Titanium Dioxide Nanotubes as Model Systems for Electrosorption Studies

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Fabrication of Titanium Dioxide Nanotube Electrodes
2.2. Electrochemical Tests
2.3. Electrosorption Capacity
3. Results
3.1. Surface Architecture of Anodized TiO2 Electrodes
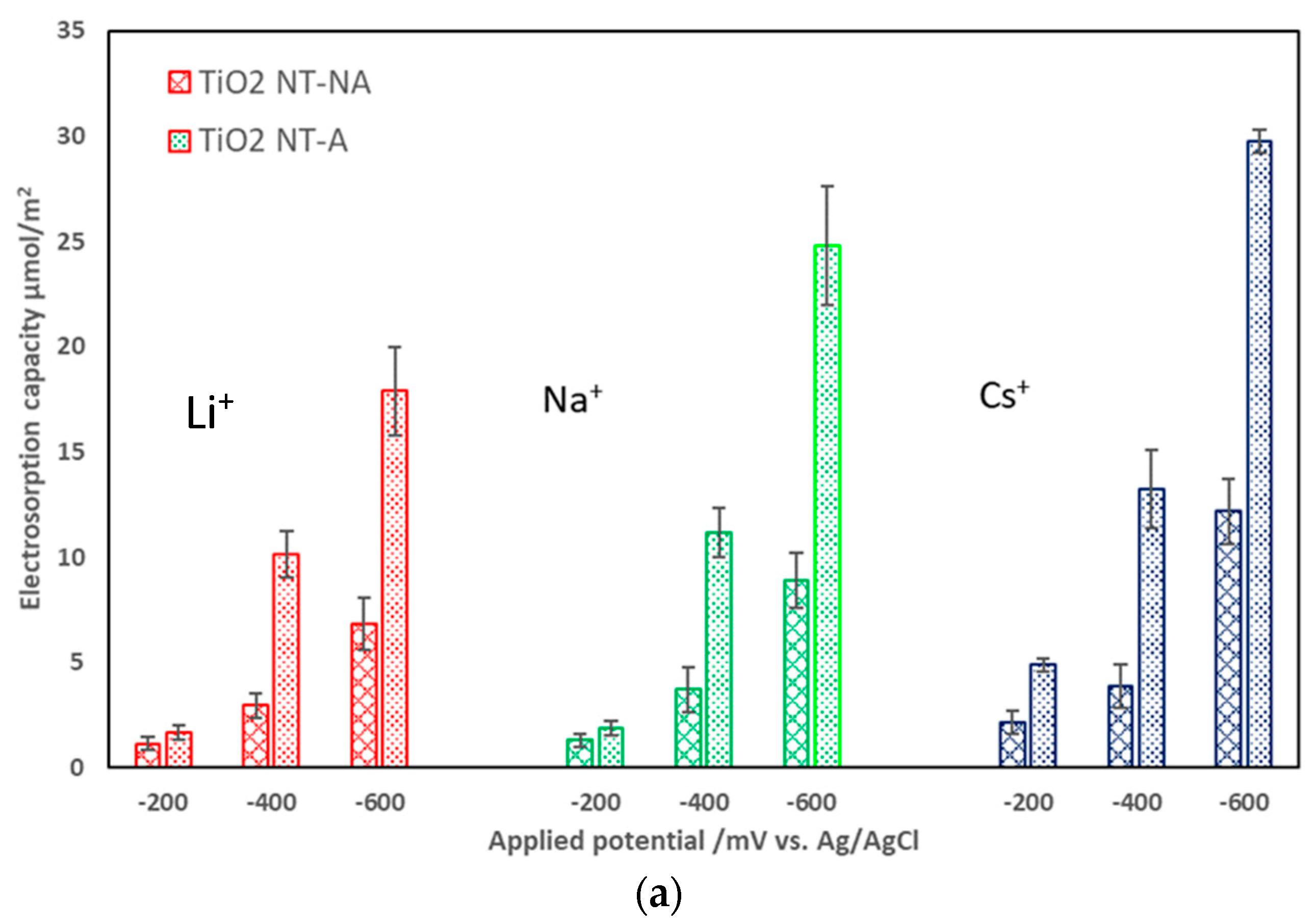
3.2. Effects of Electrode Crystalline Structure and Ionic Strength on Electrosorption
3.3. Charging Dynamics Dependence on Ion and Crystalline Structure
3.4. Electrochemically Active Surface Area
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, C.; He, D.; Ma, J.; Tang, W.; Waite, T.D. Faradaic reactions in capacitive deionization (CDI)-problems and possibilities: A review. Water Res. 2018, 128, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.U.; Yan, T.; Shi, L.; Zhang, D. Improved Capacitive Deionization by Using 3D Intercalated Graphene Sheet-Sphere Nanocomposite Architectures. Environ. Sci. Nano 2018, 5, 980–991. [Google Scholar] [CrossRef]
- Yu, X.; Ma, H.; Du, W.; Qu, L.; Li, C.; Shi, G. Robust graphene composite films for multifunctional electrochemical capacitors with an ultrawide range of areal mass loading toward high-rate frequency response and ultrahigh specific capacitance. Energy Environ. Sci. 2018, 11, 559–565. [Google Scholar]
- Wang, Z.; Tan, Y.; Yang, Y.; Zhao, X.; Liu, Y.; Niu, L.; Tichnell, B.; Kong, L.; Kang, L.; Liu, Z. Pomelo peels-derived porous activated carbon microsheets dual-doped with nitrogen and phosphorus for high performance electrochemical capacitors. J. Power Sources 2018, 378, 499–510. [Google Scholar] [CrossRef]
- Foo, K.; Hameed, B. A short review of activated carbon assisted electrosorption process: An overview, current stage and future prospects. J. Hazard. Mater. 2009, 170, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Beden, B.; Morin, M.-C.; Hahn, F.; Lamy, C. “In situ” analysis by infrared reflectance spectroscopy of the adsorbed species resulting from the electrosorption of ethanol on platinum in acid medium. J. Electroanal. Chem. Interfacial Electrochem. 1987, 229, 353–366. [Google Scholar] [CrossRef]
- Sun, Y.; Zeng, W.; Sun, H.; Luo, S.; Chen, D.; Chan, V.; Liao, K. Inorganic/polymer-graphene hybrid gel as versatile electrochemical platform for electrochemical capacitor and biosensor. Carbon 2018, 132, 589–597. [Google Scholar] [CrossRef]
- Liu, P.; Yan, T.; Shi, L.; Park, H.S.; Chen, X.; Zhao, Z.; Zhang, D. Graphene-based materials for capacitive deionization. J. Mater. Chem. A 2017, 5, 13907–13943. [Google Scholar] [CrossRef]
- Suss, M.; Porada, S.; Sun, X.; Biesheuvel, P.; Yoon, J.; Presser, V. Water desalination via capacitive deionization: What is it and what can we expect from it? Energy Environ. Sci. 2015, 8, 2296–2319. [Google Scholar] [CrossRef]
- Liu, Y.; Nie, C.; Liu, X.; Xu, X.; Sun, Z.; Pan, L. Review on carbon-based composite materials for capacitive deionization. RSC Adv. 2015, 5, 15205–15225. [Google Scholar] [CrossRef]
