Synthetic Bio-Graphene Based Nanomaterials through Different Iron Catalysts




Abstract
1. Introduction
2. Experimental
2.1. Iron-Lignin Precursor Preparation
2.2. Catalytic Graphitization
2.3. Thermogravimetric Analyses (TGA)
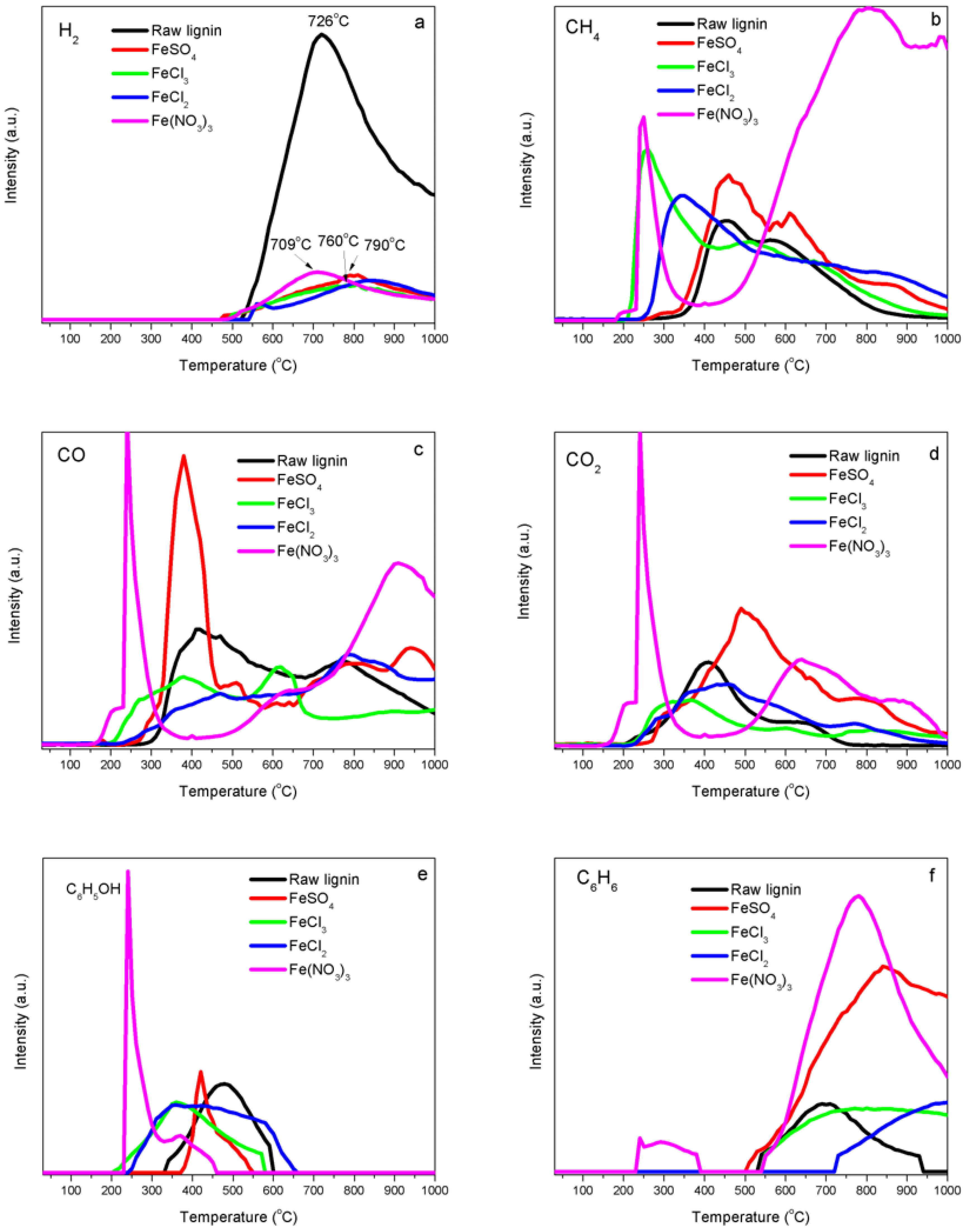
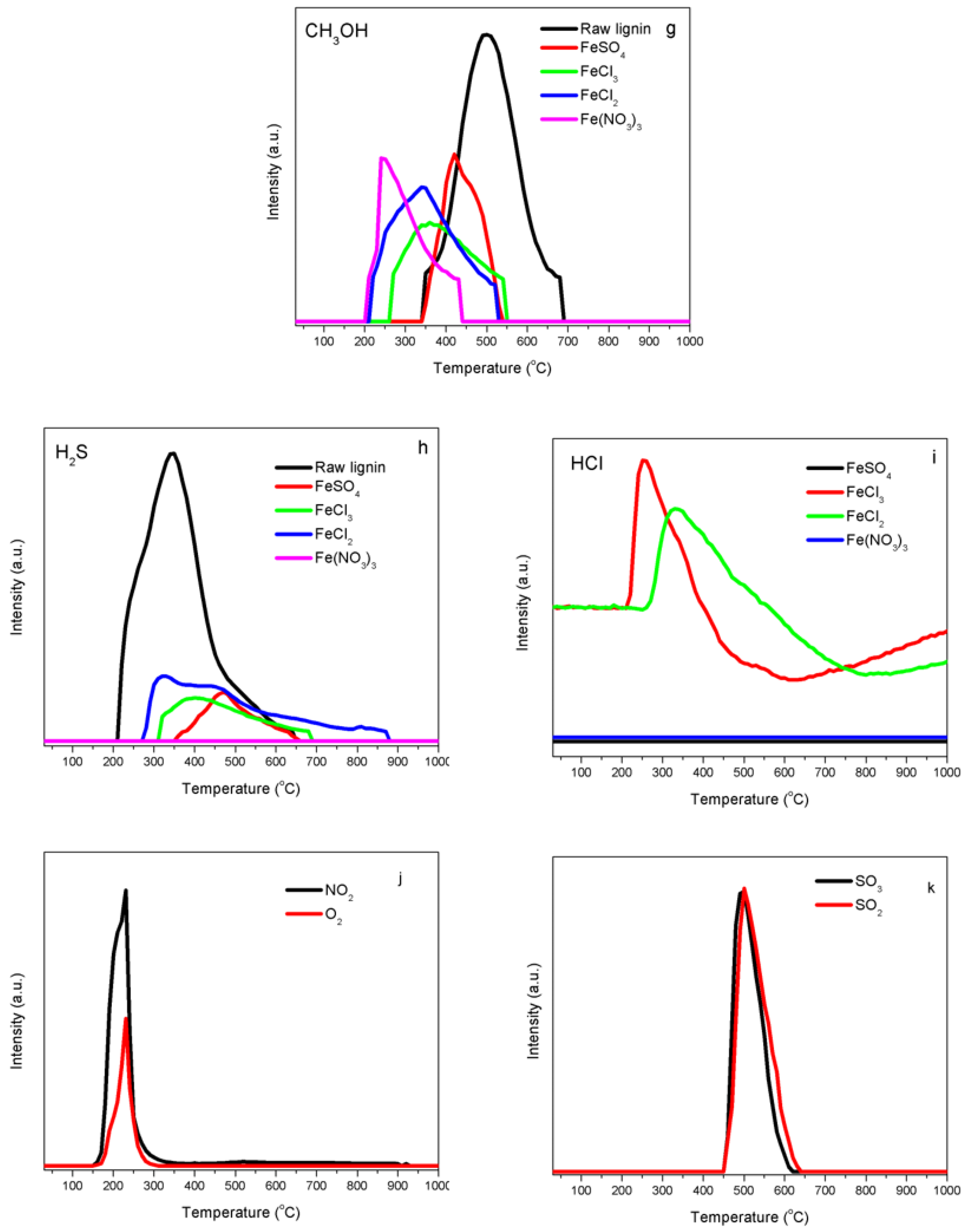
2.4. Temperature-Programmed Decomposition (TPD) Analyses
2.5. Elemental Analyses
2.6. Characterization
3. Results and Discussion
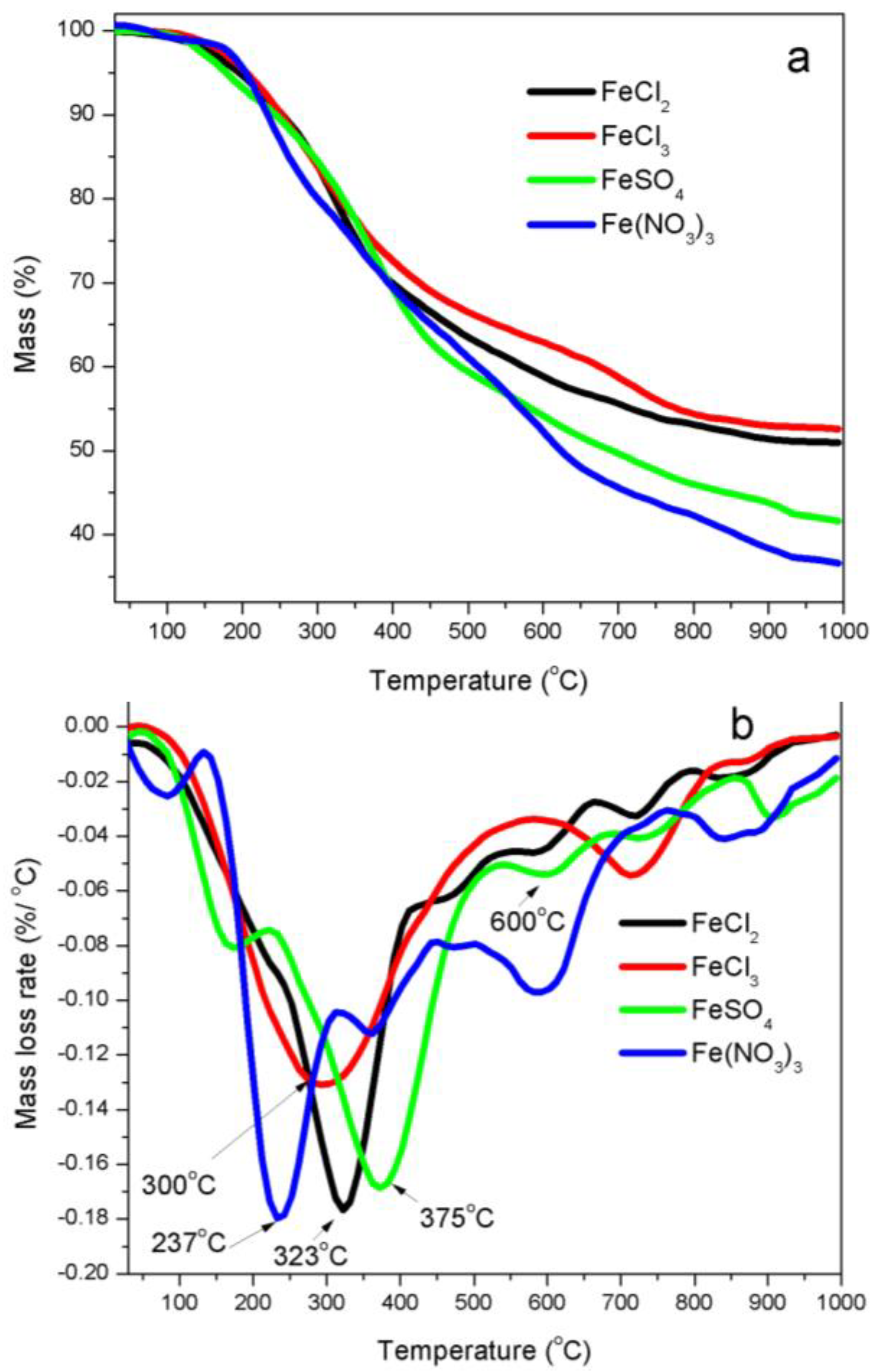
3.1. Thermal Analyses
3.2. Iron Catalyst Effects
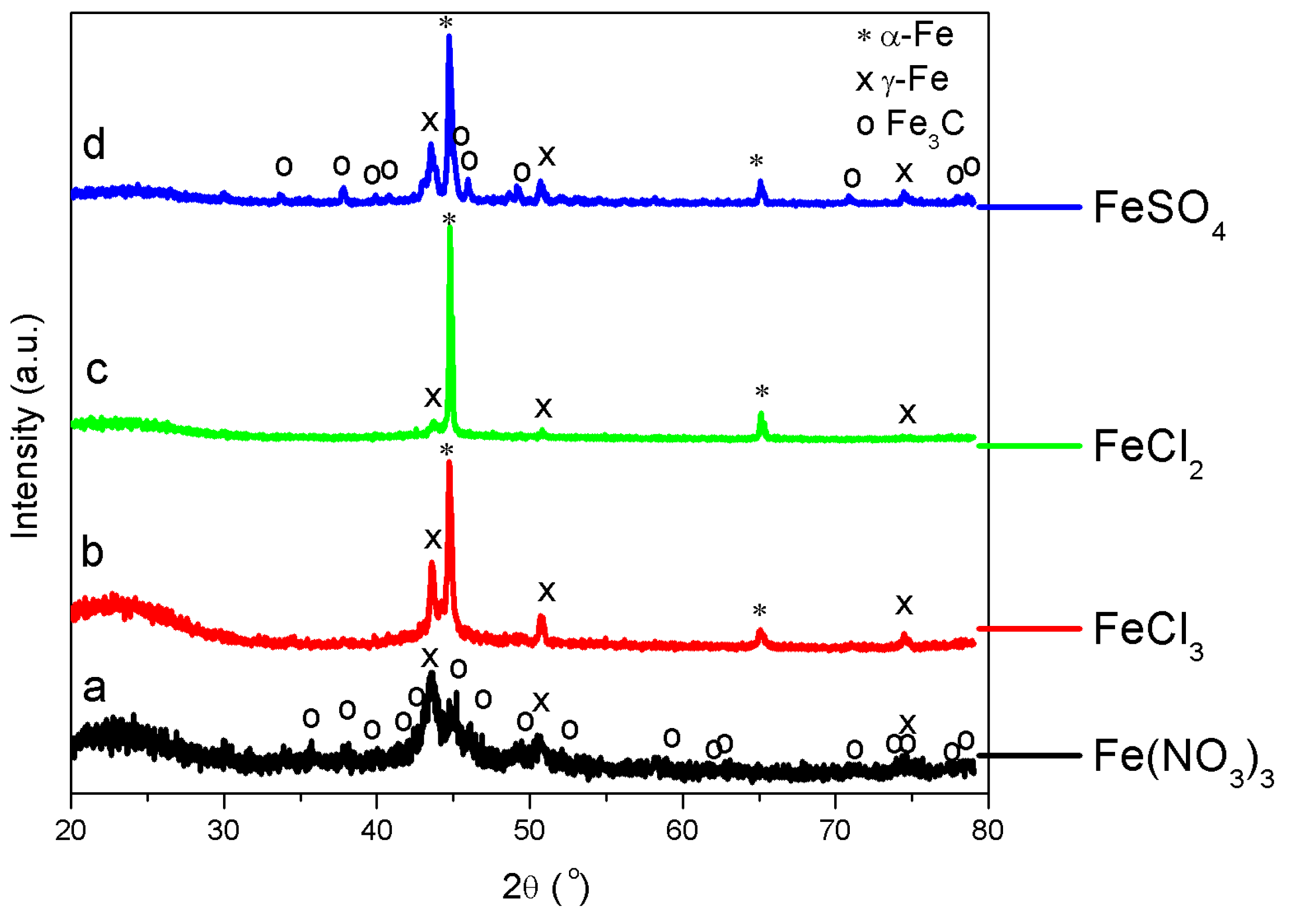
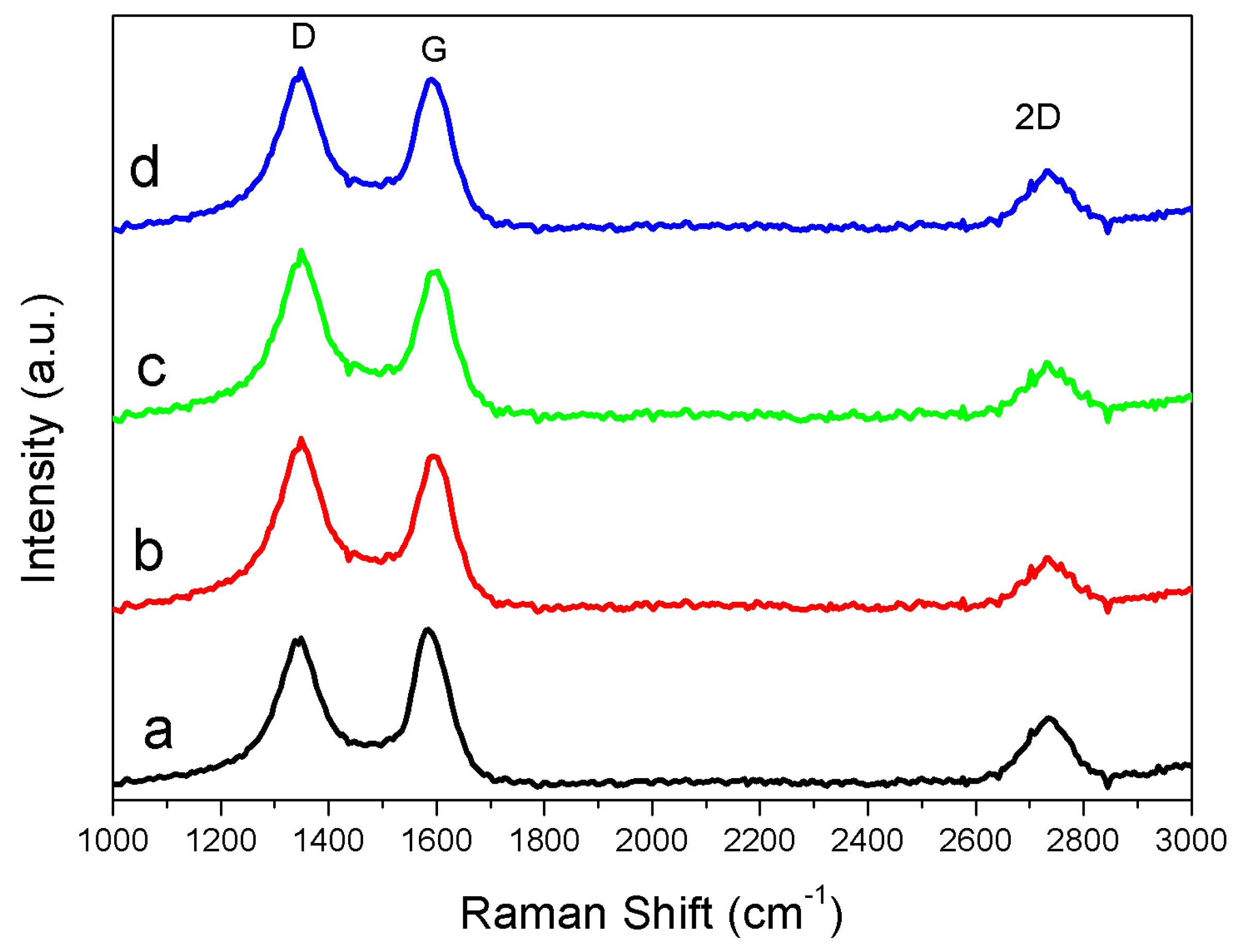
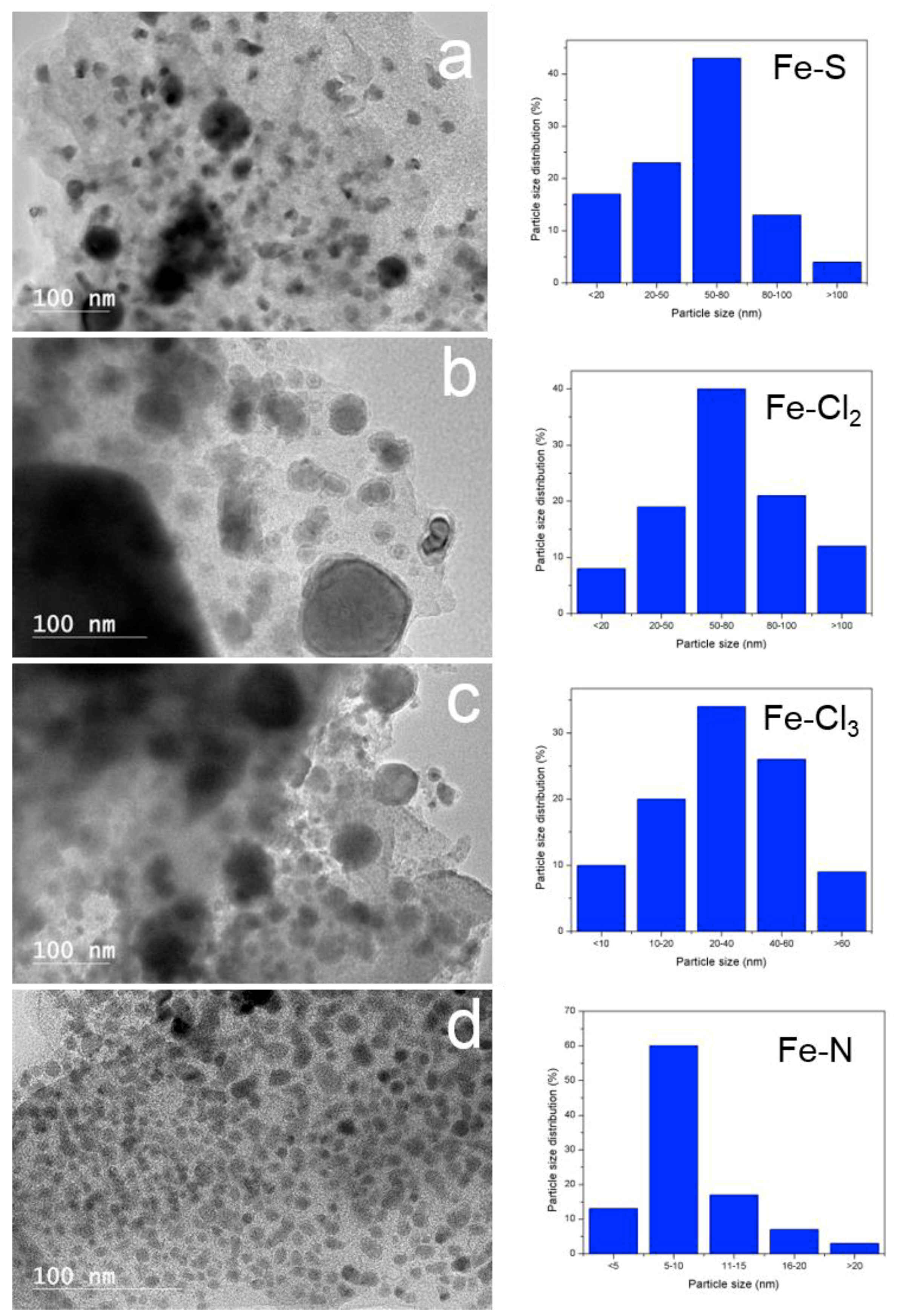
3.3. Solid Products Characterization
4. Discussion
4.1. The Solubility and the Hydration Properties of Iron Ions
4.2. Thermal Stability of Iron Precursors
4.3. Interaction of Iron with Functional Groups in Kraft Lignin
4.4. Impacts of Chlorine on Iron Catalyst
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aro, T.; Fatehi, P. Production and Application of Lignosulfonates and Sulfonated Lignin. ChemSusChem 2017, 10, 1861–1877. [Google Scholar] [CrossRef] [PubMed]
- Doherty, W.O.S.; Mousavioun, P.; Fellows, C.M. Value-adding to cellulosic ethanol: Lignin polymers. Ind. Crop. Prod. 2011, 33, 259–276. [Google Scholar] [CrossRef]
