
Soft Interaction in Liposome Nanocarriers for Therapeutic Drug Delivery

 ,
, 



Abstract
:1. Introduction
2. Basic Concepts of Liposomal Nanocarriers
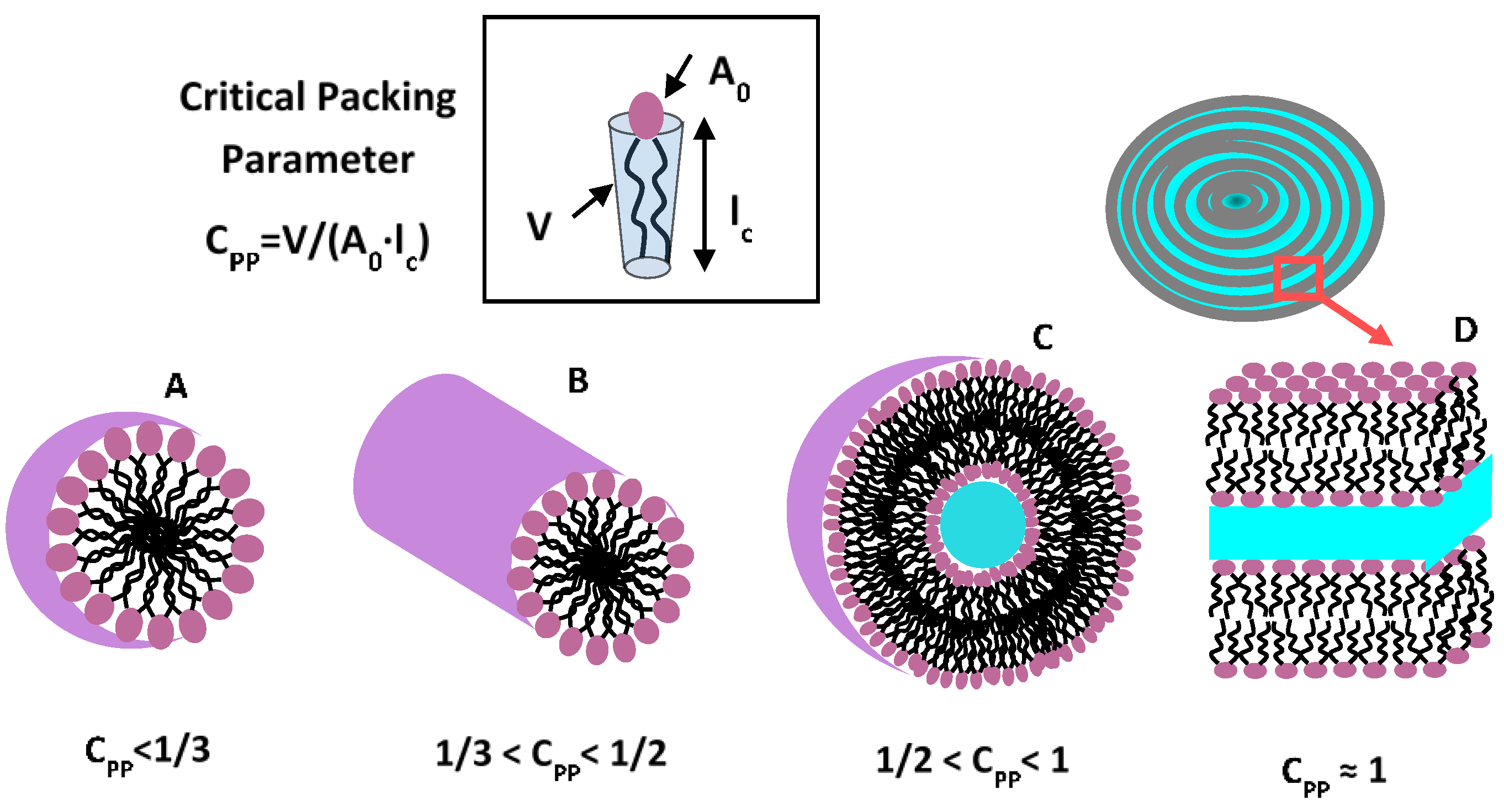
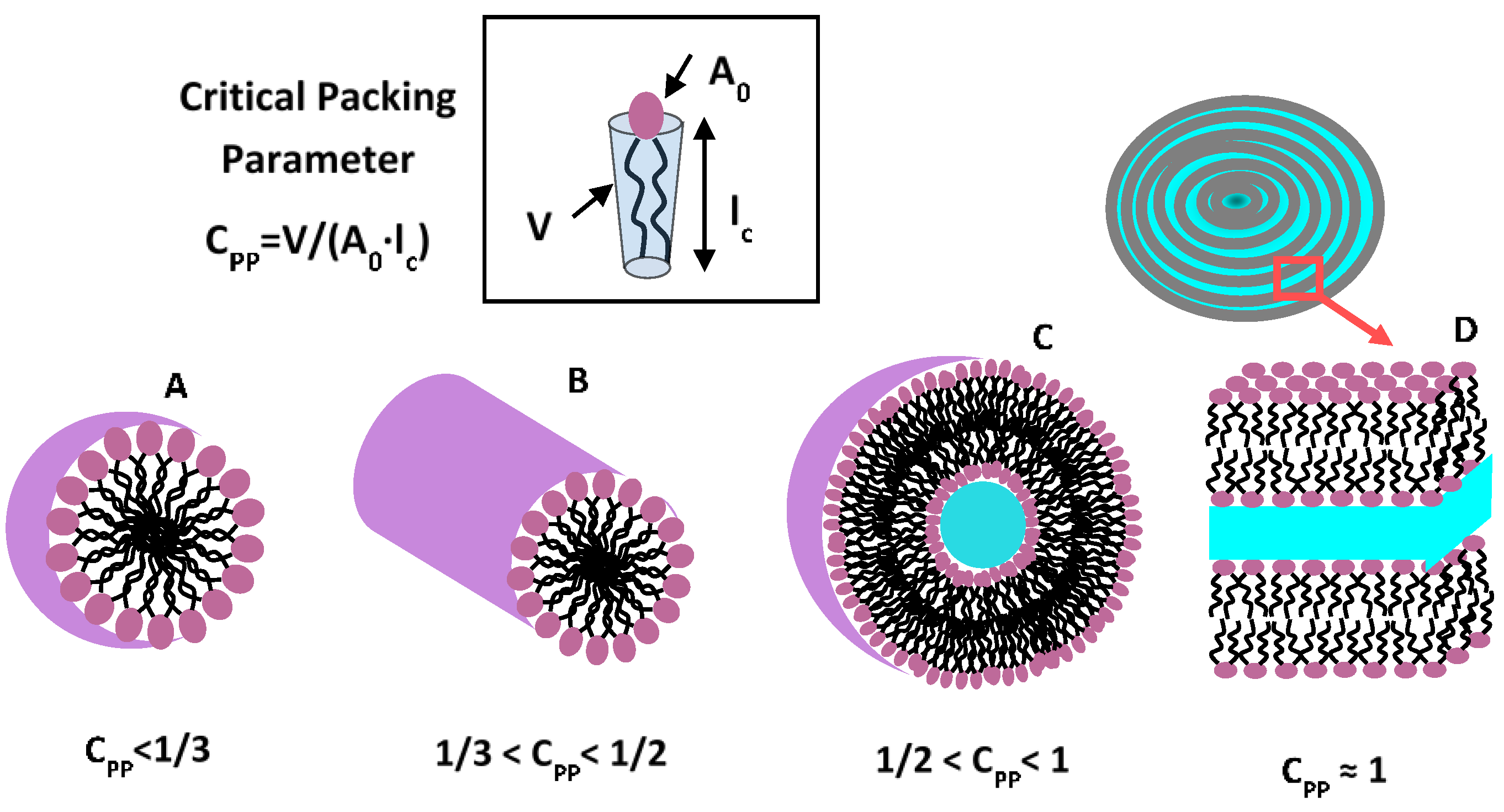
3. Amphiphilic Soft Nanocarriers: Micelles, Vesicles, and Bilayers
4. Soft Interactions Involved in Liposome Nanocarriers
