Multifunctional Platform Based on Electroactive Polymers and Silica Nanoparticles for Tissue Engineering Applications

 ,
,  ,
,  ,
,  ,
, 
 and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Silica Nanoparticles
2.2.1. Preparation of the Silica Nanoparticles
2.2.2. Characterization of the SiNPs
2.3. Nanocomposite Samples
2.3.1. Preparation of the SiNPs/PVDF Nanocomposites
2.3.2. Characterization of the Nanocomposite Samples
2.4. Cell Culture Experiments
2.4.1. Sample Sterilization
2.4.2. Cell Culture
2.4.3. Cytotoxicity Assay by the Indirect Contact
2.4.4. Direct Contact and Proliferation
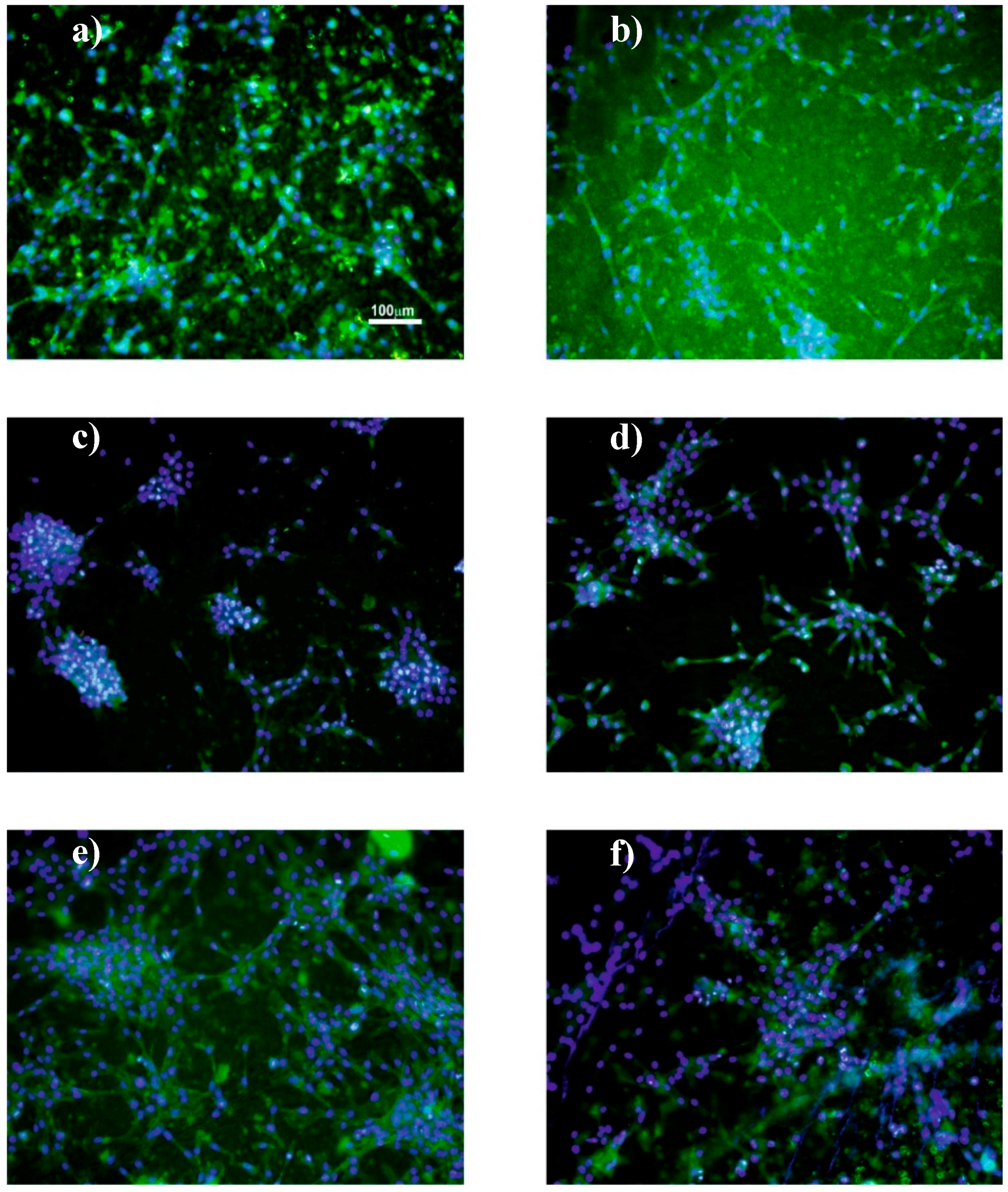
2.4.5. Immunofluorescence Staining
3. Results and Discussion
3.1. Silica Nanoparticles
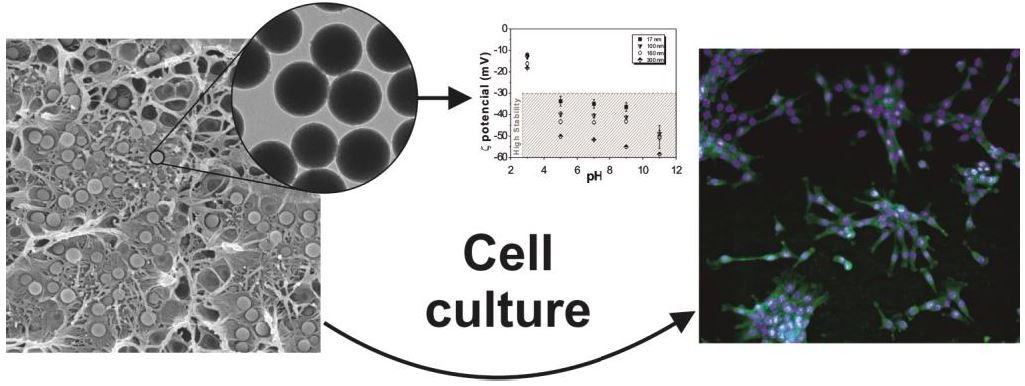
3.1.1. Morphology and Size of the Nanoparticles
3.1.2. Surface Charge of the Nanoparticles
3.2. SiNPs/PVDF Nanocomposite Samples
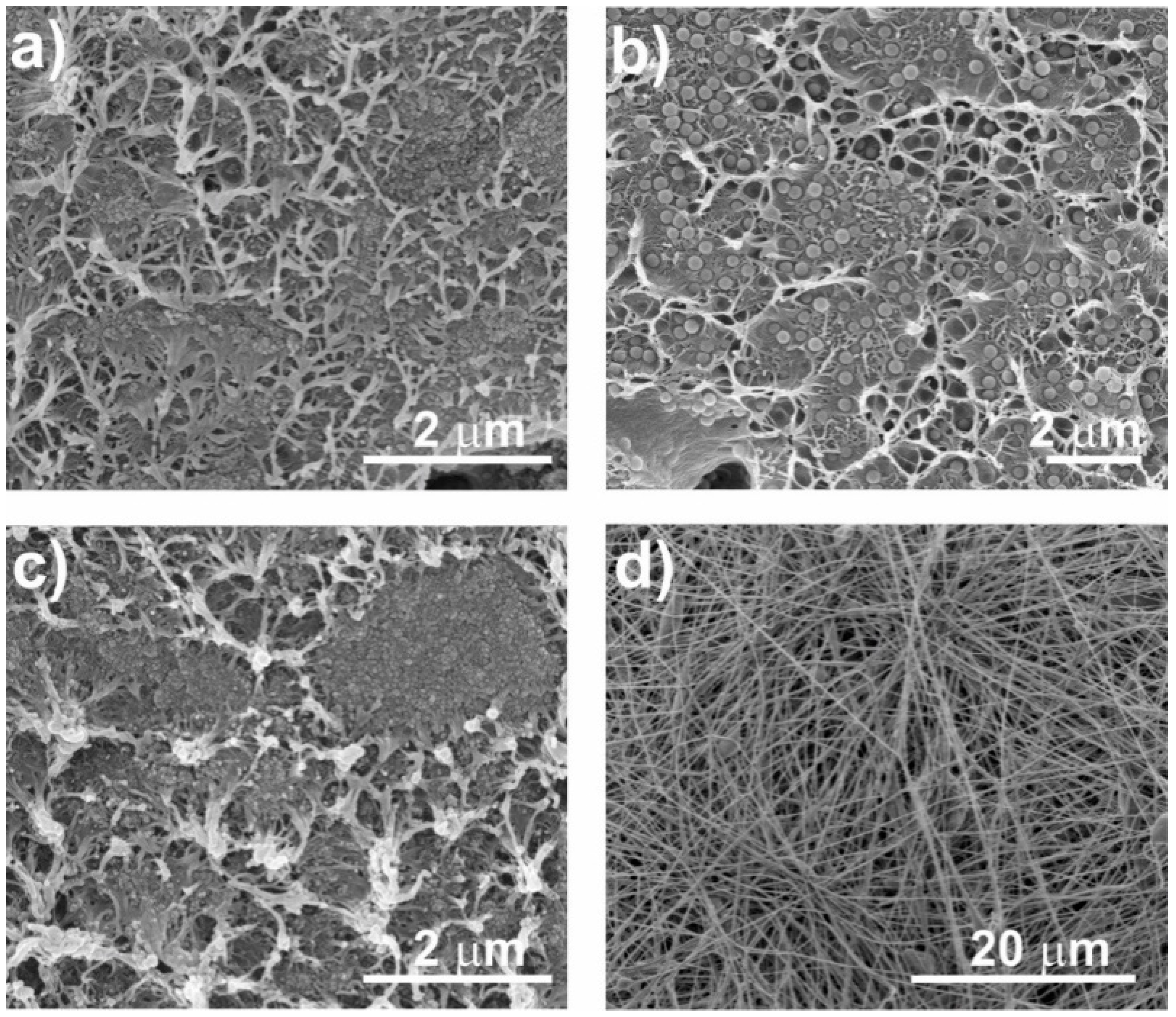
3.2.1. Morphology of the Nanocomposites
3.2.2. Confocal Fluorescence Microscopy of the Nanocomposites
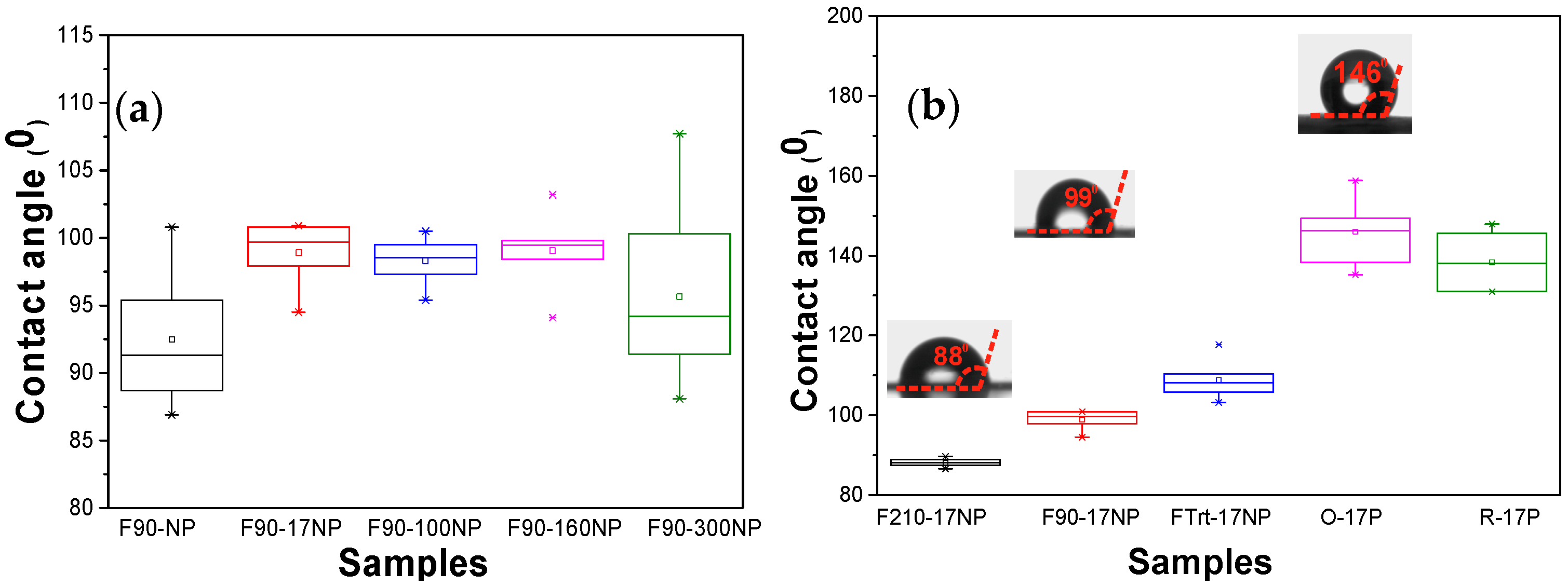
3.2.3. Wettability of the Nanocomposites
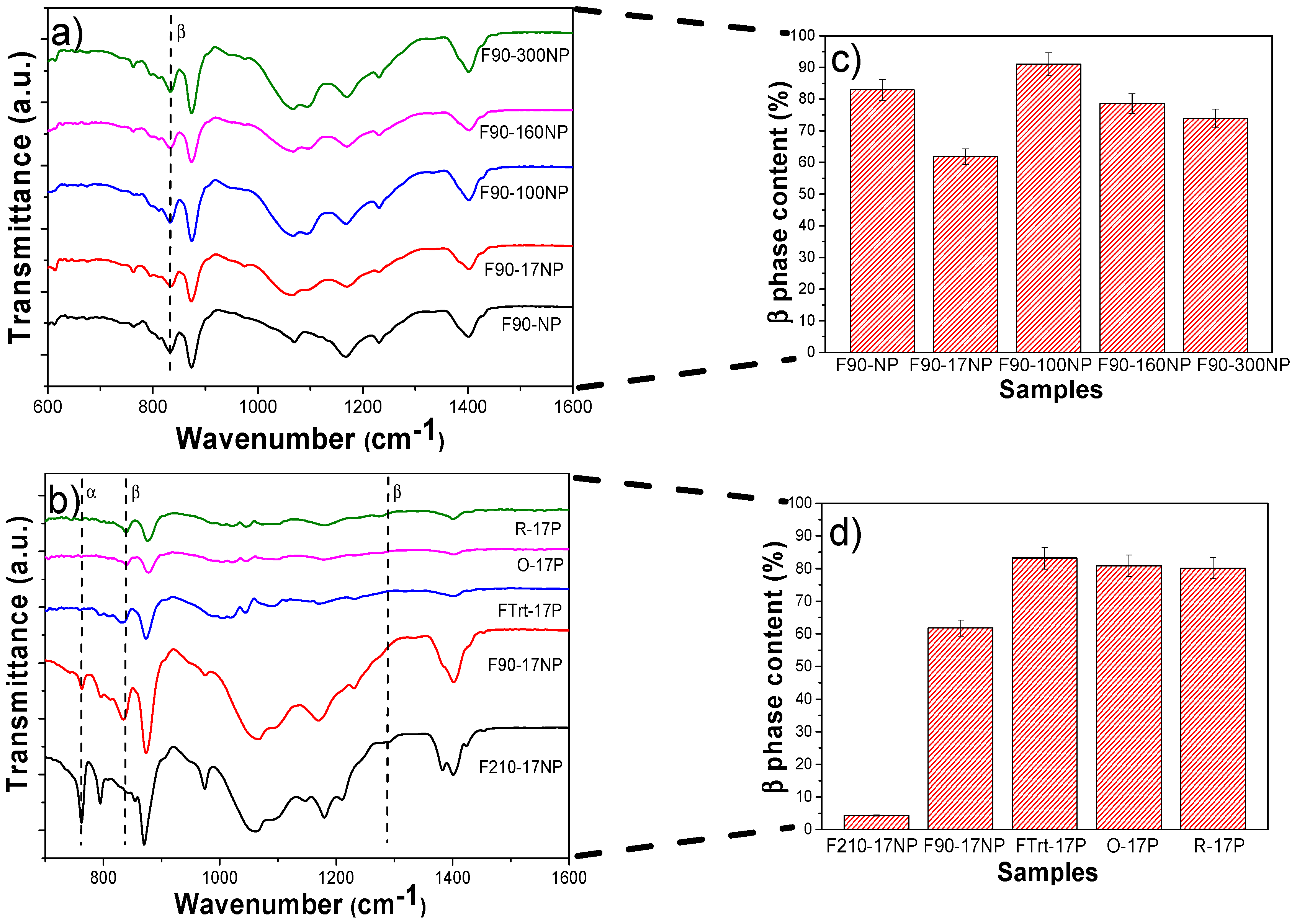
3.2.4. Structural Properties and Electroactive Phase Content of the Nanocomposites
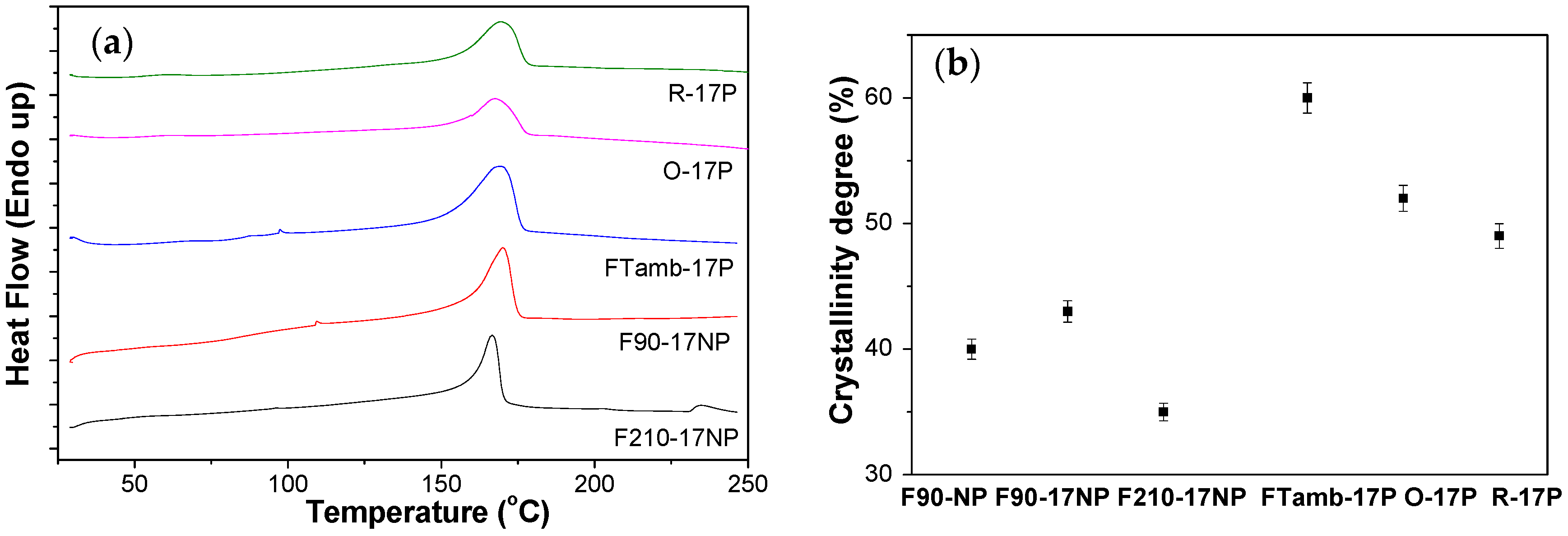
3.2.5. Thermal Behaviour of the Nanocomposites
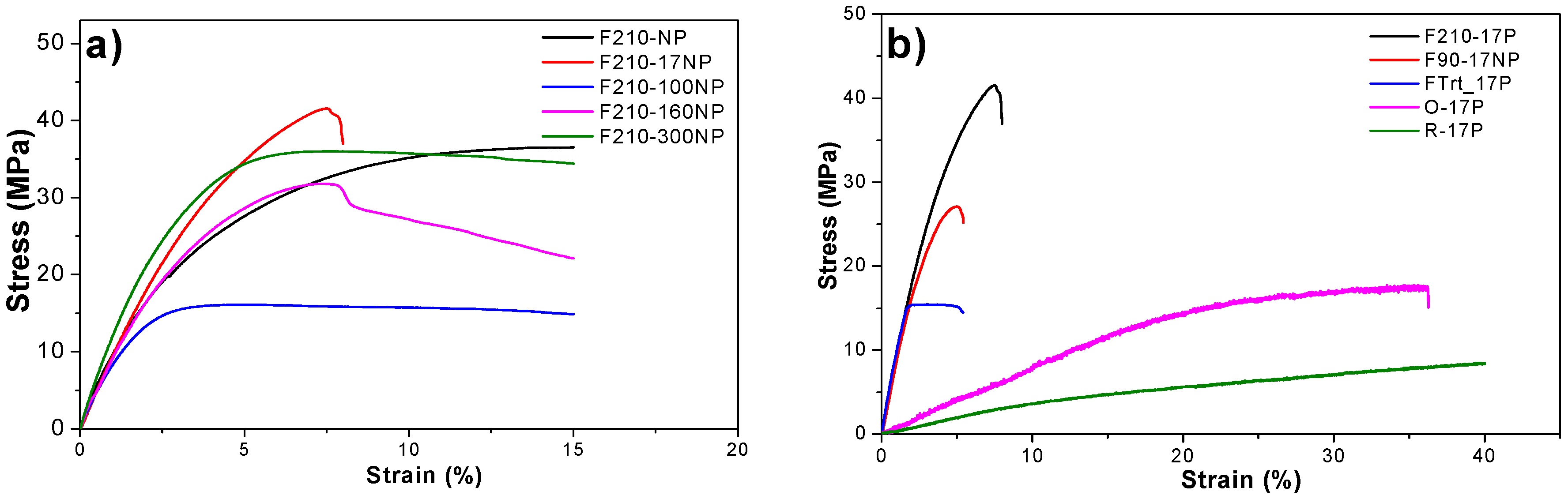
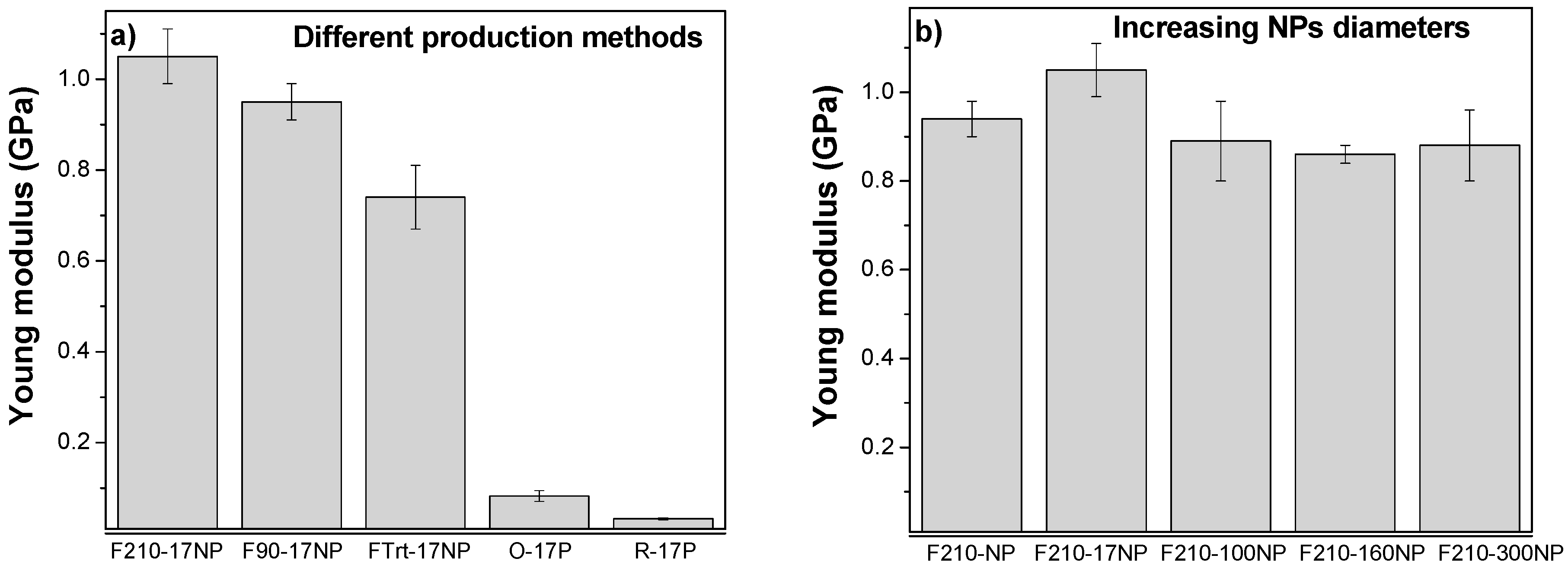
3.2.6. Mechanical Properties of the Nanocomposites
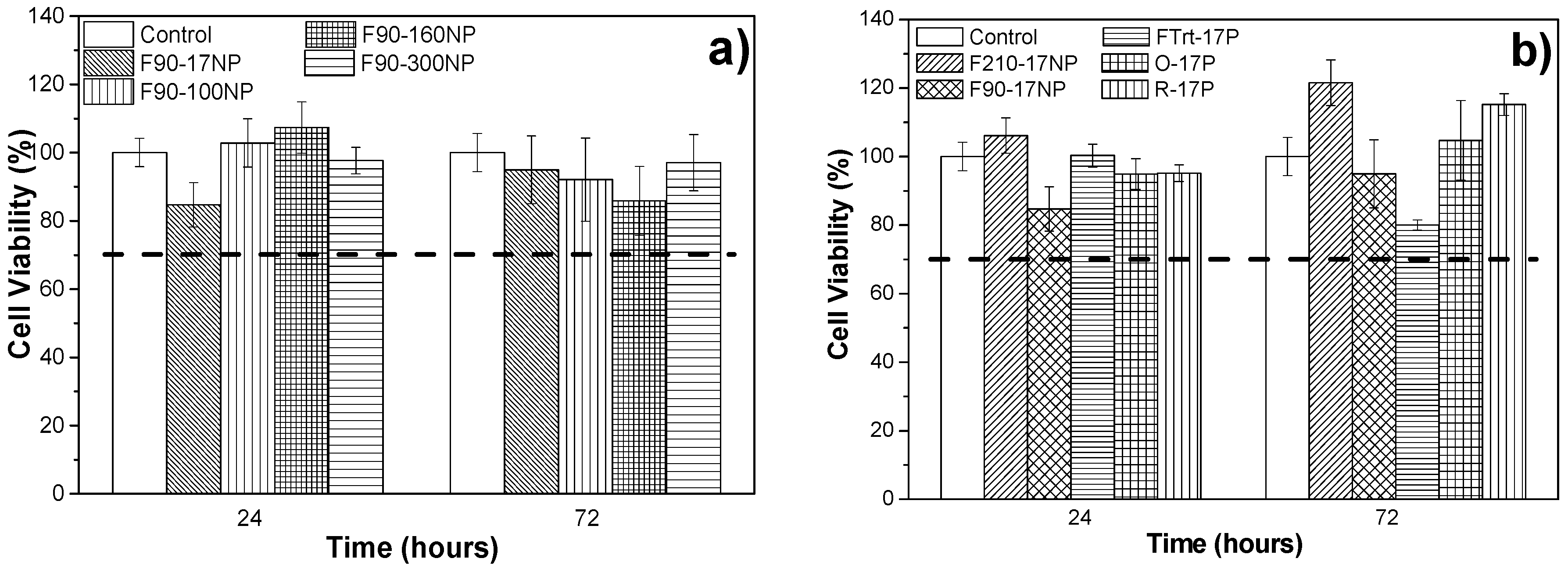
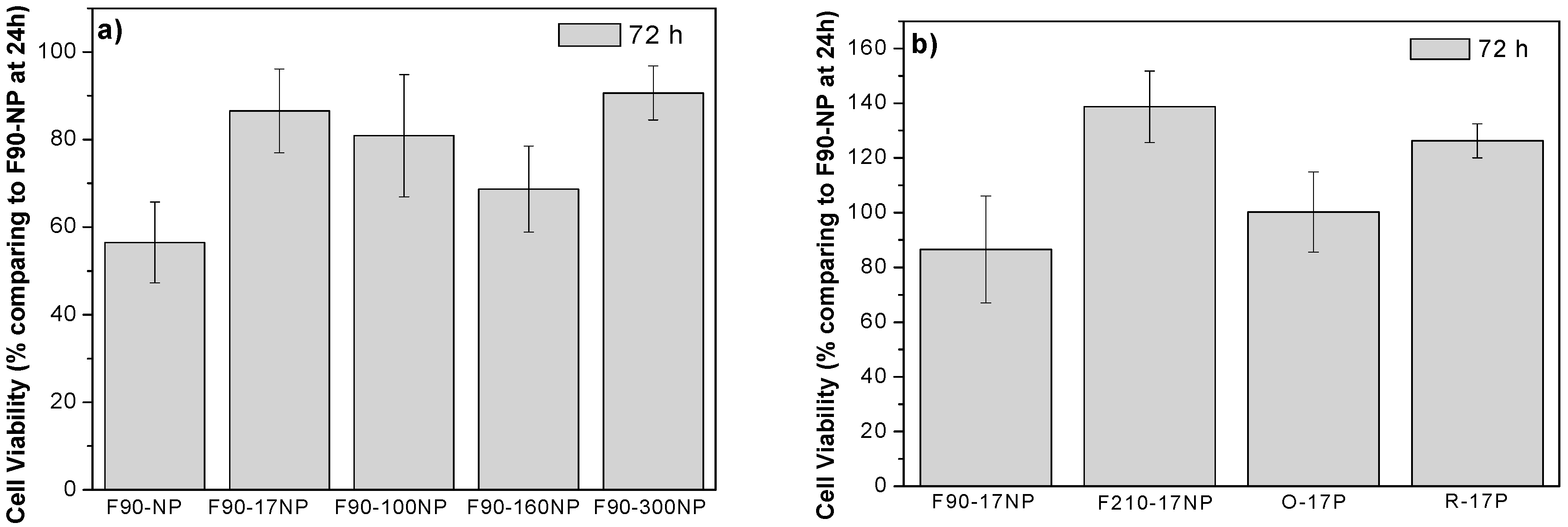
3.3. Cell Culture Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Camargo, P.H.C.; Satyanarayana, K.G.; Wypych, F. Nanocomposites: Synthesis, structure, properties and new application opportunities. Mater. Res.-Ibero-Am. J. Mater. 2009, 12, 1–39. [Google Scholar] [CrossRef]
- Muller, K.; Bugnicourt, E.; Latorre, M.; Jorda, M.; Sanz, Y.E.; Lagaron, J.M.; Miesbauer, O.; Bianchin, A.; Hankin, S.; Bolz, U.; et al. Review on the processing and properties of polymer nanocomposites and nanocoatings and their applications in the packaging, automotive and solar energy fields. Nanomaterials 2017, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Raji, M.; Mekhzoum, M.E.M.; Rodrigue, D.; Qaiss, A.E.K.; Bouhfid, R. Effect of silane functionalization on properties of polypropylene/clay nanocomposites. Compos. B Eng. 2018, 146, 106–115. [Google Scholar] [CrossRef]