- Ruiz-Rosas, R.; Valero-Romero, M.J.; Salinas-Torres, D.; Rodríguez-Mirasol, J.; Cordero, T.; Morallón, E.; Cazorla-Amorós, D. Electrochemical Performance of Hierarchical Porous Carbon Materials Obtained from the Infiltration of Lignin into Zeolite Templates. ChemSusChem 2014, 7, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Otani, S.; Fukuoka, Y.; Igarashi, B.; Sasaki, K. Method for Producing Carbonized Lignin Fiber. U.S. Patent 3461082 A, 12 August 1969. [Google Scholar]
- Suhas, P.J.M.; Carrott, M.M.L.; Carrott, R. Lignin—From natural adsorbent to activated carbon: A review. Bioresour. Technol. 2007, 98, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Leng, W.; Barnes, H.M.; Yan, Q.; Cai, Z.; Zhang, J. Low temperature synthesis of graphene-encapsulated copper nanoparticles from kraft lignin. Mater. Lett. 2016, 185, 131–134. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, Q.; Li, J.; Cai, Z.; Zhang, J. Temperature effects on formation of carbon-based nanomaterials from kraft lignin. Mater. Lett. 2017, 203, 42–45. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, Q.; Cai, Z. Methods for Synthesizing Graphene from a Lignin Source. U.S. Patent US20170113936 A1, 8 January 2017. [Google Scholar]