- Fic, K.; He, M.; Berg, E.J.; Novák, P.; Frackowiak, E. Comparative operando study of degradation mechanisms in carbon-based electrochemical capacitors with Li2SO4 and LiNO3 electrolytes. Carbon 2017, 120, 281–293. [Google Scholar] [CrossRef]
- Przygocki, P.; Abbas, Q.; Béguin, F. Capacitance enhancement of hybrid electrochemical capacitor with asymmetric carbon electrodes configuration in neutral aqueous electrolyte. Electrochim. Acta 2018, 296, 640–648. [Google Scholar] [CrossRef]
- Rasines, G.; Lavela, P.; Macías, C.; Zafra, M.C.; Tirado, J.L.; Parra, J.B.; Ania, C.O. N-doped monolithic carbon aerogel electrodes with optimized features for the electrosorption of ions. Carbon 2015, 83, 262–274. [Google Scholar] [CrossRef]
- Rasines, G.; Lavela, P.; Macías, C.; Zafra, M.; Tirado, J.; Ania, C. On the use of carbon black loaded nitrogen-doped carbon aerogel for the electrosorption of sodium chloride from saline water. Electrochim. Acta 2015, 170, 154–163. [Google Scholar] [CrossRef]
- Li, K.; Guo, D.; Lin, F.; Wei, Y.; Liu, W.; Kong, Y. Electrosorption of copper ions by poly (m-phenylenediamine)/reduced graphene oxide synthesized via a one-step in situ redox strategy. Electrochim. Acta 2015, 166, 47–53. [Google Scholar] [CrossRef]
- Kalluri, R.; Biener, M.; Suss, M.; Merrill, M.; Stadermann, M.; Santiago, J.; Baumann, T.; Biener, J.; Striolo, A. Unraveling the potential and pore-size dependent capacitance of slit-shaped graphitic carbon pores in aqueous electrolytes. Phys. Chem. Chem. Phys. 2013, 15, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Tsouris, C.; Mayes, R.; Kiggans, J.; Sharma, K.; Yiacoumi, S.; DePaoli, D.; Dai, S. Mesoporous carbon for capacitive deionization of saline water. Environ. Sci. Technol. 2011, 45, 10243–10249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, H.; Wu, C.; Wang, Y.; Li, W. A study of electrosorption selectivity of anions by activated carbon electrodes in capacitive deionization. Desalination 2015, 369, 46–50. [Google Scholar] [CrossRef]
- Tighineanu, A.; Ruff, T.; Albu, S.; Hahn, R.; Schmuki, P. Conductivity of TiO2 nanotubes: Influence of annealing time and temperature. Chem. Phys. Lett. 2010, 494, 260–263. [Google Scholar] [CrossRef]
- Nightingale Jr, E. Phenomenological theory of ion solvation. Effective radii of hydrated ions. J. Phys. Chem. 1959, 63, 1381–1387. [Google Scholar] [CrossRef]
- Richter, C.; Schmuttenmaer, C.A. Exciton-like trap states limit electron mobility in TiO2 nanotubes. Nat. Nanotechnol. 2010, 5, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Close, T.; Pustulka, S.; Pedu, S.; Xue, Y.; Richter, C.; Taboada-Serrano, P. Electrosorption of monovalent alkaline metal ions onto highly ordered mesoporous titanium dioxide nanotube electrodes. Electrochim. Acta 2017, 231, 632–640. [Google Scholar] [CrossRef]
- McCrory, C.C.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Benck, J.D.; Chen, Z.; Kuritzky, L.Y.; Forman, A.J.; Jaramillo, T.F. Amorphous molybdenum sulfide catalysts for electrochemical hydrogen production: Insights into the origin of their catalytic activity. ACS Catal. 2012, 2, 1916–1923. [Google Scholar] [CrossRef]