- Ribeiro, S.; Costa, P.; Ribeiro, C.; Sencadas, V.; Botelho, G.; Lanceros-Méndez, S. Electrospun styrene-butadiene-styrene elastomer copolymers for tissue engineering applications: Effect of butadiene/styrene ratio, block structure, hydrogenation and carbon nanotube loading on physical properties and cytotoxicity. Compos. B Eng. 2014, 67, 30–38. [Google Scholar] [CrossRef]
- Narayanan, K.B.; Han, S.S. Dual-crosslinked poly(vinyl alcohol)/sodium alginate/silver nanocomposite beads—A promising antimicrobial material. Food Chem. 2017, 234, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Sencadas, V.; Correia, D.M.; Lanceros-Mendez, S. Piezoelectric polymers as biomaterials for tissue engineering applications. Colloids Surf. B 2015, 136, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.F.; Correia, D.M.; Ribeiro, C.; Fernandes, M.M.; Lanceros-Méndez, S. Fluorinated polymers as smart materials for advanced biomedical applications. Polymers 2018, 10, 161. [Google Scholar] [CrossRef]
- Maciel, M.M.; Ribeiro, S.; Ribeiro, C.; Francesko, A.; Maceiras, A.; Vilas, J.L.; Lanceros-Méndez, S. Relation between fiber orientation and mechanical properties of nano-engineered poly(vinylidene fluoride) electrospun composite fiber mats. Compos. B Eng. 2018, 139, 146–154. [Google Scholar] [CrossRef]
- Cardoso, V.F.; Francesko, A.; Ribeiro, C.; Bañobre-López, M.; Martins, P.; Lanceros-Mendez, S. Advances in magnetic nanoparticles for biomedical applications. Adv. Healthc. Mater. 2017, 7, 1700845. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pallon, L.K.H.; Pourrahimi, A.M.; Zhang, P.; Diaz, A.; Holler, M.; Schneider, K.; Olsson, R.T.; Hedenqvist, M.S.; Yu, S.; et al. Cavitation in strained polyethylene/aluminium oxide nanocomposites. Eur. Polym. J. 2017, 87, 255–265. [Google Scholar] [CrossRef]
- Xu, D.; Cheng, X.; Banerjee, S.; Huang, S. Dielectric and electromechanical properties of modified cement/polymer based 1–3 connectivity piezoelectric composites containing inorganic fillers. Compos. Sci. Technol. 2015, 114, 72–78. [Google Scholar] [CrossRef]
- Liu, D.; Hoang, A.T.; Pourrahimi, A.M.; Pallon, L.H.; Nilsson, F.; Gubanski, S.M.; Olsson, R.T.; Hedenqvist, M.S.; Gedde, U.W. Influence of nanoparticle surface coating on electrical conductivity of LDPE/Al2O3 nanocomposites for HVDC cable insulations. IEEE Trans. Dielectr. Electr. Insul. 2017, 24, 1396–1404. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.Y.; Feng, Y.; Yuan, Z.Y.; Su, B.L. One-dimensional metal oxide nanotubes, nanowires, nanoribbons, and nanorods: Synthesis, characterizations, properties and applications. Crit. Rev. Solid State Mater. Sci. 2012, 37, 1–74. [Google Scholar] [CrossRef]