- Yan, Q.; Zhang, X.; Li, J.; Wang, C.; Zhang, J.; Cai, Z. Catalytic conversion of Kraft lignin to bio-multilayer graphene materials under different atmospheres. J. Mater. Sci. 2018, 53, 8020–8029. [Google Scholar] [CrossRef]
- Yan, Q. Catalytic Thermal Conversion of Kraft Lignin to Multi-Layer Graphene Materials. Ph.D. Thesis, Mississippi State University, Starkville, MS, USA, 2017. [Google Scholar]
- Yan, Q.; Li, J.; Zhang, X.; Wang, C.; Zhang, J.; Cai, Z. Catalytic graphitization of kraft lignin to graphene-based structures with four different transitional metals. J. Nanopart. Res. 2018, 20, 223. [Google Scholar] [CrossRef]
- Takehira, K.; Shishido, T. Preparation of supported metal catalysts starting from hydrotalcites as the precursors and their improvements by adopting “memory effect”. Catal. Surv. Asia 2007, 11, 1–30. [Google Scholar] [CrossRef]
- Perry, D.L. Handbook of Inorganic Compounds; Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Fígoll, N.S.; Sad, M.R.; Beltraminl, J.N.; Jablonski, E.L.; Parera, J.M. Influence of the Chlorine Content on the Behavior of Catalysts for n-Heptane Reforming. Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 545–551. [Google Scholar] [CrossRef]
