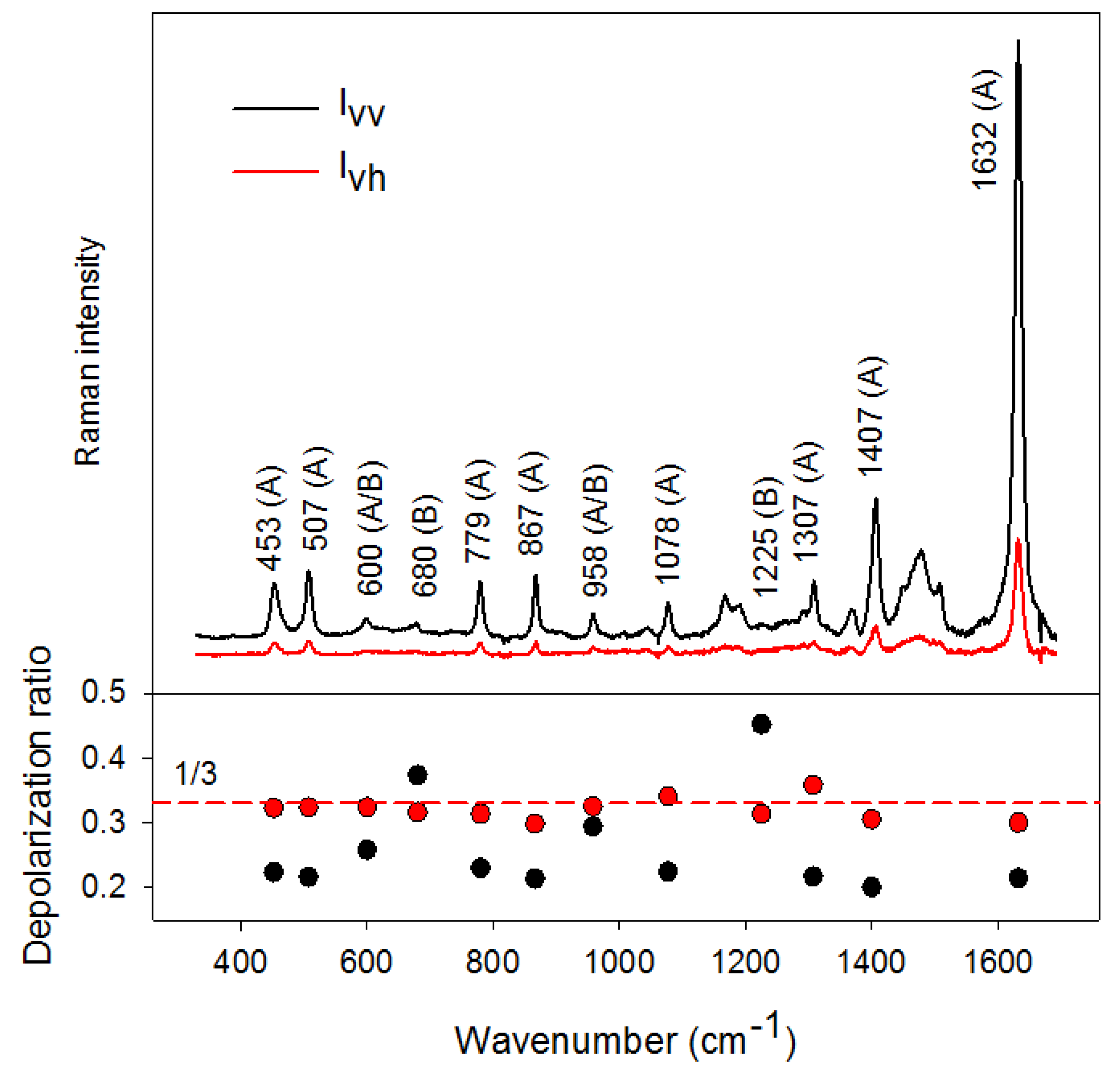
3.2. Simple Illustration of the Depolarization Ratio Measurement
3.3. Other Possible Polarization Arrangements Used
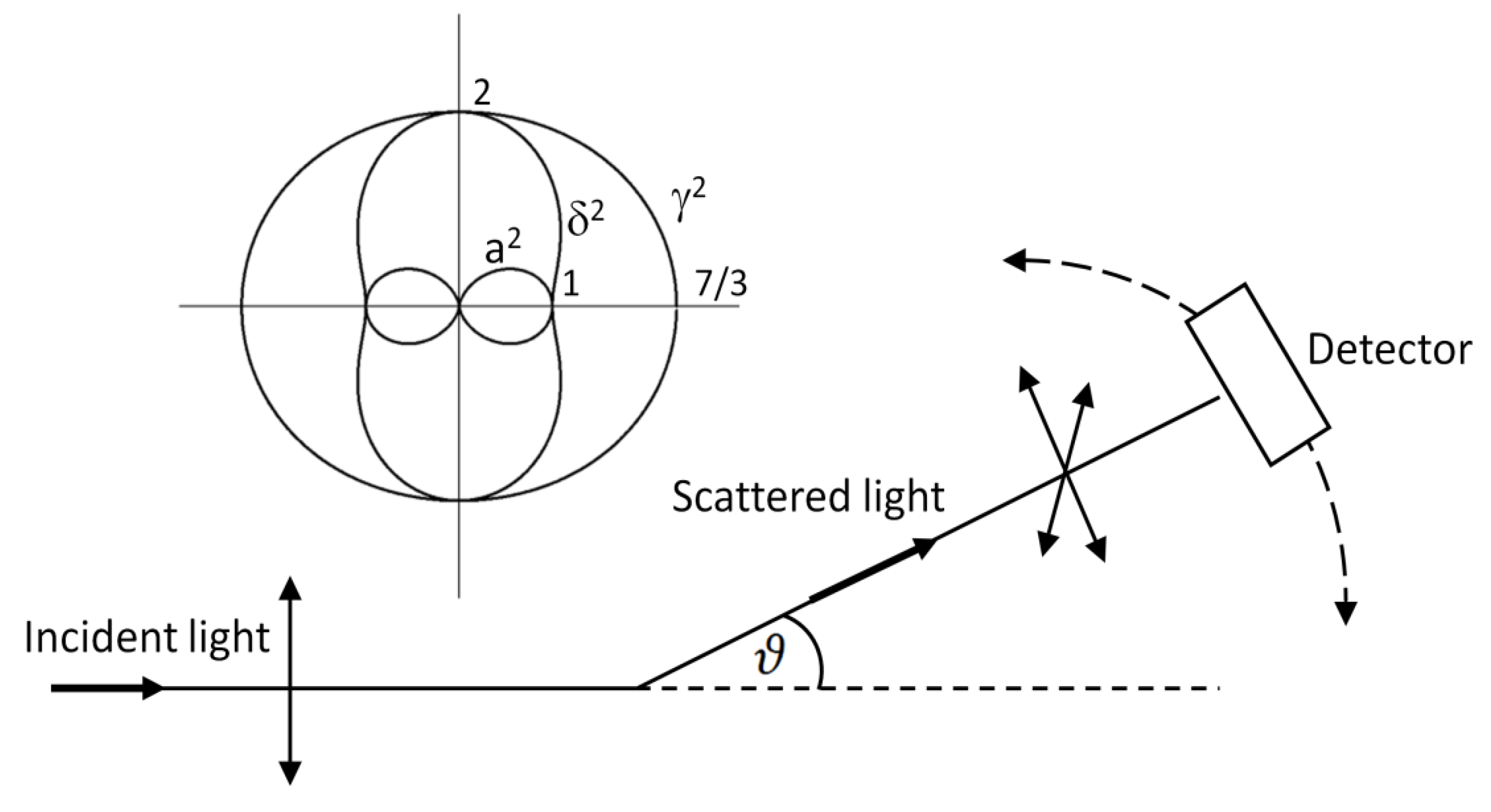
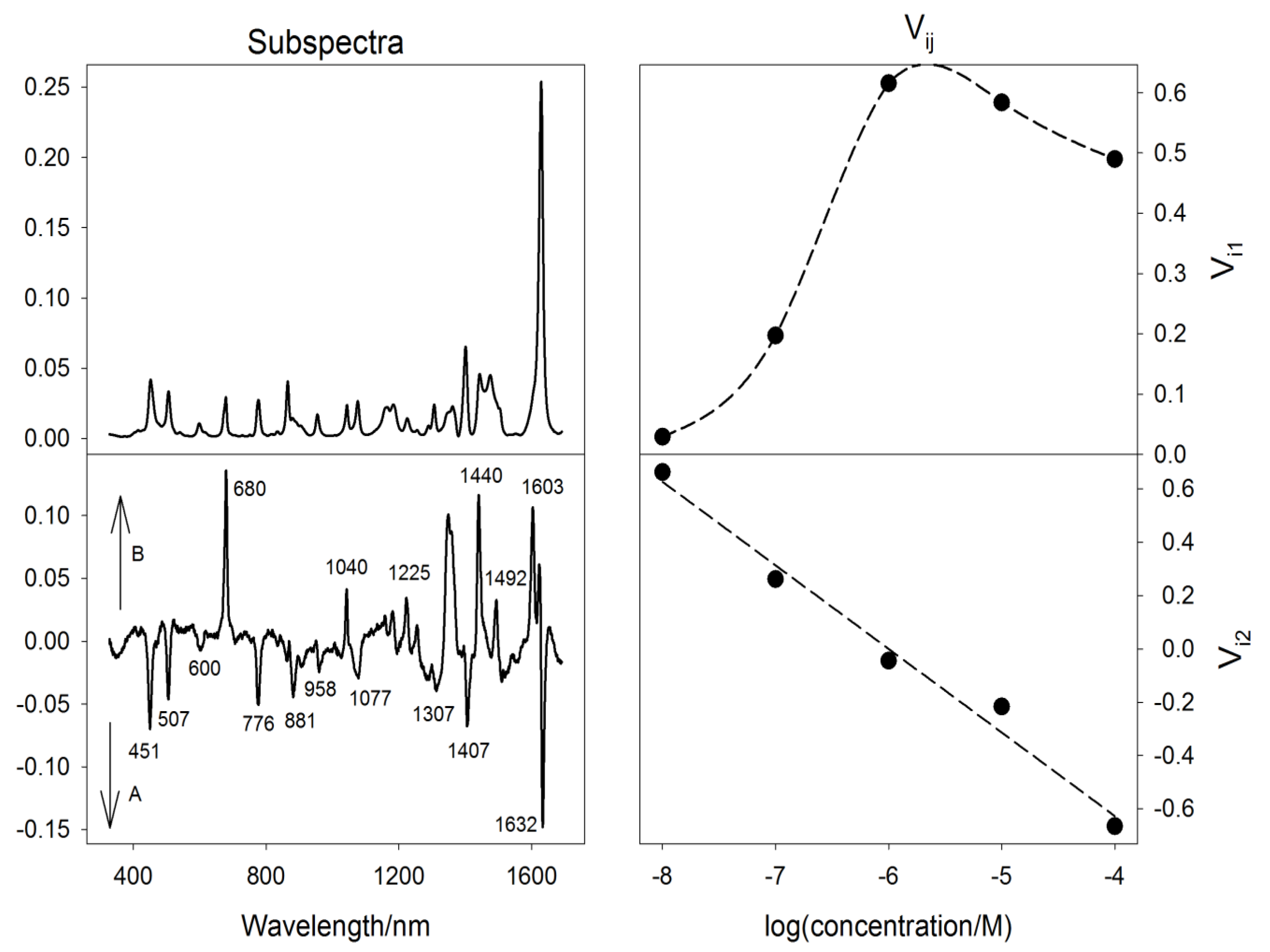
3.4. Directional Properties in Raman Spectroscopy
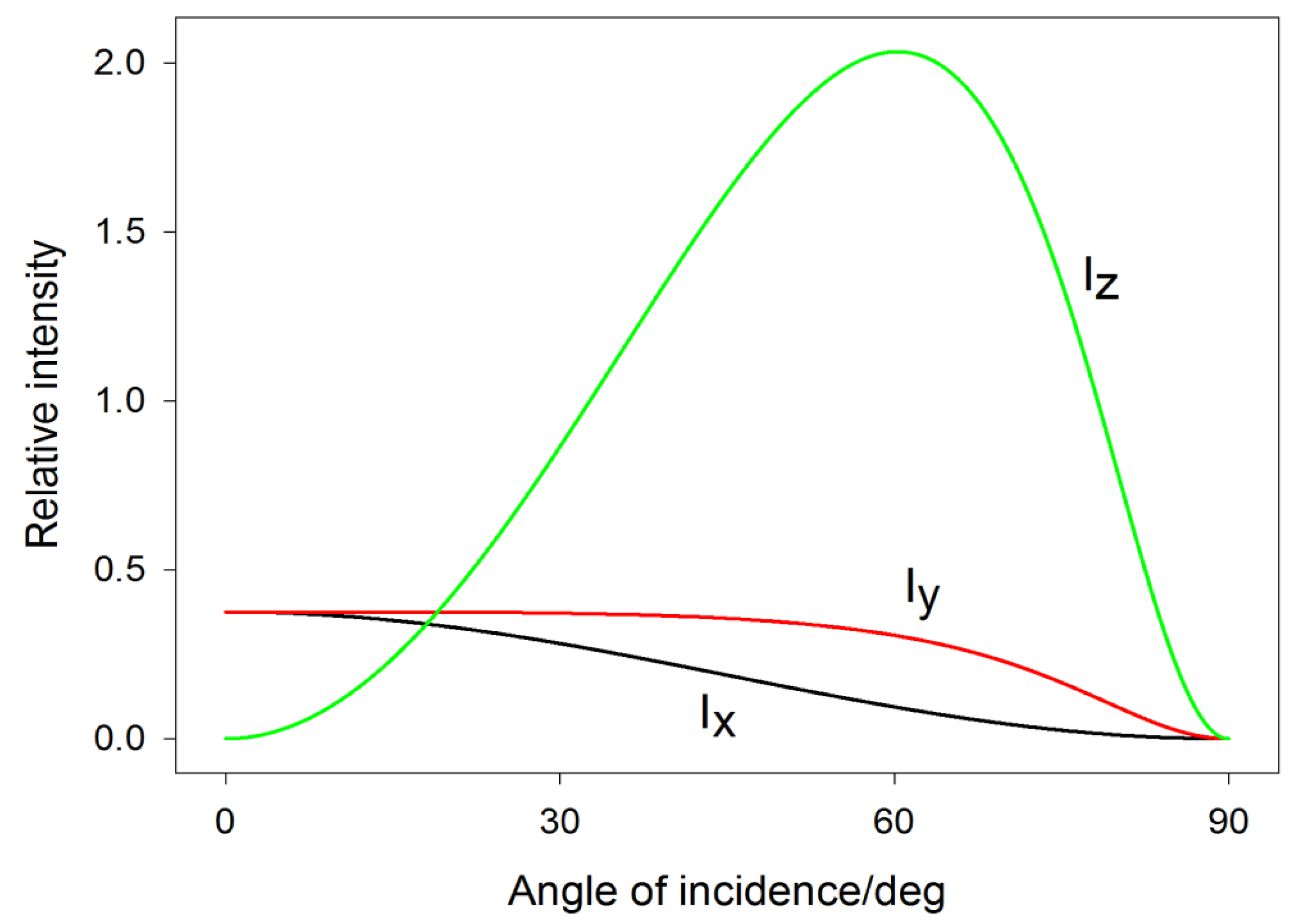
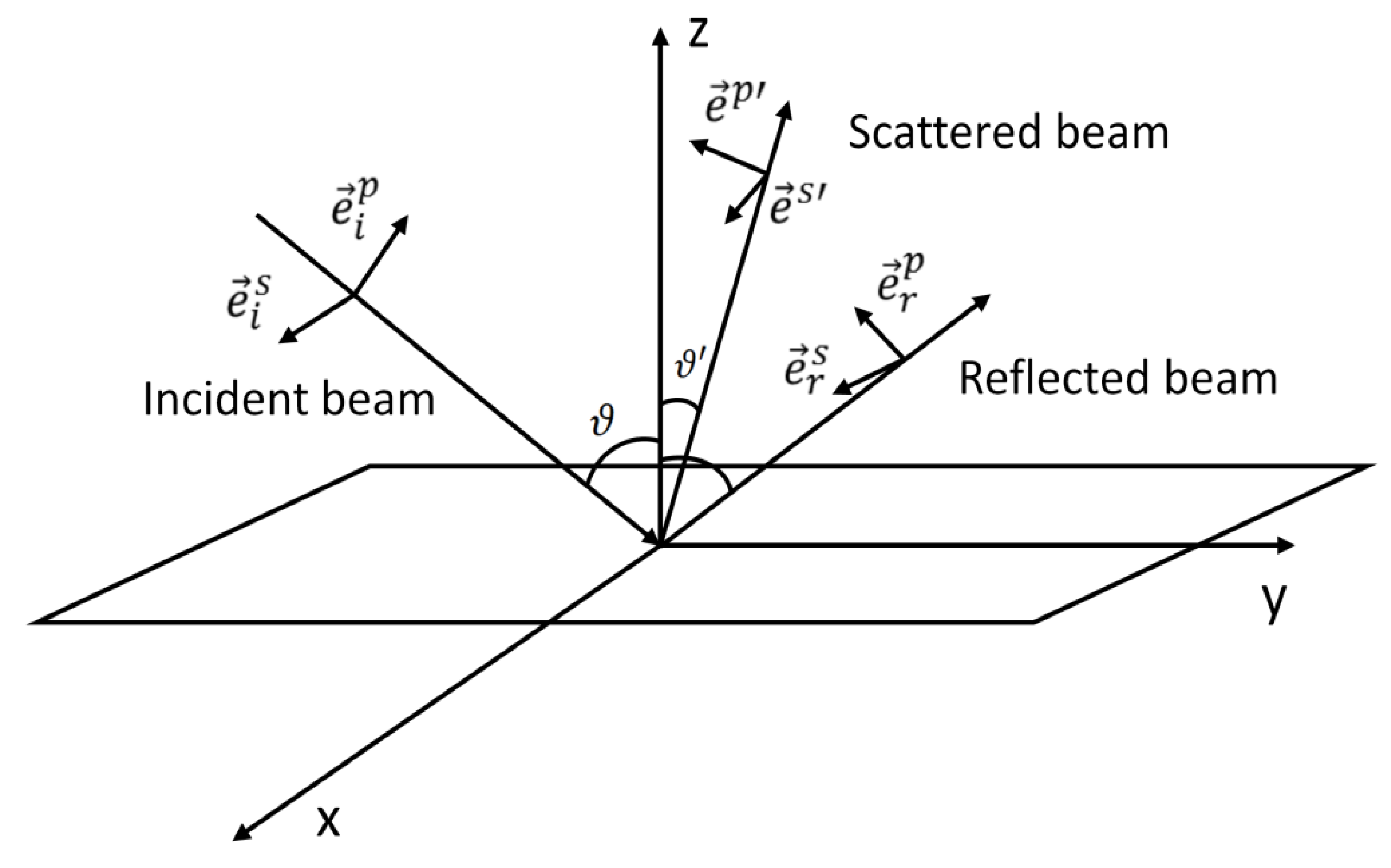
4. Polarization Effects for Crystals and Molecules Adsorbed on Smooth Surfaces
5. Polarization Effects for Molecules on Plasmonic Nanostructures
5.1. Polarization Properties of Isolated Spherical Particles
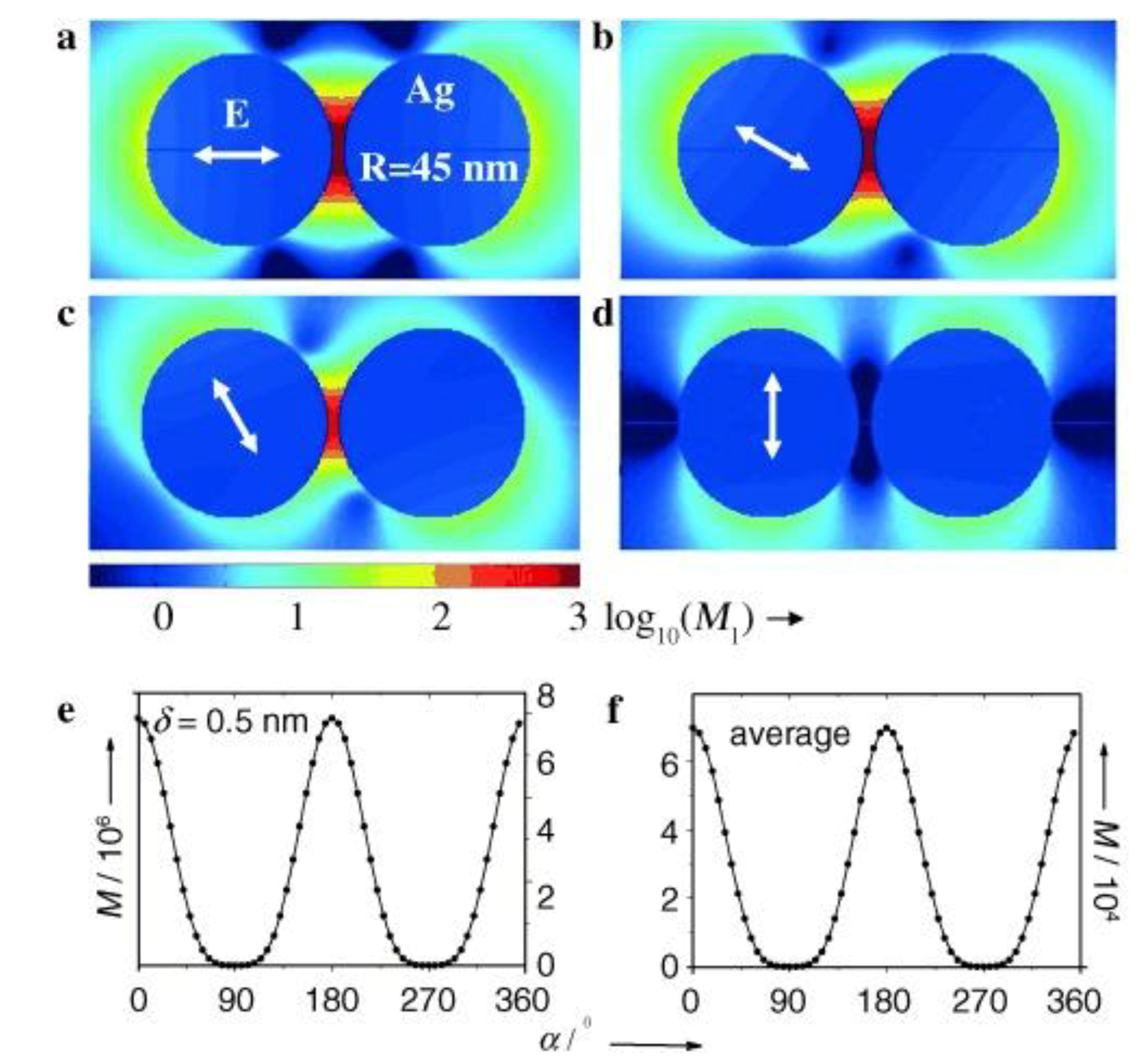
5.2. Local Field Distribution in Presence of Hot-Spots and Its Relation to Molecular Orientation
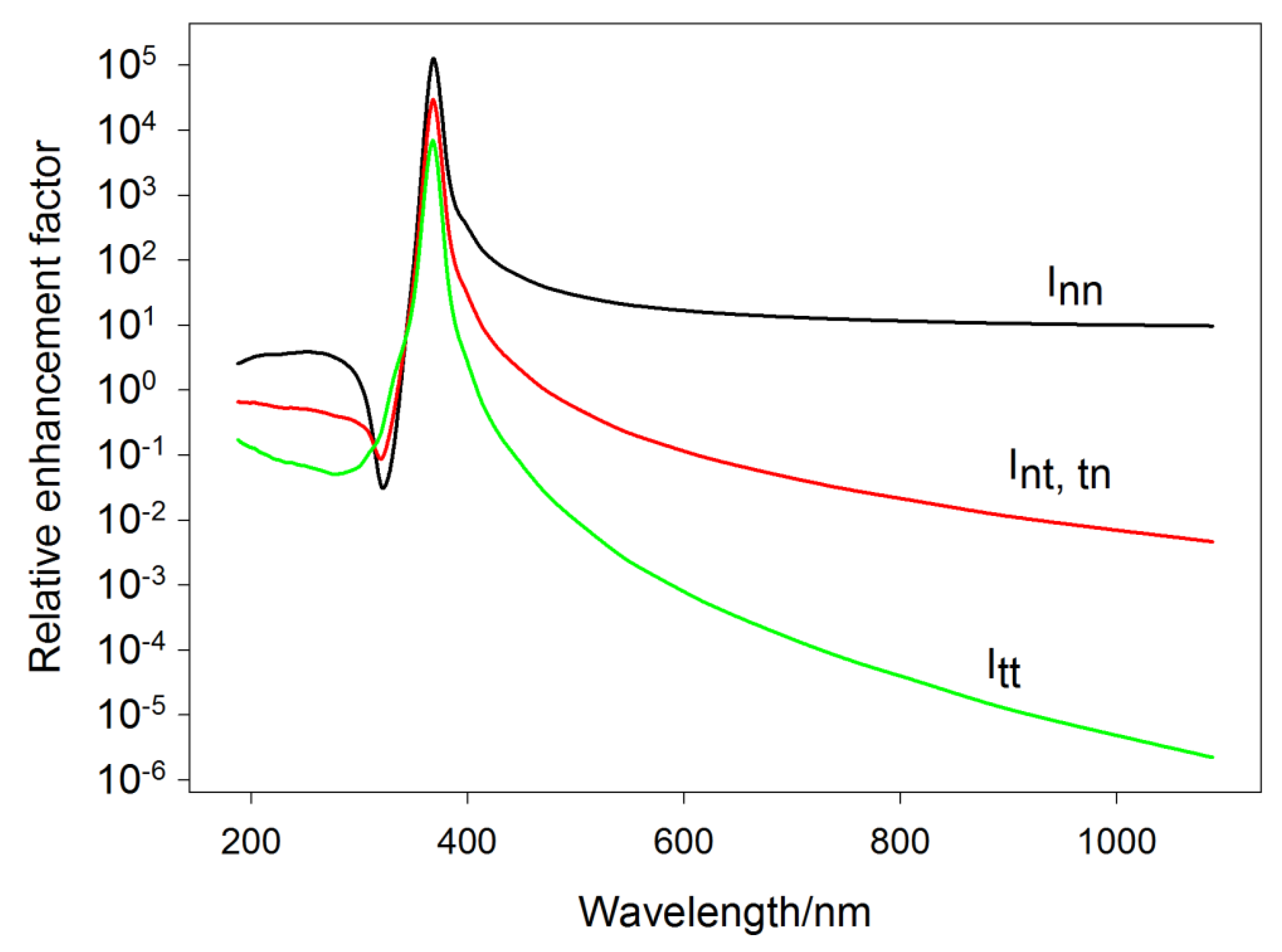
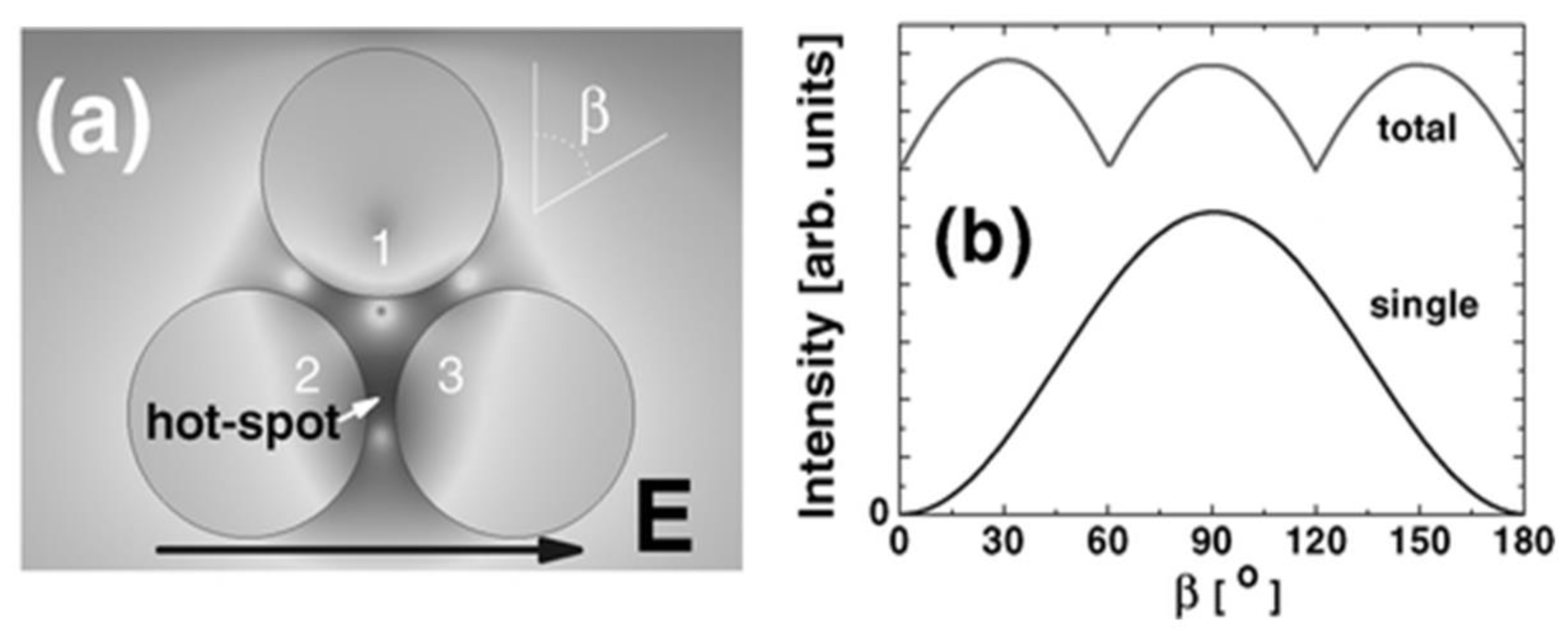
5.3. Polarization Properties of Trimers and Aggregated NP Domains
5.4. Polarization Properties of Regular Solid Plasmonic Platforms
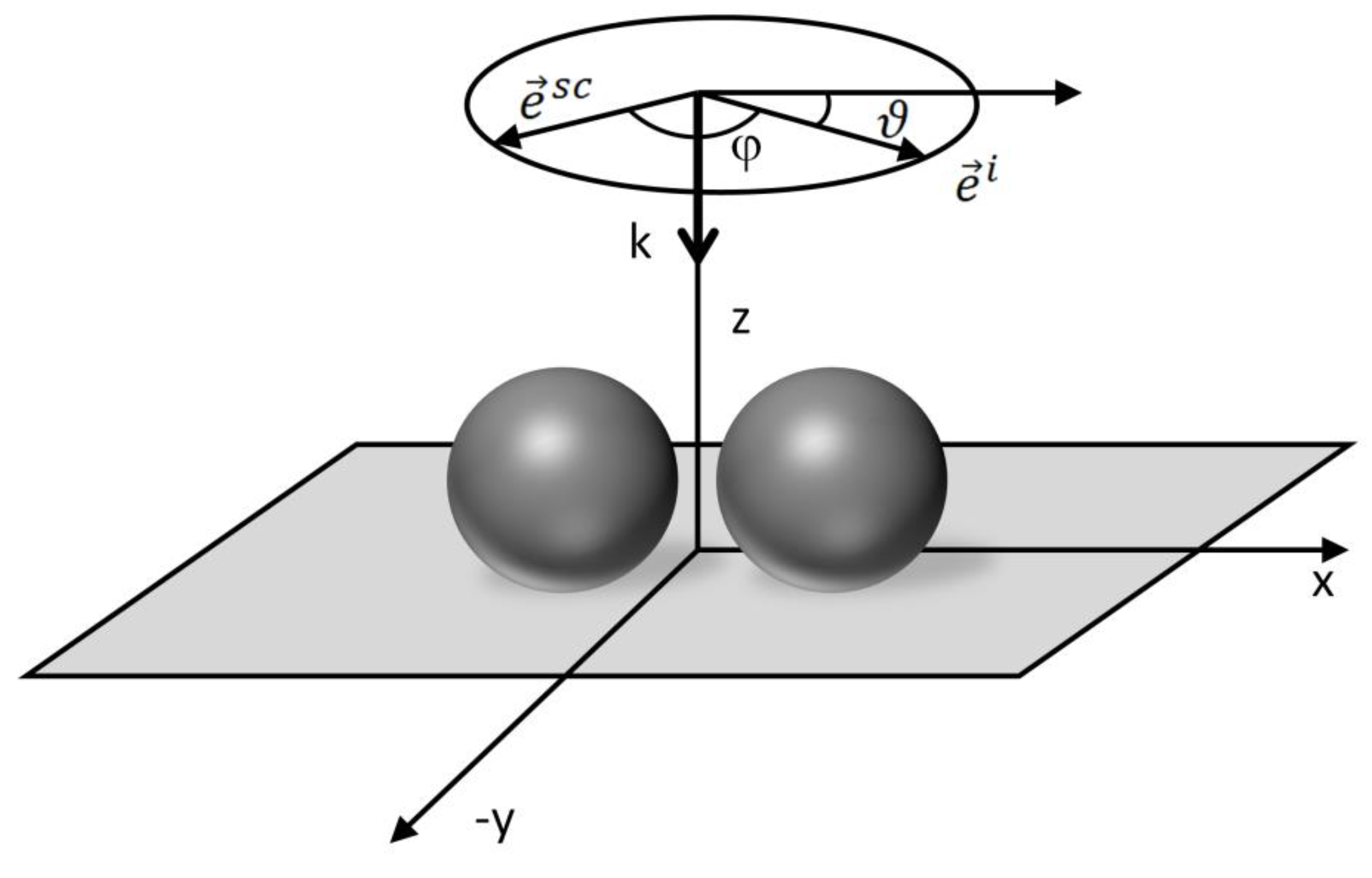
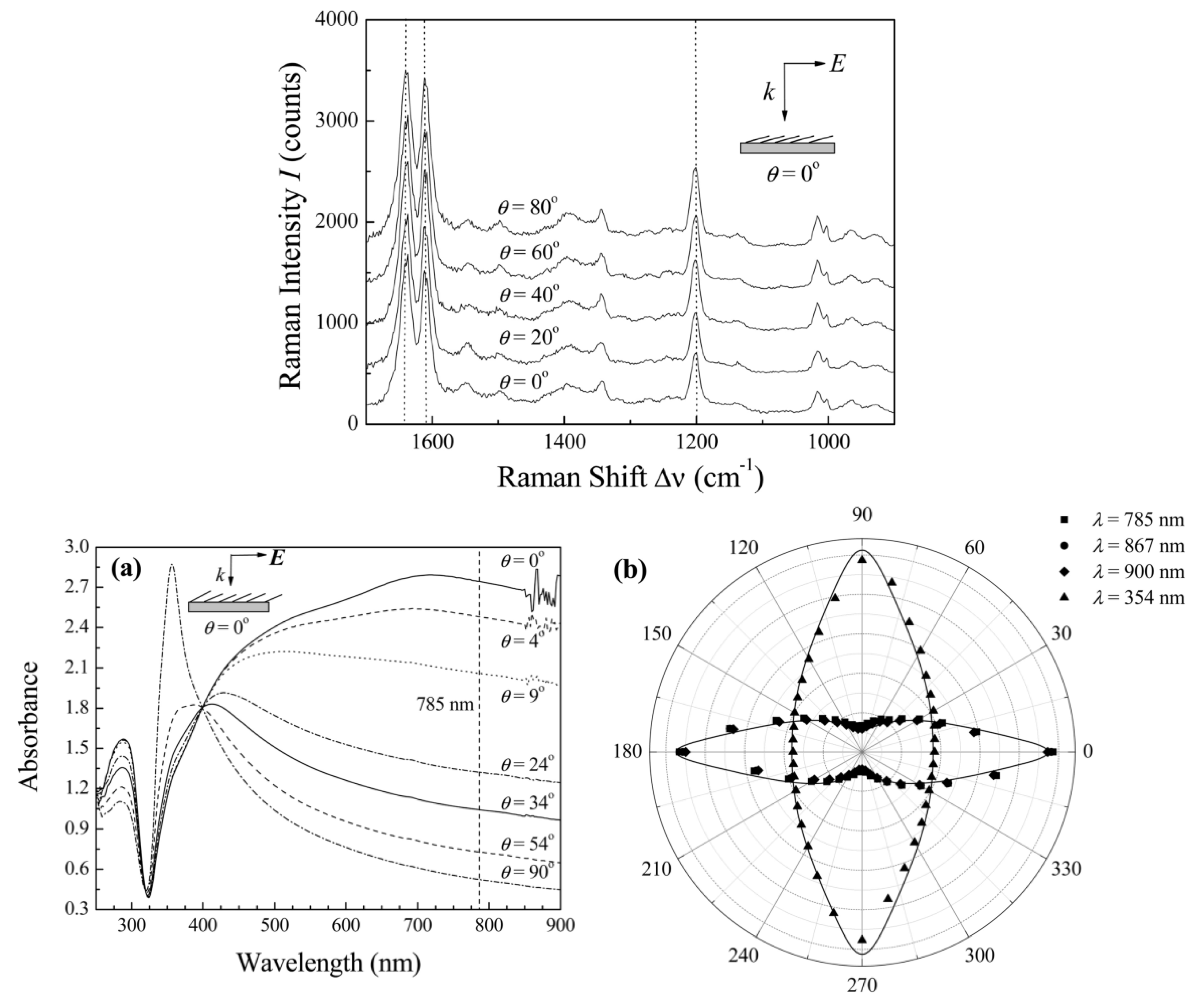
5.5. Angular Dependences in SERS
5.6. Plasmonic Anisotropy in Polarized Absorption and Emission and Their Influence on SERS
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allemand, C.D. Depolarization Ratio Measurements in Raman Spectroscopy. Appl. Spectrosc. 1970, 24, 348–353. [Google Scholar] [CrossRef]
- Dawson, P. Polarization Measurements in Raman Spectroscopy. Spectrochim. Acta Part A-Mol. Spectrosc. 1972, 28, 715–723. [Google Scholar] [CrossRef]
- Porto, S.P.S. Angular Dependence and Depolarization Ratio of the Raman Effect. J. Opt. Soc. Am. 1966, 56, 1585–1589. [Google Scholar] [CrossRef]
- Greenler, R.G. Infrared Study of Adsorbed Molecules on Metal Surfaces by Reflection Techniques. J. Chem. Phys. 1966, 44, 310–315. [Google Scholar] [CrossRef]
- Greenler, R.G.; Slager, T.L. Method for Obtaining Raman-Spectrum of a Thin-Film on a Metal Surface. Spectrochim. Acta Part A-Mol. Spectrosc. 1973, A 29, 193–201. [Google Scholar] [CrossRef]
- Pearce, H.A.; Sheppard, N. Possible Importance of a Metal-Surface Selection Rule in Interpretation of Infrared-Spectra of Molecules Adsorbed on Particulate Metals—Infrared-Spectra from Ethylene Chemisorbed on Silica-Supported Metal-Catalysts. Surf. Sci. 1976, 59, 205–209. [Google Scholar] [CrossRef]
- Murphy, C.J.; Sau, T.K.; Gole, A.M.; Orendorff, C.J.; Gao, J.; Gou, L.; Hunyadi, S.E.; Li, T. Anisotropic Metal Nanoparticles: Synthesis, Assembly, and Optical Applications. J. Phys. Chem. B 2005, 109, 13857–13870. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, P.X.; Astruc, D. Anisotropic Gold Nanoparticles: Synthesis, Properties, Applications, and Toxicity. Angew. Chem. Int. Ed. 2014, 53, 1756–1789. [Google Scholar] [CrossRef] [PubMed]
- Thorkelsson, K.; Bai, P.; Xu, T. Self-assembly and applications of anisotropic nanomaterials: A review. Nano Today 2015, 10, 48–66. [Google Scholar] [CrossRef]
- Paramasivam, G.; Kayambu, N.; Rabel, A.M.; Sundramoorthy, A.K.; Sundaramurthy, A. Anisotropic noble metal nanoparticles: Synthesis, surface functionalization and applications in biosensing, bioimaging, drug delivery and theranostics. Acta Biomater. 2017, 49, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.D. The development of biomolecular Raman optical activity spectroscopy. Biomed. Spectrosc. Imaging 2015, 4, 223–253. [Google Scholar] [CrossRef]
- Born, M.; Wolf, E. Principles of Optics, 7th ed.; Cambridge University Press: Cambridge, UK, 1999; ISBN 0-521-642221. [Google Scholar]
- Le Ru, E.C.; Etchegoin, P.G. Principles of Surface-Enhanced Raman Spectroscopy and Related Plasmonic Effects; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 978-0-444-52779-0. [Google Scholar]
- Procházka, M. Surface-Enhanced Raman Spectroscopy: Bioanalytical, Biomolecular and Medical Applications; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-23990-3. [Google Scholar]