- Chen, L.; Jia, Z.; Tang, Y.; Wu, L.; Luo, Y.; Jia, D. Novel functional silica nanoparticles for rubber vulcanization and reinforcement. Compos. Sci. Technol. 2017, 144, 11–17. [Google Scholar] [CrossRef]
- Meinardi, F.; Ehrenberg, S.; Dhamo, L.; Carulli, F.; Mauri, M.; Bruni, F.; Simonutti, R.; Kortshagen, U.; Brovelli, S. Highly efficient luminescent solar concentrators based on earth-abundant indirect-band gap silicon quantum dots. Nat. Photonics 2017, 11, 177–185. [Google Scholar] [CrossRef]
- Bergren, M.R.; Makarov, N.S.; Ramasamy, K.; Jackson, A.; Guglielmetti, R.; McDaniel, H. High-Performance CuInS2 Quantum Dot Laminated Glass Luminescent Solar Concentrators for Windows. Energy Lett. 2018, 3, 520–525. [Google Scholar] [CrossRef]
- Marinins, A.; Shafagh, R.Z.; van der Wijngaart, W.; Haraldsson, T.; Linnros, L.; Veinot, J.G.C.; Popov, S.; Sychugov, I. Light-Converting Polymer/Si Nanocrystal Composites with Stable 60–70% Quantum Efficiency and Their Glass Laminates. Appl. Mater. Interfaces 2017, 9, 30267–30272. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, T.; Baleizao, C.; Farinha, J.P.S. Functional films from silica/polymer nanoparticles. Materials 2014, 7, 3881–3900. [Google Scholar] [CrossRef] [PubMed]
- Asefa, T.; Tao, Z.M. Biocompatibility of mesoporous silica nanoparticles. Chem. Res. Toxicol. 2012, 25, 2265–2284. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.S.; Ribeiro, T.; Fernandes, F.; Farinha, J.P.S.; Baleizao, C. Intrinsically fluorescent silica nanocontainers: A promising theranostic platform. Microsc. Microanal. 2013, 19, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Liu, C.; Li, X.; Liu, J.; Di, D.; Zhang, Y.; Zhao, Q.; Wang, S. Fluorescent carbon dot modified mesoporous silica nanocarriers for redox-responsive controlled drug delivery and bioimaging. J. Colloid Interface Sci. 2016, 483, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Ow, H.; Wiesner, U. Fluorescent core-shell silica nanoparticles: Towards “Lab on a particle” Architectures for nanobiotechnology. Chem. Soc. Rev. 2006, 35, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Hableel, G.; Zhao, E.R.; Jokerst, J.V. Multifunctional nanomedicine with silica: Role of silica in nanoparticles for theranostic, imaging, and drug monitoring. J. Colloid Interface Sci. 2018, 521, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Estevez, M.C.; Smith, J.E.; Wang, K.; He, X.; Wang, L.; Tan, W. Dye-doped nanoparticles for bioanalysis. Nano Today 2007, 2, 44–50. [Google Scholar] [CrossRef]