- Patterson, A. The Scherrer Formula for X-ray Particle Size Determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Jenkins, R.; Snyder, R.L. Introduction to X-ray Powder Diffractometry; John Wiley & Sons Inc.: New York, NY, USA, 1996; pp. 89–91. ISBN 0-471-51339-3. [Google Scholar]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cançado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Bassilakis, R.; Carangelo, R.M.; Wójtowicz, M.A. TG-FTIR analysis of biomass pyrolysis. Fuel 2001, 80, 1765–1786. [Google Scholar] [CrossRef]
- Freudenberg, K.; Neish, A.C. Constitution and Biosynthesis of Lignin; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1968. [Google Scholar]
- Melnikov, P.; Nascimento, V.A.; Arkhangelsky, I.V.; Zanoni Consolo, L.Z.; de Oliveira, L.C.S. Thermal decomposition mechanism of iron(III) nitrate and characterization of intermediate products by the technique of computerized modeling. J. Therm. Anal. Calorim. 2014, 115, 145–151. [Google Scholar] [CrossRef]
- Gallagher, P.K.; Johnson, D.W.; Schrey, F. Thermal Decomposition of Iron(II) Sulfates. J. Am. Ceram. Soc. 1970, 53, 666–670. [Google Scholar] [CrossRef]
- King, H.-H.; Solomon, P.R.; Avni, E.; Coughlin, R.W. Modeling Tar Composition in Lignin Pyrolysis. In Proceedings of the Symposium on Mathematical Modeling of Biomass Pyrolysis Phenomena, Washington, DC, USA, 1 January 1983; pp. 319–329. [Google Scholar]