- Moore, T.J.; Moody, A.S.; Payne, T.D.; Sarabia, G.M.; Daniel, A.R.; Sharma, B. In Vitro and In Vivo SERS Biosensing for Disease Diagnosis. Biosensors 2018, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Štolcová, L.; Peksa, V.; Proška, J.; Procházka, M. Gold film over very small (107 nm) spheres as efficient substrate for sensitive and reproducible surface-enhanced Raman scattering (SERS) detection of biologically important molecules. J. Raman Spectrosc. 2017, 49, 499–505. [Google Scholar] [CrossRef]
- Zhu, Q.; Yu, X.; Wu, Z.; Lu, F.; Yuan, Y. Antipsychotic drug poisoning monitoring of clozapine in urine by using coffee ring effect based surface-enhanced Raman spectroscopy. Anal. Chim. Acta 2018, 1014, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Mukanova, Z.; Gudun, K.; Elemessova, Z.; Khamkhash, L.; Ralchenko, E.; Bukasov, R. Detection of Paracetamol in Water and Urea in Artifical Urine with Gold Nanoparticle@Al Foil Cost-efficient SERS Substrate. Anal. Sci. 2018, 34, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Peksa, V.; Jahn, M.; Štolcová, L.; Schulz, V.; Proška, J.; Procházka, M.; Weber, K.; Cialla-May, D.; Popp, J. Quantitative SERS Analysis of Azorubine (E 122) in Sweet Drinks. Anal. Chem. 2015, 87, 2840–2844. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.-J.; Liang, P.; Wu, Y.-X.; Dong, Q.-M.; Li, J.-B.; Bai, Y.; Xu, B.-J.; Yu, Z.; Ni, D. Rapid qualitative and quantitative determination of food colorants by both Raman spectra and Surface-enhanced Raman Scattering (SERS). Food Chem. 2018, 241, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.; Wang, F.C.; Andersson, P.O.; Wingfors, H.; Rindzevicius, T.; Schmidt, M.S.; Soma, V.R.; Xu, S.; Li, Y.Q.; Boisen, A.; et al. Hand-Held Femtogram Detection of Hazardous Picric Acid with Hydrophobic Ag Nanopillar SERS Substrates and Mechanism of Elasto-Capillarity. ACS Sens. 2017, 2, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, T.; Rael, A.; Schaffer, S.; Zaidi, S.; Goodpaster, J.V.; Sardar, R. Fabrication of a self-assembled and flexible SERS nanosesor for explosive detection at parts-per-quadrillion levels from fingerprints. Analyst 2018, 143, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-W.; Huan, S.-Y.; Yu, R.-Q. Nanoparticle-based substrates for surface-enhanced Raman scattering detection of bacterial spores. Analyst 2012, 137, 3601–3608. [Google Scholar] [CrossRef] [PubMed]
- Morelli, L.; Zor, K.; Jendersen, C.B.; Rindzevicius, T.; Schmidt, M.S.; Nielsen, A.T.; Boisent, A. Surface Enhanced Raman Scattering for Quantification of p-Coumaric Acid Produced by Escherichia coli. Anal. Chem. 2017, 89, 3981–3987. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Purra, M.; Carre-Camps, M.; de Puig, H.; Bosch, I.; Gehrke, L.; Hamad-Schifferli, K. Surface-Enhanced Raman Spectroscopy-Based Sandwich Immunoassays for Multiplexed Detection of Zika and Dengue Viral Biomarkers. ACS Infect. Dis. 2017, 3, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Placzek, G. Rayleigh-Streuung und Raman-Effekt. In Handbuch der Radiologie; Marx, E., Ed.; Academische Verlag: Leipzig, Germany, 1934; Volume 6, pp. 205–374. [Google Scholar]
- Long, D.A. The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; John Wiley & Sons Ltd.: Chichester, UK, 2002; ISBN 0-471-49028-8. [Google Scholar]
- Smith, E.; Dent, G. Modern Raman Spectroscopy—A Practical Approach; John Wiley & Sons Ltd.: Chichester, UK, 2005; ISBN 0-471-49668-5. [Google Scholar]
- Cotton, F.A. Chemical Applications of Group Theory, 3rd ed.; John Wiley & Sons Ltd.: Chichester, UK, 1990; ISBN 0-471-51094-7. [Google Scholar]
- Ferraro, J.R.; Nakamoto, K.; Brown, C.W. Introductory Raman Spectroscopy, 2nd ed.; Elsevier: New York, NY, USA, 2003; ISBN 978-0-12-254105-6. [Google Scholar]
- Gucciardi, P.G.; de la Chapelle, M.L.; Lidgi-Guigui, N. Handbook of Enhanced Spectroscopy; Pan Stanford Publishing Pte. Ltd.: Singapore, 2016; ISBN 978-981-4613-32-3. [Google Scholar]
- Strommen, D.P. Specific Values of the Depolarization Ratio in Raman-Spectroscopy—Their Origins and Significance. J. Chem. Educ. 1992, 69, 803–807. [Google Scholar] [CrossRef]
- Damen, T.C.; Porto, S.P.S.; Tell, B. Raman Effect in Zinc Oxide. Phys. Rev. 1966, 142, 570–574. [Google Scholar] [CrossRef]