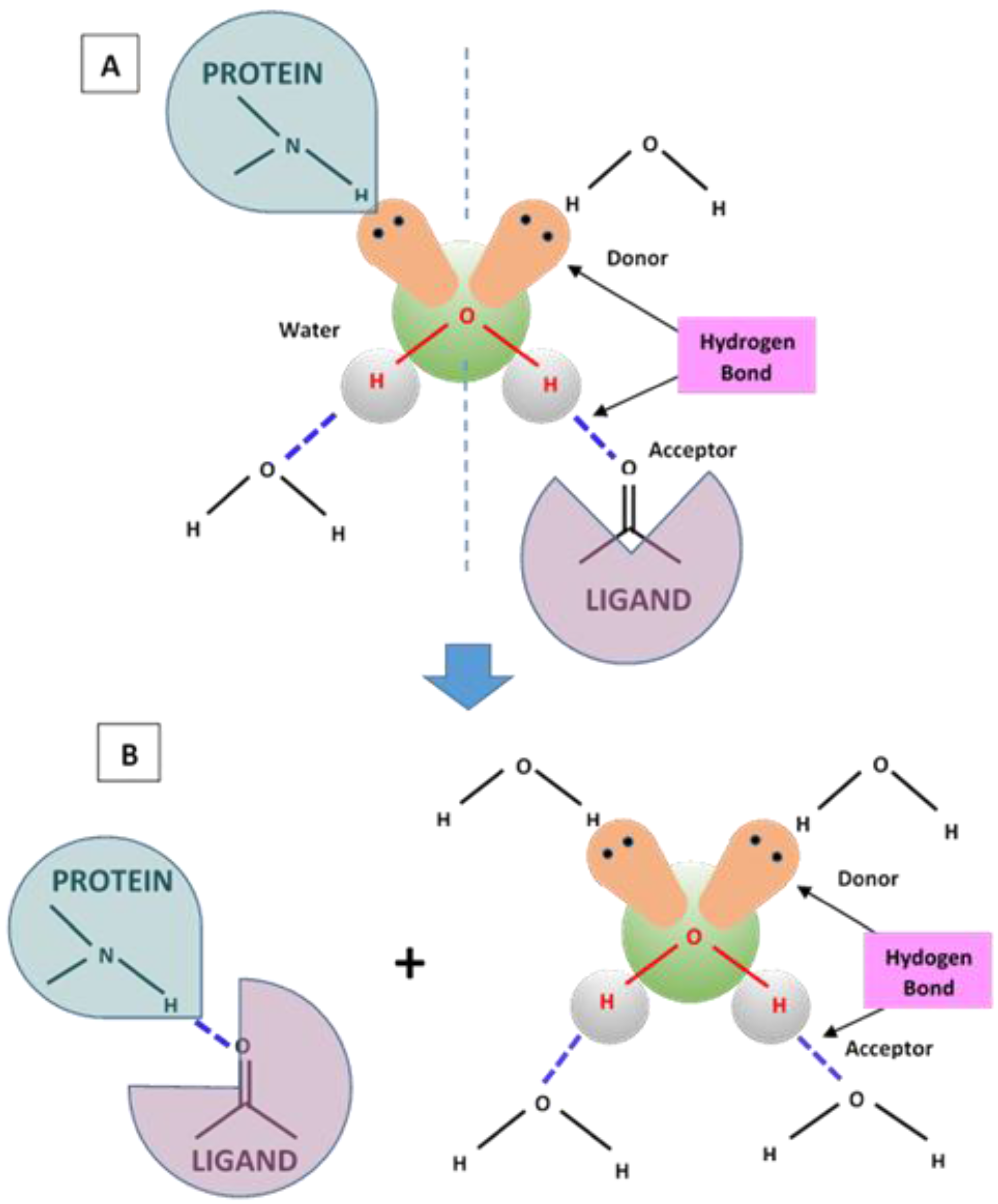
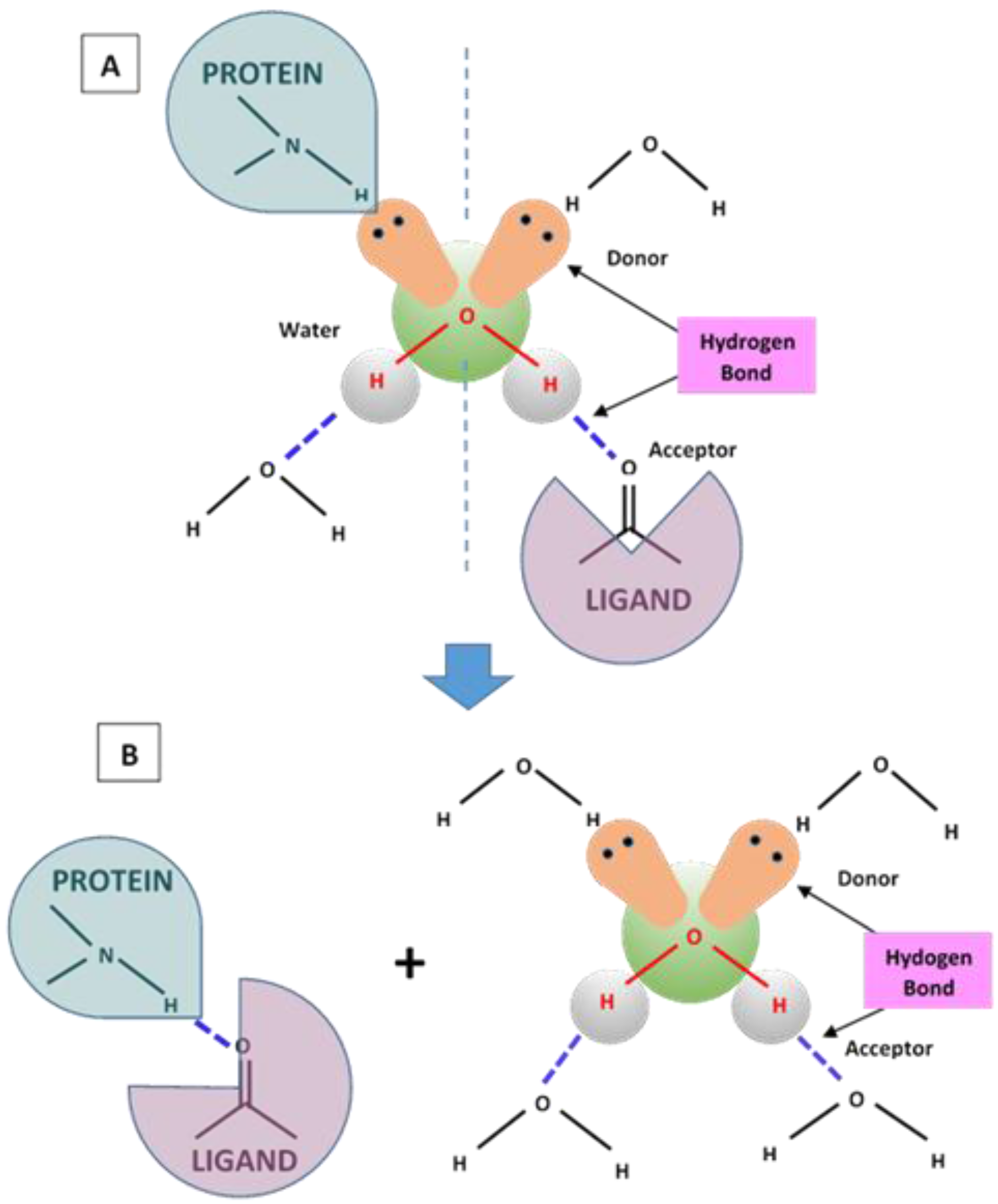
4.1. Hydrogen Bonding and Hydrophobic Forces
4.2. Van der Waals Forces