- Hirano, K.; Kouzu, M.; Okada, T.; Kobayashi, M.; Ikenaga, N.; Suzuki, T. Catalytic activity of iron compounds for coal liquefaction. Fuel 1999, 78, 1867–1873. [Google Scholar] [CrossRef]
- Demir, M.; Kahveci, Z.; Aksoy, B.; Palapati, N.K.R.; Subramanian, A.; Cullinan, H.T.; El-Kaderi, H.M.; Harris, C.T.; Gupta, R.B. Graphitic Biocarbon from Metal-Catalyzed Hydrothermal Carbonization of Lignin. Ind. Eng. Chem. Res. 2015, 54, 10731–10739. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G.; Knozinger, H.; Schüth, F.; Weitkamp, J. Handbook of Heterogeneous Catalysis, 2nd ed.; Wiley: Hoboken, NJ, USA, 2008; Volume 8, pp. 4270–4278. ISBN 3527312412, 9783527312412. [Google Scholar]
- Ogden, M.I.; Beer, P.D. Water & O-Donor Ligands. In Encyclopedia of Inorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Lincoln, S.F.; Richens, D.T.; Sykes, A.G. Metal Aqua Ions. In Comprehensive Coordination Chemistry II; Elsevier Science: London, UK, 1987; Volume 1, pp. 515–555. [Google Scholar]
- Sham, T.K.; Hastings, J.B.; Perlman, M.L. Structure and dynamic behavior of transition-metal ions in aqueous solution: An EXAFS study of electron-exchange reactions. J. Am. Chem. Soc. 1980, 102, 5904–5906. [Google Scholar] [CrossRef]
- Böhm, F.; Sharma, V.; Schwaab, G.; Havenith, M. The low frequency modes of solvated ions and ion pairs in aqueous electrolyte solutions: Iron(II) and iron(III) chloride. Phys. Chem. Chem. Phys. 2015, 7, 19582–19591. [Google Scholar] [CrossRef] [PubMed]