- Moskovits, M. Surface Selection-Rules. J. Chem. Phys. 1982, 77, 4408–4416. [Google Scholar] [CrossRef]
- Moskovits, M. Surface-Enhanced Spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826. [Google Scholar] [CrossRef]
- Creighton, J.A. Surface Raman Electromagnetic Enhancement Factors for Molecules at the Surface of Small Isolated Metal Spheres—The Determination of Adsorbate Orientation from Sers Relative Intensities. Surf. Sci. 1983, 124, 209–219. [Google Scholar] [CrossRef]
- Aroca, R. Surface-Enhanced Vibrational Spectroscopy; John Wiley & Sons Ltd.: Chichester, UK, 2006; ISBN 0-471-60731-2. [Google Scholar]
- Johnson, P.B.; Christy, R.W. Optical Constants of Noble Metals. Phys. Rev. B 1972, 6, 4370–4379. [Google Scholar] [CrossRef]
- Foti, A.; D’Andrea, C.; Messina, E.; Irrera, A.; Marago, O.M.; Fazio, B.; Gucciardi, P.G. On the SERS depolarization ratio. Nanospectroscopy 2015, 1, 26–62. [Google Scholar] [CrossRef]
- Etchegoin, P.G.; Galloway, C.; Le Ru, E.C. Polarization-dependent effects in surface-enhanced Raman scattering (SERS). Phys. Chem. Chem. Phys. 2006, 8, 2624–2628. [Google Scholar] [CrossRef] [PubMed]
- Le Ru, E.C.; Meyer, M.; Blackie, E.; Etchegoin, P.G. Advanced aspects of electromagnetic SERS enhancement factors at a hot spot. J. Raman Spectrosc. 2008, 39, 1127–1134. [Google Scholar] [CrossRef]
- Moskovits, M. Surface-enhanced Raman spectroscopy: A brief retrospective. J. Raman Spectrosc. 2005, 36, 485–496. [Google Scholar] [CrossRef]
- Fazio, B.; D’Andrea, C.; Bonaccorso, F.; Irrera, A.; Calogero, G.; Vasi, C.; Gucciardi, P.G.; Allegrini, M.; Toma, A.; Chiappe, D.; et al. Re-radiation Enhancement in Polarized Surface-Enhanced Resonant Raman Scattering of Randomly Oriented Molecules on Self-Organized Gold Nanowires. ACS Nano 2011, 5, 5945–5956. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Chew, H.; Kerker, M. Enhanced Raman-Scattering at the Surface (SERS) of a Spherical-Particle. Appl. Opt. 1980, 19, 2256–2257. [Google Scholar] [CrossRef] [PubMed]
- Moskovits, M.; Suh, J.S. Surface Selection-Rules for Surface-Enhanced Raman Spectroscopy-Calculation and Application to the Surface-Enhanced Raman Spectrum of Phthalazine on Silver. J. Phys. Chem. 1984, 88, 5526–5530. [Google Scholar] [CrossRef]
- Macdonald, I.D.G.; Smith, W.E. Orientation of Cytochrome c Adsorbed on a Citrate-Reduced Silver Colloid Surface. Langmuir 1996, 12, 706–713. [Google Scholar] [CrossRef]
- Gao, X.; Davies, J.P.; Weaver, M.J.A. Test of Surface Selection-Rules for Surface-Enhanced Raman-Scattering: The Orientation of Adsorbed Benzene and Monosubstituted Benzenes on Gold. J. Phys. Chem. 1990, 94, 6858–6864. [Google Scholar] [CrossRef]
- Carron, K.T.; Hurley, L.G. Axial and Azimuthal Angle Determination with Surface-Enhanced Raman-Spectroscopy: Thiophenol on Copper, Silver, and Gold Metal-Surfaces. J. Phys. Chem. 1991, 95, 9979–9984. [Google Scholar] [CrossRef]
- Barber, P.W.; Chang, R.K.; Massoudi, H. Surface-Enhanced Electric Intensities on Large Silver Spheroids. Phys. Rev. Lett. 1983, 50, 997–1000. [Google Scholar] [CrossRef]
- Aravind, P.K.; Nitzan, A.; Metiu, H. The Interaction between Electromagnetic Resonances and Its Role in Spectroscopic Studies of Molecules Adsorbed on Colloidal Particles or Metal Spheres. Surf. Sci. 1981, 110, 189–204. [Google Scholar] [CrossRef]
- Aravind, P.K.; Metiu, H. The Effects of the Interaction between Resonances in the Electromagnetic Response of a Sphere-Plane Structure; Applications to Surface Enhanced Spectroscopy. Surf. Sci. 1983, 124, 506–528. [Google Scholar] [CrossRef]
- Xu, H.; Bjerneld, E.J.; Käll, M.; Borjesson, L. Spectroscopy of single hemoglobin molecules by surface enhanced Raman scattering. Phys. Rev. Lett. 1999, 83, 4357–4360. [Google Scholar] [CrossRef]
- Xu, H.; Aizpurua, J.; Käll, M.; Apell, P. Electromagnetic contributions to single-molecule sensitivity in surface-enhanced Raman scattering. Phys. Rev. E 2000, 62, 4318–4324. [Google Scholar] [CrossRef]