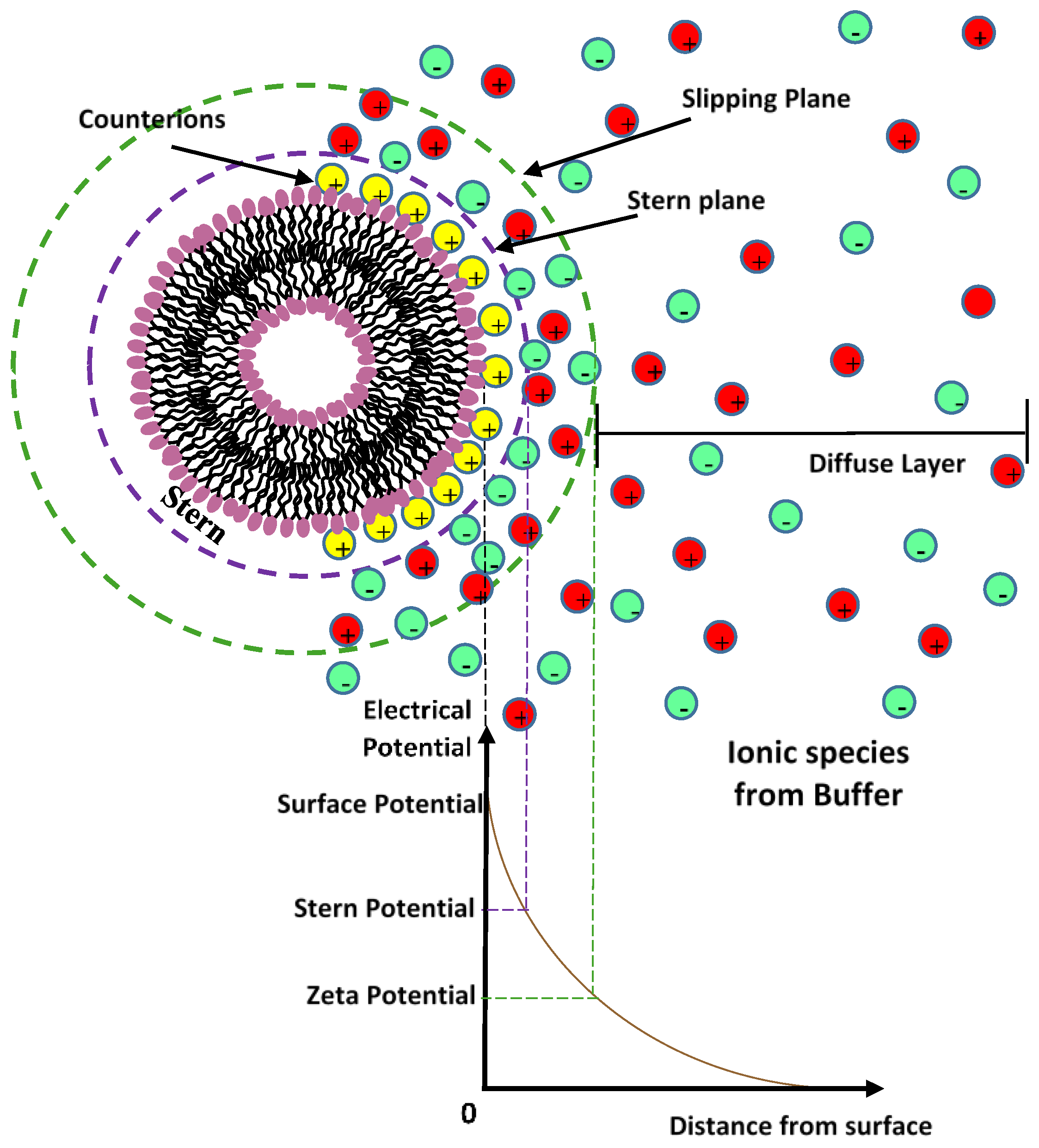
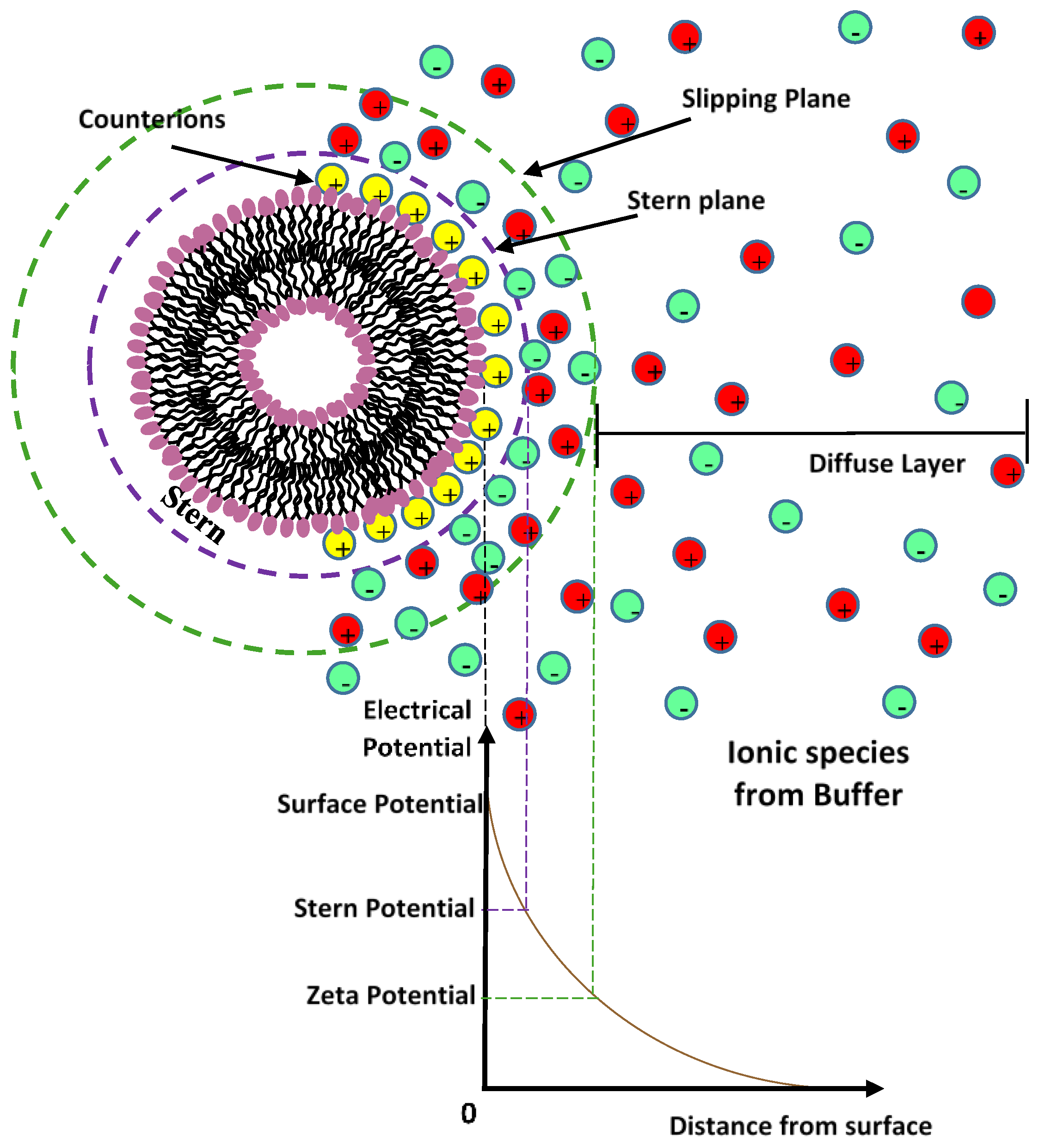
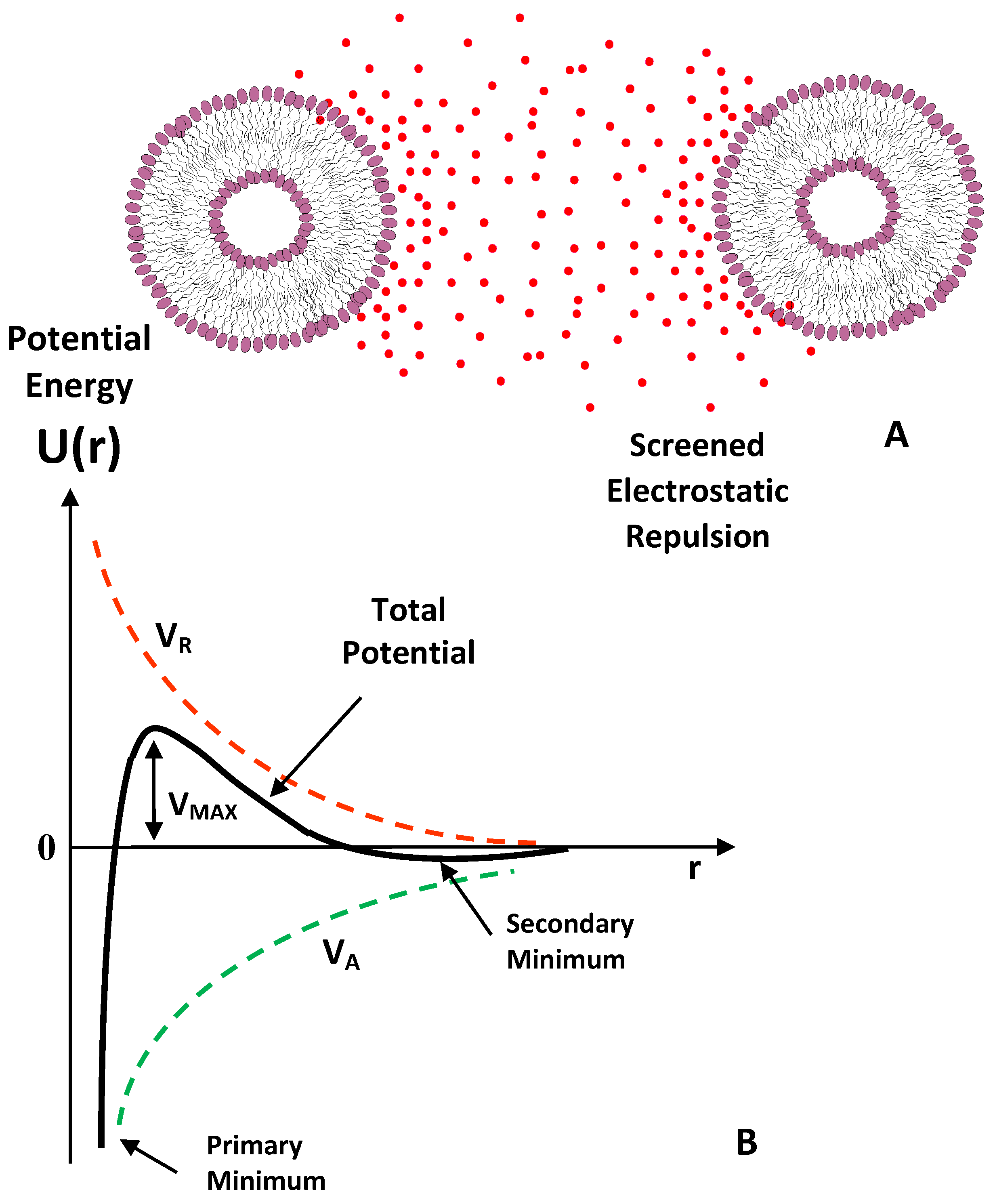
4.3. Elctrostatic Interaction: Electrical Double Layer (EDL)