- Sietsma, J.R.A.; Meeldijk, J.D.; den Breejen, J.P.; Versluijs-Helder, M.; van Dillen, A.J.; de Jongh, P.E.; de Jong, K.P. The Preparation of Supported NiO and Co3O4 Nanoparticles by the Nitric Oxide Controlled Thermal Decomposition of Nitrates. Angew. Chem. Int. Ed. 2007, 46, 4547–4549. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Gallagher, P.K. Kinetics of the decomposition of freeze-dried iron(II) sulfate. J. Phys. Chem. 1971, 75, 1179–1185. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Marchetti, G.S.; do Carmo Rangel, M. The effect of the starting material on the thermal decomposition of iron oxyhydroxides. J. Therm. Anal. Calorim. 2003, 73, 233–240. [Google Scholar] [CrossRef]
- Wang, L.; Guo, C.; Zhu, Y.; Zhou, J.; Fan, L.; Qian, Y. A FeCl2-graphite sandwich composite with Cl doping in graphite layers: A new anode material for high-performance Li-ion batteries. Nanoscale 2014, 6, 14174–14179. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, S.B.; Mishra, S.K. Thermal dehydration and decomposition of FeCl3·xH2O. J. Therm. Anal. 1996, 46, 1487–1500. [Google Scholar] [CrossRef]
- Nihira, Y.; Nomura, T. Magnetite Formation by Thermal Decomposition of FeCl2. J. Jpn. Soc. Powder Powder Metall. 1995, 42, 598–602. [Google Scholar] [CrossRef]