- Xu, H.; Käll, M. Polarization-Dependent Surface-Enhanced Raman Spectroscopy of Isolated Silver Nanoaggregates. ChemPhysChem 2003, 4, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Le Ru, E.C.; Blackie, E.; Meyer, M.; Etchegoin, P.G. Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study. J. Phys. Chem. C 2007, 111, 13794–13803. [Google Scholar] [CrossRef]
- Camden, J.P.; Dieringer, J.A.; Wang, Y.; Masiello, D.J.; Marks, L.D.; Schatz, G.C.; Van Duyne, R.P. Probing the Structure of Single-Molecule Surface-Enhanced Raman Scattering Hot Spots. J. Am. Chem. Soc. 2008, 130, 12616–12617. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.; Svedendahl, M.; Ogier, R.; Yang, Z.-J.; Lodewijks, K.; Verre, R.; Shegai, T.; Andersson, P.O.; Käll, M. Dimer-on-mirror SERS substrates with attogram sensitivity fabricated by colloidal lithography. Nanoscale 2015, 7, 9405–9410. [Google Scholar] [CrossRef] [PubMed]
- Wustholz, K.L.; Henry, A.I.; McMahon, J.M.; Freeman, R.G.; Valley, N.; Piotti, M.E.; Natan, M.J.; Schatz, G.C.; Van Duyne, R.P. Structure-Activity Relationships in Gold Nanoparticle Dimers and Trimers for Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2010, 132, 10903–10910. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Shi, H.; Huang, Y.; Zeng, X.; Pan, L.; Wang, S.; Wen, W. SERS polarization dependence of Ag nanorice dimer on metal and dielectric film. Chem. Phys. Lett. 2017, 684, 373–377. [Google Scholar] [CrossRef]
- Tao, A.R.; Yang, P.D. Polarized surface-enhanced Raman spectroscopy on coupled metallic nanowires. J. Phys. Chem. B 2005, 109, 15687–15690. [Google Scholar] [CrossRef] [PubMed]
- Le Ru, E.C.; Etchegoin, P.G.; Meyer, M. Enhancement factor distribution around a single surface-enhanced Raman scattering hot spot and its relation to single molecule detection. J. Chem. Phys. 2006, 125, 204701. [Google Scholar] [CrossRef] [PubMed]
- Moskovits, M. Persistent misconceptions regarding SERS. Phys. Chem. Chem. Phys. 2013, 15, 5301–5311. [Google Scholar] [CrossRef] [PubMed]
- Leopold, N.; Lendl, B. A New Method for Fast Preparation of Highly Surface-Enhanced Raman Scattering (SERS) Active Silver Colloids at Room Temperature by Reduction of Silver Nitrate with Hydroxylamine Hydrochloride. J. Phys. Chem. B 2003, 107, 5723–5727. [Google Scholar] [CrossRef]
- Roy, S.D.; Ghosh, M.; Chowdhury, J. Adsorptive parameters and influence of hot geometries on the SER(R)S spectra of methylene blue molecules adsorbed on gold nanocolloidal particles. J. Raman Spectrosc. 2015, 46, 451–461. [Google Scholar] [CrossRef]
- Malinowski, E.R. FactorAnalysis in Chemistry, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2002; ISBN 0-471-13479-1. [Google Scholar]
- Zhong, L.; Hu, Y.J.; Xing, D.; Gu, H.M. Adsorption of Methylene Blue on Colloidal Silver-A Surface-Enhanced Raman Spectroscopy Study Combined with Density Functional Theory Calculations. Spectrosc. Spectr. Anal. 2010, 30, 90–94. [Google Scholar] [CrossRef]
- Timmermans, F.J.; Lenferink, A.T.M.; van Wolferen, H.A.G.M.; Otto, C. Correlative SEM SERS for quantitative analysis of dimer nanoparticles. Analyst 2016, 141, 6455–6462. [Google Scholar] [CrossRef] [PubMed]
- Sergiienko, S.; Moor, K.; Gudun, K.; Yelemessova, Z.; Bukasov, R. Nanoparticle-nanoparticle vs. nanoparticle-substrate hot spot contributes to the SERS signal: Studying Raman labelled monomers, dimers and trimers. Phys. Chem. Chem. Phys. 2017, 19, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Li, Z.; Zheng, M.; Liu, Q.; Chen, Y.; Yang, L.; Jiang, T.; Duan, H. Sensitive SERS detection at the single-particle level based on nanometer-separated mushroom-shaped plasmonic dimers. Nanotechnology 2018, 29, 105301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, G.-C.; Lo, T.W.; Lei, D.Y. Polarization-resolved optical response of plasmonic particle-on-film nanocavities. J. Opt. 2018, 20, 024010. [Google Scholar] [CrossRef]
- Cao, Z.; He, Y.; Cheng, Y.; Zhao, J.; Li, G.; Gong, Q.; Lu, G. Nano-gap between a gold tip and nanorod for polarization dependent surface enhanced Raman scattering. Appl. Phys. Lett. 2016, 109, 233103. [Google Scholar] [CrossRef]