- Cardoso, V.F.; Irusta, S.; Navascues, N.; Lanceros-Mendez, S. Comparative study of sol-gel methods for the facile synthesis of tailored magnetic silica spheres. Mater. Res. Express 2016, 3, 075402. [Google Scholar] [CrossRef]
- Slowing, I.I.; Vivero-Escoto, J.L.; Trewyn, B.G.; Lin, V.S.Y. Mesoporous silica nanoparticles: Structural design and applications. J. Mater. Chem. 2010, 20, 7924–7937. [Google Scholar] [CrossRef]
- Liong, M.; Lu, J.; Kovochich, M.; Xia, T.; Ruehm, S.G.; Nel, A.E.; Tamanoi, F.; Zink, J.I. Multifunctional inorganic nanoparticles for imaging, targeting, and drug delivery. ACS Nano 2008, 2, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Bharti, C.; Nagaich, U.; Pal, A.K.; Gulati, N. Mesoporous silica nanoparticles in target drug delivery system: A review. Int. J. Pharm. Investig. 2015, 5, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Parssinen, J.; Hammaren, H.; Rahikainen, R.; Sencadas, V.; Ribeiro, C.; Vanhatupa, S.; Miettinen, S.; Lanceros-Mendez, S.; Hytonen, V.P. Enhancement of adhesion and promotion of osteogenic differentiation of human adipose stem cells by poled electroactive poly(vinylidene fluoride). J. Biomed. Mater. Res. A 2015, 103, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.R.; Meissner, G. Sodium-calcium ion-exchange in skeletal-muscle sarcolemmal vesicles. J. Membr. Biol. 1982, 69, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Brito-Pereira, R.; Ribeiro, C.; Lanceros-Mendez, S.; Martins, P. Magnetoelectric response on Terfenol-D/P(VDF-TrFE) two-phase composites. Compos. B Eng. 2017, 120, 97–102. [Google Scholar] [CrossRef]
- Ribeiro, C.; Correia, V.; Martins, P.; Gama, F.M.; Lanceros-Mendez, S. Proving the suitability of magnetoelectric stimuli for tissue engineering applications. Colloids Surf. B 2016, 140, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Correia, D.M.; Ribeiro, C.; Lanceros-Méndez, S. Electrospun polymeric smart materials for tissue engineering applications. In Electrospun Biomaterials and Related Technologies; Almodovar, J., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 251–282. [Google Scholar]
- Cardoso, V.F.; Ribeiro, C.; Lanceros-Mendez, S. Metamorphic biomaterials. In Bioinspired Materials for Medical Applications; Woodhead Publishing: Cambridge, UK, 2016; pp. 69–99. [Google Scholar]