- Bockris, J.M.; Srinivasan, S. Electrode kinetic aspects of electrochemical energy conversion. J. Electroanal. Chem. 1966, 11, 350–389. [Google Scholar] [CrossRef]
- Boggio, R.; Carugati, A.; Trasatti, S. Electrochemical surface properties of Co3O4 electrodes. J. Appl. Electrochem. 1987, 17, 828–840. [Google Scholar] [CrossRef]
- Orazem, M.E.; Tribollet, B. Electrochemical Impedance Spectroscopy; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 233–237. [Google Scholar]
- Brug, G.; Van Den Eeden, A.; Sluyters-Rehbach, M.; Sluyters, J. The analysis of electrode impedances complicated by the presence of a constant phase element. J. Electroanal. Chem. Interfacial Electrochem. 1984, 176, 275–295. [Google Scholar] [CrossRef]
- Taboada-Serrano, P.; Yiacoumi, S.; Tsouris, C. Behavior of mixtures of symmetric and asymmetric electrolytes near discretely charged planar surfaces: A Monte Carlo study. J. Chem. Phys. 2005, 123, 054703. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.-H. Monte Carlo simulation of electrical double-layer formation from mixtures of electrolytes inside nanopores. J. Chem. Phys. 2008, 128, 044705. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.-H.; Taboada-Serrano, P.; Yiacoumi, S.; Tsouris, C. Electrosorption selectivity of ions from mixtures of electrolytes inside nanopores. J. Chem. Phys. 2008, 129, 224703. [Google Scholar] [CrossRef] [PubMed]
- Ney, E.M.; Hou, C.-H.; Taboada-Serrano, P. Calculation of electrical double layer potential profiles in nanopores from grand canonical Monte Carlo simulations. J. Chem. Eng. Data 2018. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Target Ion | Rct/Ω | EASA-CV/m2/g | EASA-EIS/m2/g | EASA-CV/Specific Area/% | EASA-EIS/Specific Area/% |
|---|---|---|---|---|---|---|
| TiO2 NT-A | Li+ | 10.3 | 14.1 | 13.7 | 44.7 | 43.6 |
| TiO2 NT-NA | Li+ | 13.9 | 21.1 | 20.6 | 67.1 | 65.7 |
| TiO2 NT-A | Na+ | 7.6 | 12.3 | 11.7 | 39.3 | 37.7 |
| TiO2 NT-NA | Na+ | 9.9 | 17.8 | 16.8 | 56.8 | 57.7 |
| TiO2 NT-A | Cs+ | 3.1 | 8.9 | 9.2 | 27.6 | 29.7 |
| TiO2 NT-NA | Cs+ | 6.2 | 16.2 | 15.9 | 51.5 | 50.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Pustulka, S.; Pedu, S.; Close, T.; Xue, Y.; Richter, C.; Taboada-Serrano, P. Titanium Dioxide Nanotubes as Model Systems for Electrosorption Studies. Nanomaterials 2018, 8, 404. https://doi.org/10.3390/nano8060404
Li X, Pustulka S, Pedu S, Close T, Xue Y, Richter C, Taboada-Serrano P. Titanium Dioxide Nanotubes as Model Systems for Electrosorption Studies. Nanomaterials. 2018; 8(6):404. https://doi.org/10.3390/nano8060404
Chicago/Turabian StyleLi, Xian, Samantha Pustulka, Scott Pedu, Thomas Close, Yuan Xue, Christiaan Richter, and Patricia Taboada-Serrano. 2018. "Titanium Dioxide Nanotubes as Model Systems for Electrosorption Studies" Nanomaterials 8, no. 6: 404. https://doi.org/10.3390/nano8060404
APA StyleLi, X., Pustulka, S., Pedu, S., Close, T., Xue, Y., Richter, C., & Taboada-Serrano, P. (2018). Titanium Dioxide Nanotubes as Model Systems for Electrosorption Studies. Nanomaterials, 8(6), 404. https://doi.org/10.3390/nano8060404