- Haidar, I.; Levi, G.; Mouton, L.; Aubard, J.; Grand, J.; Lau-Truong, S.; Neuville, D.R.; Felidj, N.; Boubekeur-Lecaque, L. Highly stable silica-coated gold nanorods dimers for solution-based SERS. Phys. Chem. Chem. Phys. 2016, 18, 32272–32280. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Li, S.; Han, F.; Liu, L.; Xu, L.; Ma, W.; Kuang, H.; Li, A.; Wang, L.; Xu, C. SERS-active Au@Ag nanorod dimers for ultrasensitive dopamine detection. Biosens. Bioelectron. 2015, 71, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Aswathy, V.G.; Patra, P.P.; Kumar, G.V.P. Geometry-dependent anti-Stokes SERS radiation patterns from gold nanorod dimers. J. Opt. 2015, 17, 114011. [Google Scholar] [CrossRef]
- Maune, H.T.; Han, S.-P.; Barish, R.D.; Bockrath, M.; Goddard, W.A., III; Rothemund, P.W.K.; Winfree, E. Self-assembly of carbon nanotubes into two-dimensional geometries using DNA origami templates. Nat. Nanotechnol. 2010, 5, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Thacker, V.V.; Herrmann, L.O.; Sigle, D.O.; Zhang, T.; Liedl, T.; Baumberg, J.J.; Keyser, U.F. DNA origami based assembly of gold nanoparticle dimers for surface-enhanced Raman scattering. Nat. Commun. 2014, 5, 3448. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.; Heck, C.; Ellerik, L.; Merk, V.; Bald, I. DNA origami based Au-Ag-core-shell nanoparticle dimers with single-molecule SERS sensitivity. Nanoscale 2016, 8, 5612–5620. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Gohil, S.; Ghosh, S.; Ayyub, P. Polarization Selection Rules for Surface-Enhanced Raman Scattering from Anisotropic Microstructured Surfaces. J. Phys. Chem. C 2012, 116, 18504–18507. [Google Scholar] [CrossRef]
- Le Ru, E.C.; Grand, J.; Félidj, N.; Aubard, J.; Lévi, G.; Hohenau, A.; Krenn, J.R.; Blackie, E.; Etchegoin, P.G. Experimental Verification of the SERS Electromagnetic Model beyond the |E|4 Approximation: Polarization Effects. J. Phys. Chem. C 2012, 116, 18504–18507. [Google Scholar] [CrossRef]
- Shegai, T.; Li, Z.; Dadosh, T.; Zhang, Z.; Xu, H.; Haran, G. Managing light polarization via plasmon-molecule interactions within an asymmetric metal nanoparticle trimer. Proc. Natl. Acad. Sci. USA 2008, 105, 16448–16453. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Zhao, Y.; Jiang, Y.J. Periodic array of regular Ag nanoparticle trimers: A reliable polarization-independent surface-enhanced Raman spectroscopy substrate. RSC Adv. 2016, 6, 83273–83279. [Google Scholar] [CrossRef]
- Shegai, T.; Brian, B.; Miljkovic, V.D.; Käll, M. Angular Distribution of Surface-Enhanced Raman Scattering from Individual Au Nanoparticle Aggregates. ACS Nano 2011, 5, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Quinten, M. Local fields close to the surface of nanoparticles and aggregates of nanoparticles. Appl. Phys. B-Lasers Opt. 2001, 73, 245–255. [Google Scholar] [CrossRef]
- Dai, Z.; Xiao, X.; Wu, W.; Liao, L.; Mei, F.; Yu, X.; Guo, S.; Ying, J.; Ren, F.; Jiang, C. Side-to-side alignment of gold nanorods with polarization-free characteristic for highly reproducible surface enhanced Raman scattering. Appl. Phys. Lett. 2014, 105, 211902. [Google Scholar] [CrossRef]
- Shegai, T.O.; Haran, G. Probing the Raman Scattering Tensors of Individual Molecules. J. Phys. Chem. B 2006, 110, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Rouxel, J.; Guillot, N.; de la Chapelle, M.L.; Toury, T. Light polarization properties of three fold symmetry gold nanoparticles: Model and experiments. C. R. Phys. 2012, 13, 830–836. [Google Scholar] [CrossRef]
- De la Chapelle, M.L.; Guillot, N.; Frémaux, B.; Shen, H.; Toury, T. Novel Apolar Plasmonic Nanostructures with Extended Optical Tunability for Sensing Applications. Plasmonics 2013, 8, 475–480. [Google Scholar] [CrossRef]