- Lee, Y.S.; Collins, G.; Arinzeh, T.L. Neurite extension of primary neurons on electrospun piezoelectric scaffolds. Acta Biomater. 2011, 7, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Moreira, S.; Correia, V.; Sencadas, V.; Rocha, J.G.; Gama, F.M.; Ribelles, J.L.G.; Lanceros-Mendez, S. Enhanced proliferation of pre-osteoblastic cells by dynamic piezoelectric stimulation. RSC Adv. 2012, 2, 11504–11509. [Google Scholar] [CrossRef]
- Ribeiro, C.; Parssinen, J.; Sencadas, V.; Correia, V.; Miettinen, S.; Hytonen, V.P.; Lanceros-Mendez, S. Dynamic piezoelectric stimulation enhances osteogenic differentiation of human adipose stem cells. J. Biomed. Mater. Res. A 2015, 103, 2172–2175. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.M.; Ribeiro, S.; Ribeiro, C.; Sencadas, V.; Gomes, A.C.; Gama, F.M.; Lanceros-Mendez, S. Effect of poling state and morphology of piezoelectric poly(vinylidene fluoride) membranes for skeletal muscle tissue engineering. RSC Adv. 2013, 3, 17938–17944. [Google Scholar] [CrossRef]
- Ribeiro, C.; Costa, C.M.; Correia, D.M.; Nunes-Pereira, J.; Oliveira, J.; Martins, P.; Gonçalves, R.; Cardoso, V.F.; Lanceros-Méndez, S. Electroactive poly(vinylidene fluoride)-based structures for advanced applications. Nat. Protoc. 2018, 13, 681–704. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, T.; Baleizão, C.; Farinha, J.P.S. Synthesis and characterization of perylenediimide labeled core-shell hybrid silica-polymer nanoparticles. J. Phys. Chem. C 2009, 113, 18082–18090. [Google Scholar] [CrossRef]
- Santiago, A.M.; Ribeiro, T.; Rodrigues, A.S.; Ribeiro, B.; Frade, R.F.M.; Baleizão, C.; Farinha, J.P.S. Multifunctional hybrid silica nanoparticles with a fluorescent core and active targeting shell for fluorescence imaging biodiagnostic applications. Eur. J. Inorg. Chem. 2015, 2015, 4579–4587. [Google Scholar] [CrossRef]
- Ribeiro, T.; Raja, S.; Rodrigues, A.S.; Fernandes, F.; Baleizão, C.; Farinha, J.P.S. Nir and visible perylenediimide-silica nanoparticles for laser scanning bioimaging. Dyes Pigm. 2014, 110, 227–234. [Google Scholar] [CrossRef]
- Lopes, A.C.; Ribeiro, C.; Sencadas, V.; Botelho, G.; Lanceros-Méndez, S. Effect of filler content on morphology and physical-chemical characteristics of poly(vinylidene fluoride)/NaY zeolite-filled membranes. J. Mater. Sci. 2014, 49, 3361–3370. [Google Scholar] [CrossRef]