- Lee, J.-F.; Lee, M.-D.; Tseng, P.-K. Fischer—Tropsch synthesis on supported iron catalysts prepared from iron(III) chloride pretreatment effects on phase changes and catalytic properties. Appl. Catal. 1989, 52, 193–209. [Google Scholar] [CrossRef]
- Arabczyk, W.; Narkiewicz, U.; Moszyński, D. Chlorine as a poison of the fused iron catalyst for ammonia synthesis. Appl. Catal. A-Gen. 1996, 134, 331–338. [Google Scholar] [CrossRef]
- Consonni, M.; Jokic, D.; Murzin, Y.D.; Touroude, R. High Performances of Pt/ZnO Catalysts in Selective Hydrogenation of Crotonaldehyde. J. Catal. 1999, 188, 165–175. [Google Scholar] [CrossRef]
- Calvin, H. Bartholomew, Mechanisms of catalyst deactivation. Appl. Catal. A-Gen. 2001, 212, 17–60. [Google Scholar]
- Lara, J.; Tysoe, W.T. The surface and tribological chemistry of carbon tetrachloride on iron. Tribol. Lett. 1999, 6, 195–198. [Google Scholar] [CrossRef]
- Forzatti, P.; Lietti, L. Catalyst deactivation. Catal. Today 1999, 52, 165–181. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe Resources | Solid Carbon | Liquid | Gas |
|---|---|---|---|
| Fe-S | 33.6 | 19.5 | 46.9 |
| Fe-Cl3 | 35.0 | 20.2 | 44.8 |
| Fe-Cl2 | 35.3 | 19.7 | 45 |
| Fe-N | 31.3 | 18 | 50.7 |
| Samples | C | H | N | S | Cl |