- Reguera, J.; Langer, J.; de Aberasturi, D.J.; Liz-Marzán, L.M. Anisotropic metal nanoparticles for surface enhanced Raman scattering. Chem. Soc. Rev. 2017, 46, 3866–3885. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.M.; Li, Z.Y.; Siekkinen, A.R.; Xia, Y. The SERS Activity of a Supported Ag Nanocube Strongly Depends on Its Orientation Relative to Laser Polarization. Nano Lett. 2007, 7, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, X.; Park, D.J.; Torabi, K.; Brown, K.A.; Jones, M.R.; Zhang, C.; Schatz, G.C.; Mirkin, C.A. Shape-Selective Deposition and Assembly of Anisotropic Nanoparticles. Nano Lett. 2014, 14, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Prezgot, D.; Ianoul, A. Probing the Anisotropy of SERS Enhancement with Spatially Separated Plasmonic Modes in Strongly Coupled Silver Nanocubes on a Dielectric Substrate. J. Phys. Chem. C 2015, 119, 3293–3301. [Google Scholar] [CrossRef]
- Luo, W.; van der Veer, W.; Chu, P.; Mills, D.L.; Penner, R.M.; Hemminger, J.C. Polarization-Dependent Surface Enhanced Raman Scattering from Silver 1D Nanoparticle Arrays. J. Phys. Chem. C 2008, 112, 11609–11613. [Google Scholar] [CrossRef]
- Hossain, M.K.; Huang, G.G.; Tanaka, Y.; Kaneko, T.; Ozaki, Y. Anisotropic gold nanoassembly: A study on polarization-dependent and polarization-selective surface-enhanced Raman scattering. Phys. Chem. Chem. Phys. 2015, 17, 4268–4276. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.Y.; Brolo, A.G. Polarization-dependent surface-enhanced Raman scattering (SERS) from microarrays. Anal. Chim. Acta 2017, 972, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Farcau, C.; Vallée, R.A.L.; Boca, S.; Astilean, S. Polarized SERS on linear arrays of silver half-shells: SERS re-radiation modulated by local density of optical states. J. Opt. 2015, 17, 114017. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, J.; Zhao, Y.P.; Shanmukh, S.; Dluhy, R.A. Angle dependent surface enhanced Raman scattering obtained from a Ag nanorod array substrate. Appl. Phys. Lett. 2006, 89, 173134. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Chaney, S.B.; Shanmukh, S.; Dluhy, R.A. Polarized Surface Enhanced Raman and Absorbance Spectra of Aligned Silver Nanorod Arrays. J. Phys. Chem. B 2006, 110, 3153–3157. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Maekita, W.; Wada, Y.; Nakajima, K.; Kimura, K.; Fukuota, T.; Mori, Y. In-line aligned and bottom-up Ag nanorods for surface-enhanced Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 203121. [Google Scholar] [CrossRef]
- Ming, T.; Zhao, L.; Yang, Z.; Chen, H.; Sun, L.; Wang, J.; Yan, C. Polarization Dependence of Plasmon-Enhanced Fluorescence on Single Gold Nanorods. Nano Lett. 2009, 9, 3896–3903. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Zhang, Y.X.; Moskovits, M. Polarized Surface Enhanced Raman Scattering from Aligned Silver Nanowire Rafts. J. Phys. Chem. B 2004, 108, 12724–12728. [Google Scholar] [CrossRef]
- Zhou, W.; Hu, A.; Bai, S.; Ma, Y.; Bridges, D. Anisotropic optical properties of large-scale aligned silver nanowire films via controlled coffee ring effects. RSC Adv. 2015, 5, 39103–39109. [Google Scholar] [CrossRef]
- Chen, W.; Shi, H.; Wan, F.; Wang, P.; Gu, Z.; Li, W.; Ke, L.; Huang, Y. Substrate influence on the polarization dependence of SERS in crossed metal nanowires. J. Mater. Chem. C 2017, 5, 7028–7034. [Google Scholar] [CrossRef]
- Yoon, I.; Kang, T.; Choi, W.; Kim, J.; Yoo, Y.; Joo, S.-W.; Park, Q.-H.; Ihee, H.; Kim, B. Single Nanowire on a Film as an Efficient SERS-Active Platform. J. Am. Chem. Soc. 2009, 131, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhai, T.; Zhang, X.; Unger, C.; Koch, J.; Chichkov, B.N.; Klar, P.J. Polarization-dependent SERS effects of laser-generated sub-100 nm antenna structures. Nanotechnology 2014, 25, 265302. [Google Scholar] [CrossRef] [PubMed]
- Kessentini, S.; Barchiesi, D.; D’Andrea, C.; Toma, A.; Guillot, N.; Di Fabrizio, E.; Fazio, B.; Marago, O.M.; Gucciardi, P.G.; de la Chapelle, M.L. Gold Dimer Nanoantenna with Slanted Gap for Tunable LSPR and Improved SERS. J. Phys. Chem. C 2014, 118, 3209–3219. [Google Scholar] [CrossRef]