- Martins, P.; Lopes, A.C.; Lanceros-Mendez, S. Electroactive phases of poly(vinylidene fluoride): Determination, processing and applications. Prog. Polym. Sci. 2014, 39, 683–706. [Google Scholar] [CrossRef]
- Stober, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Martins, P.; Caparros, C.; Gonçalves, R.; Martins, P.M.; Benelmekki, M.; Botelho, G.; Lanceros-Mendez, S. Role of nanoparticle surface charge on the nucleation of the electroactive β-poly(vinylidene fluoride) nanocomposites for sensor and actuator applications. J. Phys. Chem. C 2012, 116, 15790–15794. [Google Scholar] [CrossRef]
- Ferreira, J.C.C.; Monteiro, T.S.; Lopes, A.C.; Costa, C.M.; Silva, M.M.; Machado, A.V.; Lanceros-Mendez, S. Variation of the physicochemical and morphological characteristics of solvent casted poly(vinylidene fluoride) along its binary phase diagram with dimethylformamide. J. Non-Cryst. Solids 2015, 412, 16–23. [Google Scholar] [CrossRef]
- Sencadas, V.; Martins, P.; Pitães, A.; Benelmekki, M.; Gómez Ribelles, J.L.; Lanceros-Mendez, S. Influence of ferrite nanoparticle type and content on the crystallization kinetics and electroactive phase nucleation of poly(vinylidene fluoride). Langmuir 2011, 27, 7241–7249. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Sencadas, V.; Ribelles, J.L.G.; Lanceros-Méndez, S. Influence of processing conditions on polymorphism and nanofiber morphology of electroactive poly(vinylidene fluoride) electrospun membranes. Soft Mater. 2010, 8, 274–287. [Google Scholar] [CrossRef]
- Ribeiro, T.; Raja, S.; Rodrigues, A.S.; Fernandes, F.; Farinha, J.P.S.; Baleizao, C. High performance nir fluorescent silica nanoparticles for bioimaging. RSC Adv. 2013, 3, 9171–9174. [Google Scholar] [CrossRef]
- Sebastian, M.S.; Larrea, A.; Goncalves, R.; Alejo, T.; Vilas, J.L.; Sebastian, V.; Martins, P.; Lanceros-Mendez, S. Understanding nucleation of the electroactive beta-phase of poly(vinylidene fluoride) by nanostructures. RSC Adv. 2016, 6, 113007–113015. [Google Scholar] [CrossRef]
- Martins, P.; Costa, C.M.; Lanceros-Mendez, S. Nucleation of electroactive β-phase poly(vinilidene fluoride) with CoFe2O4 and NiFe2O4 nanofillers: A new method for the preparation of multiferroic nanocomposites. Appl. Phys. A 2011, 103, 233–237. [Google Scholar] [CrossRef]
- Costa, C.M.; Rodrigues, L.C.; Sencadas, V.; Silva, M.M.; Lanceros-Méndez, S. Effect of the microsctructure and lithium-ion content in poly[(vinylidene fluoride)-co-trifluoroethylene]/lithium perchlorate trihydrate composite membranes for battery applications. Solid State Ionics 2012, 217, 19–26. [Google Scholar] [CrossRef]