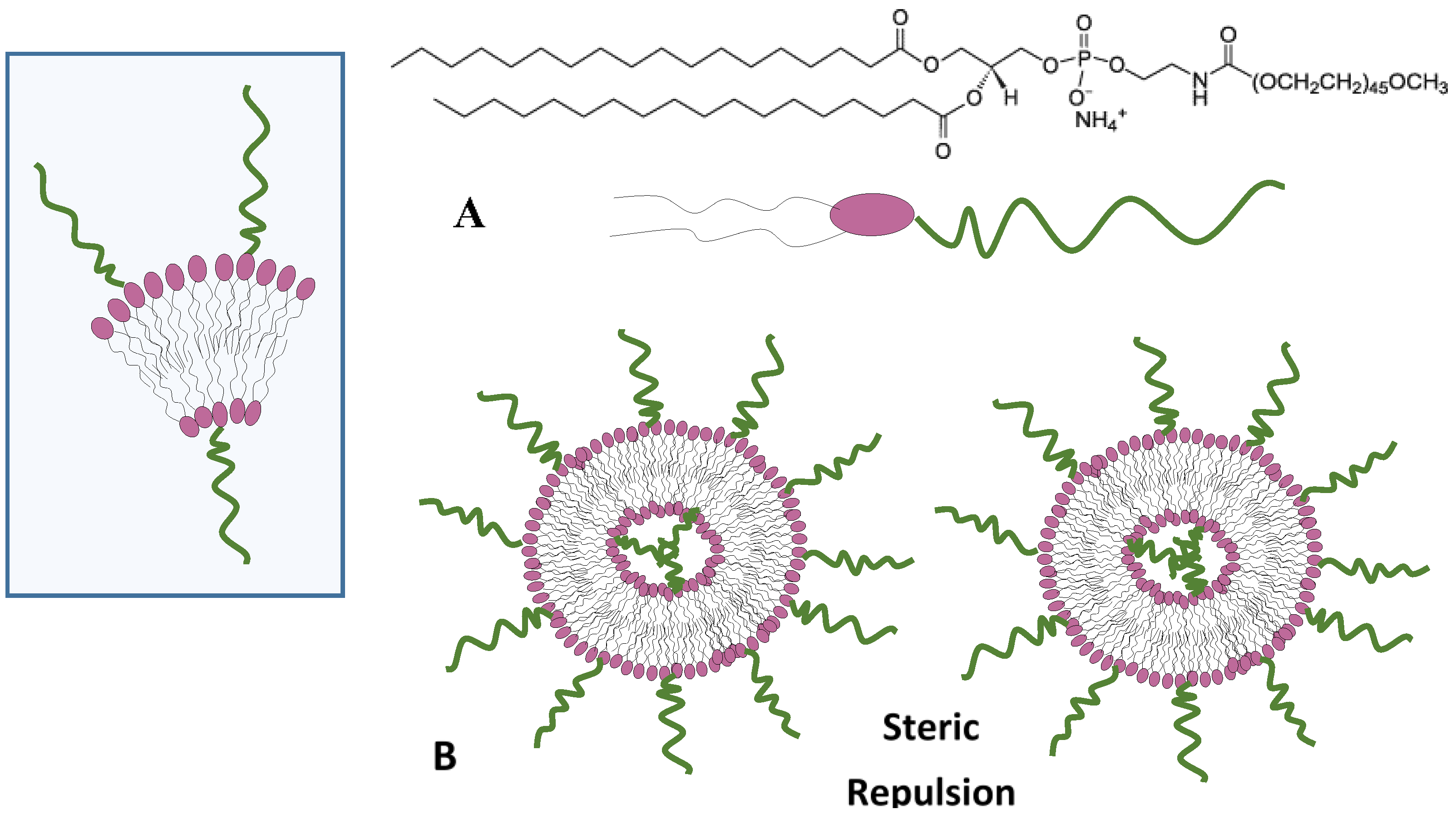
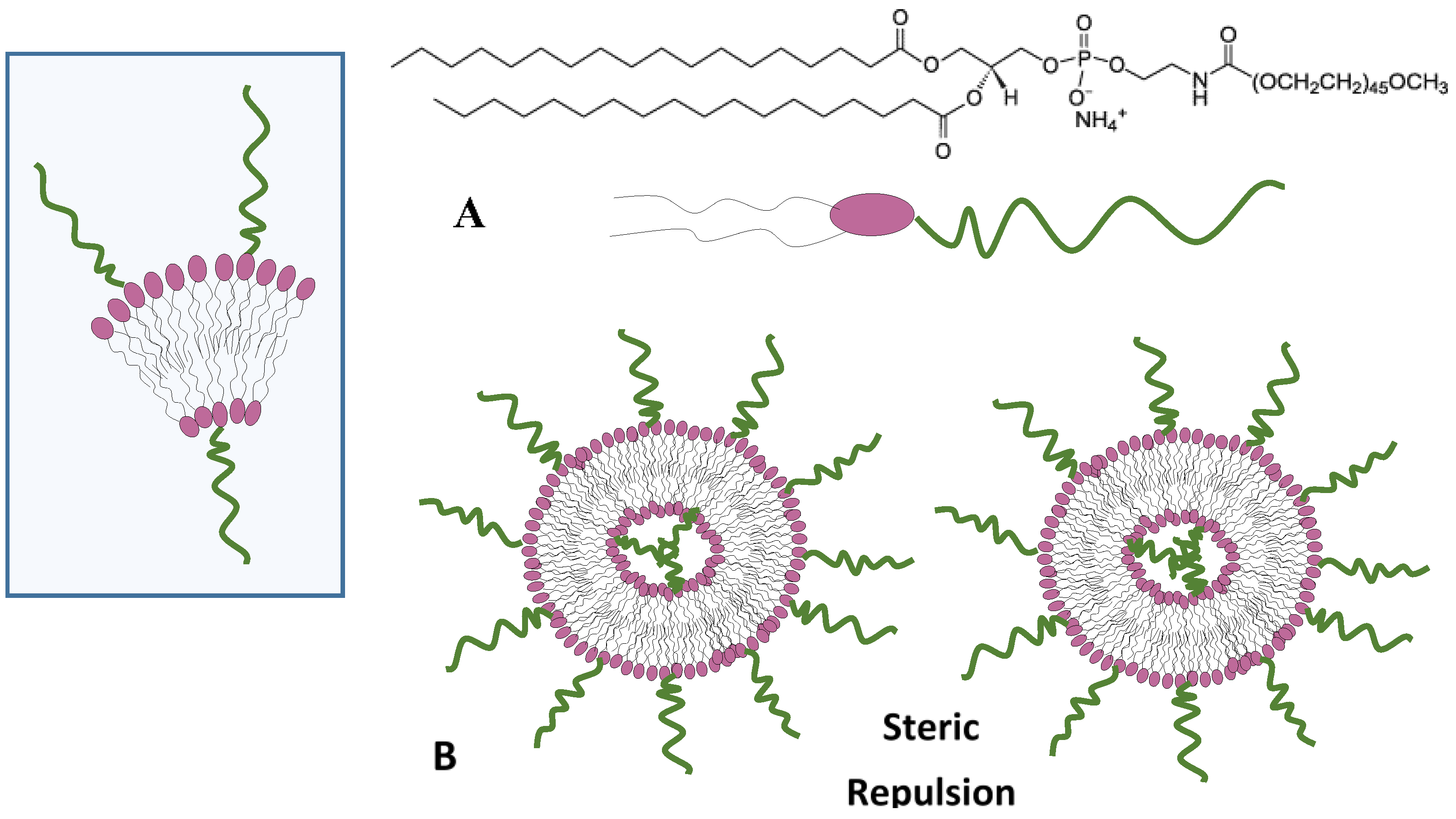
4.4. Steric Stabilization Forces
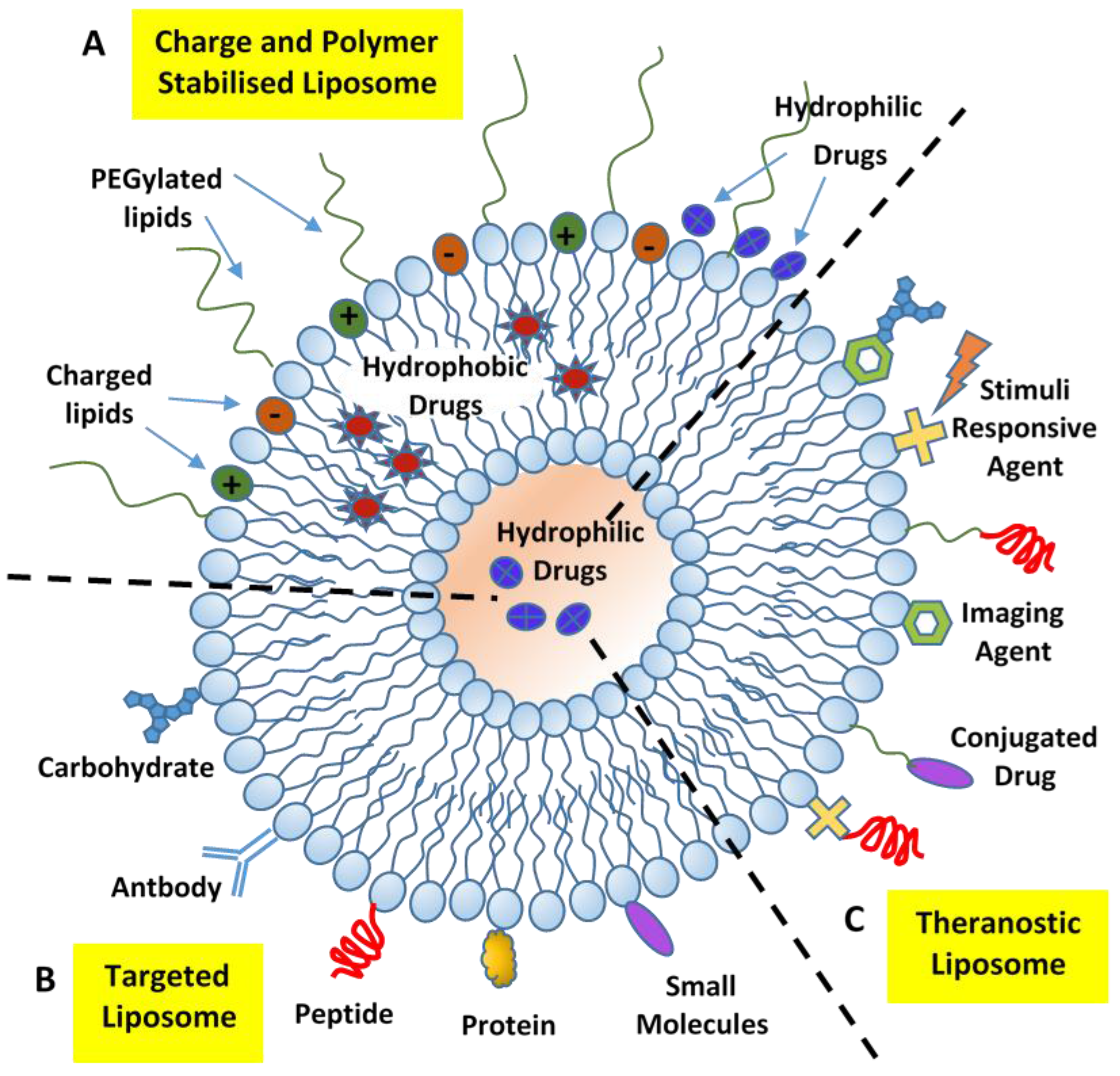