- Liu, K.K.; Tadepalli, S.; Kumari, G.; Banerjee, P.; Tian, L.; Jain, P.K.; Singamaneni, S. Polarization-Dependent Surface-Enhanced Raman Scattering Activity of Anisotropic Plasmonic Nanorattles. J. Phys. Chem. C 2016, 120, 16899–16906. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, T.; Li, Y.; Fu, Y.; Guo, L. Optimally designed gold nanorattles with strong built-in hotspots and weak polarization dependence. Nanotechnology 2017, 28, 495201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Q.; Zhang, C.-H.; Weng, G.-J.; Zhao, J.; Li, J.-J.; Zhao, J.-W. Synthesis of colloidal gold nanobones with tunable negative curvatures at end surface and their application in SERS. J. Nanopart. Res. 2017, 19, 364. [Google Scholar] [CrossRef]
- Lee, S.J.; Baik, J.M.; Moskovits, M. Polarization-Dependent Surface-Enhanced Raman Scattering from a Silver-Nanoparticle-Decorated Single Silver Nanowire. Nano Lett. 2008, 8, 3244–3247. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.G.; Xiao, X.H.; Liao, L.; Ying, J.J.; Mei, F.; Wu, W.; Ren, F.; Li, W.Q.; Jiang, C.Z. Enhanced and polarization dependence of surface-enhanced Raman scattering in silver nanoparticle array-nanowire systems. Appl. Phys. Lett. 2015, 107, 129901. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W.; Lei, H.; Li, B. Ag nanowire/nanoparticle-decorated MoS2 monolayers for surface-enhanced Raman scattering applications. Nano Res. 2018, 11, 2181–2189. [Google Scholar] [CrossRef]
- Šubr, M.; Petr, M.; Kylián, O.; Štěpánek, J.; Veis, M.; Procházka, M. Anisotropic Optical Response of Silver Nanorod Arrays: Surface Enhanced Raman Scattering Polarization and Angular Dependences Confronted with Ellipsometric Parameters. Sci. Rep. 2017, 7, 4293. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Yamaguchi, T.; Kajikawa, K. Angular-Resolved Polarized Surface Enhanced Raman Spectroscopy. J. Phys. Chem. C 2012, 116, 9716–9723. [Google Scholar] [CrossRef]
- Ranjan, M.; Facsko, S. Anisotropic surface enhanced Raman scattering in nanoparticle and nanowire arrays. Nanotechnology 2012, 23. [Google Scholar] [CrossRef] [PubMed]
- Khlebtsov, B.N.; Khlebtsov, N.G. Multipole Plasmons in Metal Nanorods: Scaling Properties and Dependence on Particle Size, Shape, Orientation, and Dielectric Environment. J. Phys. Chem. C 2007, 111, 11516–11527. [Google Scholar] [CrossRef]
- Gkogkou, D.; Schreiber, B.; Shaykhutdinov, T.; Ly, H.K.; Kuhlmann, U.; Gernert, U.; Facsko, S.; Hildebrandt, P.; Esser, N.; Hinrichs, K.; et al. Polarization- and Wavelength-Dependent Surface-Enhanced Raman Spectroscopy Using Optically Anisotropic Rippled Substrates for Sensing. ACS Sens. 2016, 1, 318–323. [Google Scholar] [CrossRef]
- Le Ru, E.C.; Meyer, S.A.; Artur, C.; Etchegoin, P.G.; Grand, J.; Lang, P.; Maurel, F. Experimental demonstration of surface selection rules for SERS on flat metallic surfaces. Chem. Commun. 2011, 47, 3903–3905. [Google Scholar] [CrossRef] [PubMed]
- Peyskens, F.; Dhakal, A.; Van Dorpe, P.; Le Thomas, N.; Baets, N. Surface Enhanced Raman Spectroscopy Using a Single Mode Nanophotonic-Plasmonic Platform. ACS Photonics 2016, 3, 102–108. [Google Scholar] [CrossRef]
- Dhakal, A.; Peyskens, F.; Clemmen, S.; Raza, A.; Wuytens, P.; Zhao, H.; Le Thomas, N.; Baets, R. Single mode waveguide platform for spontaneous and surface-enhanced on-chip Raman spectroscopy. Interface Focus 2016, 6, 20160015. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Zhao, Y.P. Simple model for surface-enhanced Raman scattering from tilted silver nanorod array substrates. Phys. Rev. B 2008, 78, 075436. [Google Scholar] [CrossRef]
- Benson, M.; Shah, P.; Marciniak, M.; Sarangan, A.; Urbas, A. Optical Characterization of Silver Nanorod Thin Films Grown Using Oblique Angle Deposition. J. Nanomater. 2014, 694982. [Google Scholar] [CrossRef]
- Perera, M.N.M.N.; Schmidt, D.; Gibbs, W.E.K.; Juodkazis, S.; Stoddart, P.R. Effective optical constants of anisotropic silver nanoparticle films with plasmonic properties. Opt. Lett. 2016, 41, 5495–5498. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Sturm, C.; Kranert, C.; Richter, S.; Schmidt-Grund, R.; Deparis, C.; Zuniga-Perez, J. Optically anisotropic media: New approaches to the dielectric function, singular axes, microcavity modes and Raman scattering intensities. Phys. Status Solidi-Rapid Res. Lett. 2017, 11, 1600295. [Google Scholar] [CrossRef]