- Zeng, F.; Liu, Y.; Sun, Y. Mechanical properties and fracture behavior of poly(vinylidene fluoride)-polyhedral oligomeric silsesquioxane nanocomposites by nanotensile testing. In Proceedings of the 13th International Conference on Fracture 2013 (ICF 2013), Beijing, China, 6–12 June 2013; pp. 4862–4869. [Google Scholar]
- Xu, Y.L.; Yu, L.Y.; Han, L.F. Polymer-nanoinorganic particles composite membranes: A brief overview. Front. Chem. Eng. China 2009, 3, 318–329. [Google Scholar] [CrossRef]
- Beck, G.R.; Ha, S.W.; Camalier, C.E.; Yamaguchi, M.; Li, Y.; Lee, J.K.; Weitzmann, M.N. Bioactive silica-based nanoparticles stimulate bone-forming osteoblasts, suppress bone-resorbing osteoclasts, and enhance bone mineral density in vivo. Nanomedicine 2012, 8, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, X.X.; Wang, K.M.; He, C.M.; Shi, H.; Jian, L.X. Biocompatible silica nanoparticles-insulin conjugates for mesenchymal stem cell adipogenic differentiation. Bioconjug. Chem. 2010, 21, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Poussard, S.; Decossas, M.; Le Bihan, O.; Mornet, S.; Naudin, G.; Lambert, O. Internalization and fate of silica nanoparticles in c2c12 skeletal muscle cells: Evidence of a beneficial effect on myoblast fusion. Int. J. Nanomed. 2015, 10, 1479–1492. [Google Scholar]
- Nunes-Pereira, J.; Ribeiro, S.; Ribeiro, C.; Gombek, C.J.; Gama, F.M.; Gomes, A.C.; Patterson, D.A.; Lanceros-Mendez, S. Poly(vinylidene fluoride) and copolymers as porous membranes for tissue engineering applications. Polym. Test. 2015, 44, 234–241. [Google Scholar] [CrossRef]
- Mehrasa, M.; Asadollahi, M.A.; Nasri-Nasrabadi, B.; Ghaedi, K.; Salehi, H.; Dolatshahi-Pirouz, A.; Arpanaei, A. Incorporation of mesoporous silica nanoparticles into random electrospun plga and plga/gelatin nanofibrous scaffolds enhances mechanical and cell proliferation properties. Mater. Sci. Eng. C 2016, 66, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mehrasa, M.; Anarkoli, A.O.; Rafienia, M.; Ghasemi, N.; Davary, N.; Bonakdar, S.; Naeimi, M.; Agheb, M.; Salamat, M.R. Incorporation of zeolite and silica nanoparticles into electrospun pva/collagen nanofibrous scaffolds: The influence on the physical, chemical properties and cell behavior. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 457–465. [Google Scholar] [CrossRef]