|---|---|---|---|---|---|
| Raw kraft lignin | 65.2 ± 0.2 | 6.1 ± 0.2 | 0.1 ± 0.05 | 0.8 ± 0.2 | N/A |
| Fe-S | 97.3 ± 0.5 | 0.4 ± 0.2 | N/A | 0.7 ± 0.1 | N/A |
| Fe-Cl3 | 95.7 ± 0.9 | 0.6 ± 0.3 | N/A | 0.4 ± 0.1 | 0.5 ± 0.3 |
| Fe-Cl2 | 95.0 ± 0.7 | 0.8 ± 0.2 | N/A | 0.2 ± 0.2 | 0.8 ± 0.4 |
| Fe-N | 98.5 ± 0.5 | 0.1 ± 0.05 | N/A | 1.2 ± 0.3 | N/A |
| Samples | Sg (m2 g−1) |
|---|---|
| Fe-N | 108 |
| Fe-Cl2 | 55 |
| Fe-Cl3 | 60 |
| Fe-S | 79 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Q.; Li, J.; Zhang, X.; Zhang, J.; Cai, Z. Synthetic Bio-Graphene Based Nanomaterials through Different Iron Catalysts. Nanomaterials 2018, 8, 840. https://doi.org/10.3390/nano8100840
Yan Q, Li J, Zhang X, Zhang J, Cai Z. Synthetic Bio-Graphene Based Nanomaterials through Different Iron Catalysts. Nanomaterials. 2018; 8(10):840. https://doi.org/10.3390/nano8100840
Chicago/Turabian StyleYan, Qiangu, Jinghao Li, Xuefeng Zhang, Jilei Zhang, and Zhiyong Cai. 2018. "Synthetic Bio-Graphene Based Nanomaterials through Different Iron Catalysts" Nanomaterials 8, no. 10: 840. https://doi.org/10.3390/nano8100840
APA StyleYan, Q., Li, J., Zhang, X., Zhang, J., & Cai, Z. (2018). Synthetic Bio-Graphene Based Nanomaterials through Different Iron Catalysts. Nanomaterials, 8(10), 840. https://doi.org/10.3390/nano8100840