- Oates, T.W.H.; Ranjan, M.; Facsko, S.; Arwin, H. Highly anisotropic effective dielectric functions of silver nanoparticle arrays. Opt. Express 2011, 19, 2014–2028. [Google Scholar] [CrossRef] [PubMed]
- Jellison, G.E. Spectroscopic ellipsometry data analysis: Measured versus calculated quantities. Thin Solid Films 1998, 313, 33–39. [Google Scholar] [CrossRef]
- Jayawardhana, S.; Rosa, L.; Juodkazis, S.; Stoddart, P.R. Additional Enhancement of Electric Field in Surface-Enhanced Raman Scattering due to Fresnel Mechanism. Sci. Rep. 2013, 3, 2335. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.N.M.N.; Gibbs, W.E.K.; Juodkazis, S.; Stoddart, P.R. Wavelength and refractive index dependence of the geometrical enhancement in surface-enhanced Raman scattering. J. Raman Spectrosc. 2017, 48, 1182–1189. [Google Scholar] [CrossRef]
- Ming, T.; Chen, H.; Jiang, R.; Li, Q.; Wang, J. Plasmon-Controlled Fluorescence: Beyond the Intensity Enhancement. J. Phys. Chem. Lett. 2012, 3, 191–202. [Google Scholar] [CrossRef]
- Liaw, J.-W.; Tsai, H.-Y.; Huang, C.-H. Size-Dependent Surface Enhanced Fluorescence of Gold Nanorod: Enhancement or Quenching. Plasmonics 2012, 7, 543–553. [Google Scholar] [CrossRef]
- Song, M.; Chen, G.X.; Liu, Y.; Wu, E.; Wu, B.T.; Zeng, H.P. Polarization properties of surface plasmon enhanced photoluminescence from a single Ag nanowire. Opt. Express 2012, 20, 22290–22297. [Google Scholar] [CrossRef] [PubMed]
- Šubr, M.; Petr, M.; Kylián, O.; Kratochvíl, J.; Procházka, M. Large-scale Ag nanoislands stabilized by a magnetron-sputtered polytetrafluoroethylene film as substrates for highly sensitive and reproducible surface-enhanced Raman scattering (SERS). J. Mater. Chem. C 2015, 3, 11478–11485. [Google Scholar] [CrossRef]
- Le Ru, E.C.; Galloway, C.; Etchegoin, P.G. On the connection between optical absorption/extinction and SERS enhancements. Phys. Chem. Chem. Phys. 2006, 8, 3083–3087. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, S.L.; Sharma, B.; Blaber, M.G.; Henry, A.I.; Valley, N.; Freeman, R.G.; Natan, M.J.; Schatz, G.C.; Van Duyne, R.P. Structure Enhancement Factor Relationships in Single Gold Nanoantennas by Surface-Enhanced Raman Excitation Spectroscopy. J. Am. Chem. Soc. 2013, 135, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua, J.; Bryant, G.W.; Richter, L.J.; de Abajo, F.J.G.; Kelley, B.K.; Mallouk, T. Optical properties of coupled metallic nanorods for field-enhanced spectroscopy. Phys. Rev. B 2005, 71, 235420. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Chaney, S.B.; Zhang, Z.Y. Absorbance spectra of aligned Ag nanorod arrays prepared by oblique angle deposition. J. Appl. Phys. 2006, 100, 063527. [Google Scholar] [CrossRef]
- Baumberg, J.J.; Kelf, T.A.; Sugawara, Y.; Cintra, S.; Abdelsalam, M.E.; Bartlett, P.N.; Russell, A.E. Angle-Resolved Surface-Enhanced Raman Scattering on Metallic Nanostructured Plasmonic Crystals. Nano Lett. 2005, 5, 2262–2267. [Google Scholar] [CrossRef] [PubMed]



















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šubr, M.; Procházka, M. Polarization- and Angular-Resolved Optical Response of Molecules on Anisotropic Plasmonic Nanostructures. Nanomaterials 2018, 8, 418. https://doi.org/10.3390/nano8060418
Šubr M, Procházka M. Polarization- and Angular-Resolved Optical Response of Molecules on Anisotropic Plasmonic Nanostructures. Nanomaterials. 2018; 8(6):418. https://doi.org/10.3390/nano8060418
Chicago/Turabian StyleŠubr, Martin, and Marek Procházka. 2018. "Polarization- and Angular-Resolved Optical Response of Molecules on Anisotropic Plasmonic Nanostructures" Nanomaterials 8, no. 6: 418. https://doi.org/10.3390/nano8060418
APA StyleŠubr, M., & Procházka, M. (2018). Polarization- and Angular-Resolved Optical Response of Molecules on Anisotropic Plasmonic Nanostructures. Nanomaterials, 8(6), 418. https://doi.org/10.3390/nano8060418