- Gessner, A.; Lieske, A.; Paulke, B.R.; Müller, R.H. Influence of surface charge density on protein adsorption on polymeric nanoparticles: Analysis by two-dimensional electrophoresis. Eur. J. Pharm. Biopharm. 2002, 54, 165–170. [Google Scholar] [CrossRef]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Diameter (nm) | EtOH (g) | H2O (g) | PDI Solution (mL) | NH3 (mL) | TEOS (mL) | Reaction Temperature (°C) |
|---|---|---|---|---|---|---|
| 17 | 84.13 | 7.99 | 3 | 1.51 | 4.46 | 50 |
| 100 | 105.73 | 4.65 | 4 | 6.68 | 9.00 | 30 |
| 160 | 53.18 | 11.03 | 4 | 2.67 | 4.46 | 50 |
| 300 | 53.18 | 11.03 | 4 | 2.67 | 4.46 | 30 |
| Morphology | Temperature (°C) | Time to Melt/Dry | Diameter of the Nanoparticles | Samples Morphology (P: Porous; NP: Non-Porous) | Denomination |
|---|---|---|---|---|---|
| Films (F) | 90 | 30 | --- | NP | F90-NP |
| 17 | NP | F90-17NP | |||
| 100 | NP | F90-100NP | |||
| 160 | NP | F90-160NP | |||
| 300 | NP | F90-300NP | |||
| 210 | 10 | 17 | NP | F210-17NP | |
| Room temperature (Trt) | ---- | P | FTrt-17P | ||
| Oriented fibres (O) | ---- | ---- | P | O-17P | |
| Random fibres (R) | ---- | ---- | P | R-17P |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, S.; Ribeiro, T.; Ribeiro, C.; Correia, D.M.; Farinha, J.P.S.; Gomes, A.C.; Baleizão, C.; Lanceros-Méndez, S. Multifunctional Platform Based on Electroactive Polymers and Silica Nanoparticles for Tissue Engineering Applications. Nanomaterials 2018, 8, 933. https://doi.org/10.3390/nano8110933
Ribeiro S, Ribeiro T, Ribeiro C, Correia DM, Farinha JPS, Gomes AC, Baleizão C, Lanceros-Méndez S. Multifunctional Platform Based on Electroactive Polymers and Silica Nanoparticles for Tissue Engineering Applications. Nanomaterials. 2018; 8(11):933. https://doi.org/10.3390/nano8110933
Chicago/Turabian StyleRibeiro, Sylvie, Tânia Ribeiro, Clarisse Ribeiro, Daniela M. Correia, José P. Sequeira Farinha, Andreia Castro Gomes, Carlos Baleizão, and Senentxu Lanceros-Méndez. 2018. "Multifunctional Platform Based on Electroactive Polymers and Silica Nanoparticles for Tissue Engineering Applications" Nanomaterials 8, no. 11: 933. https://doi.org/10.3390/nano8110933
APA StyleRibeiro, S., Ribeiro, T., Ribeiro, C., Correia, D. M., Farinha, J. P. S., Gomes, A. C., Baleizão, C., & Lanceros-Méndez, S. (2018). Multifunctional Platform Based on Electroactive Polymers and Silica Nanoparticles for Tissue Engineering Applications. Nanomaterials, 8(11), 933. https://doi.org/10.3390/nano8110933