5. Structural Characterization of Liposome Nanocarriers
6. Passive Targeting in Nanocarrier Drug Delivery: Beyond the Biological Barriers
7. Drug Delivery Mechanism
8. Strategies to Prolong the Circulation Time
8.1. Electrostatic Stabilization
8.2. Steric Stabilization
9. Liposome-Liposome Interaction and Molecular Recognition Processes
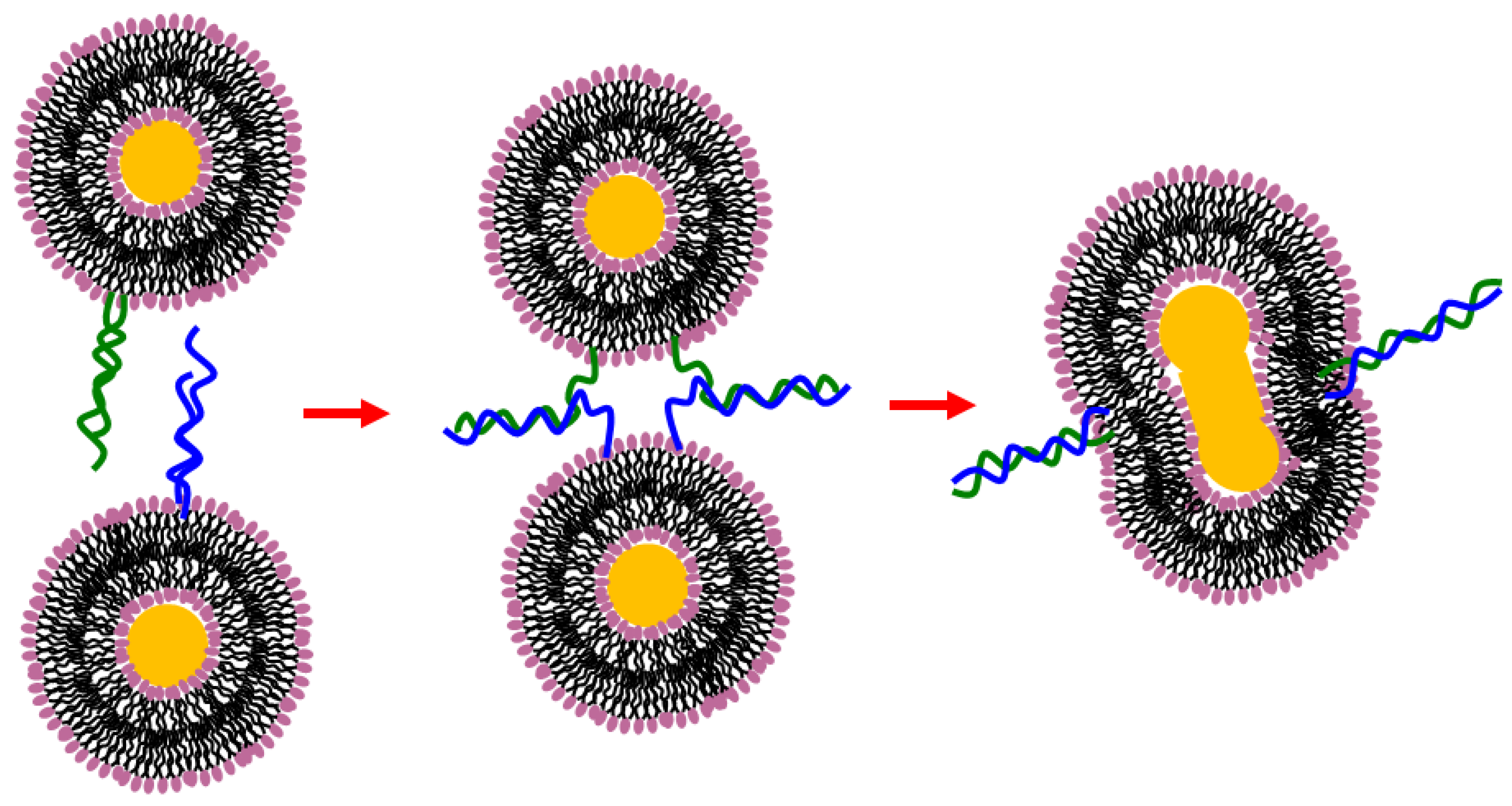
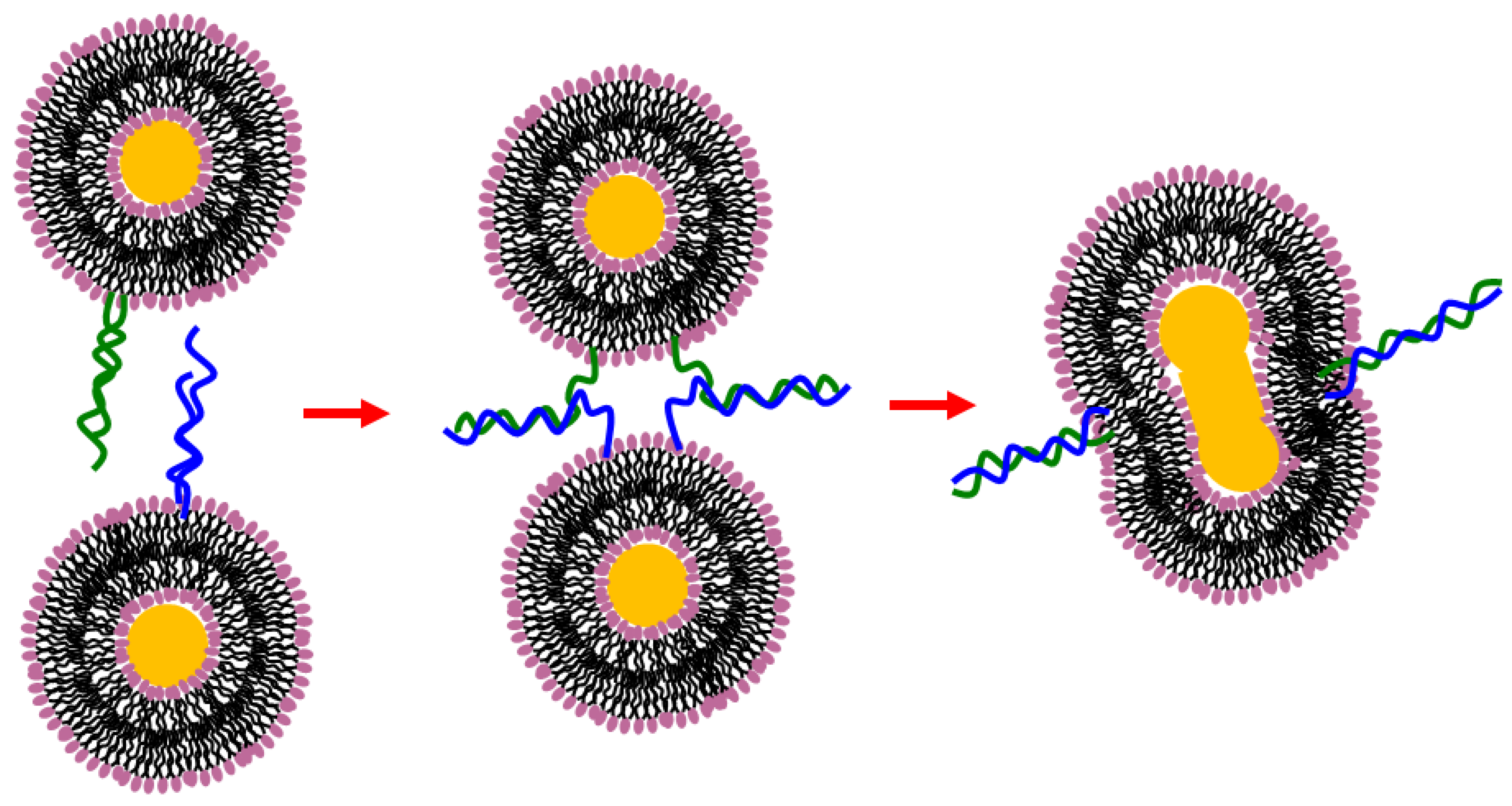
9.1. Liposome Aggregation-Fusion
9.2. Governing the Colloidal Stability in Bio-Systems
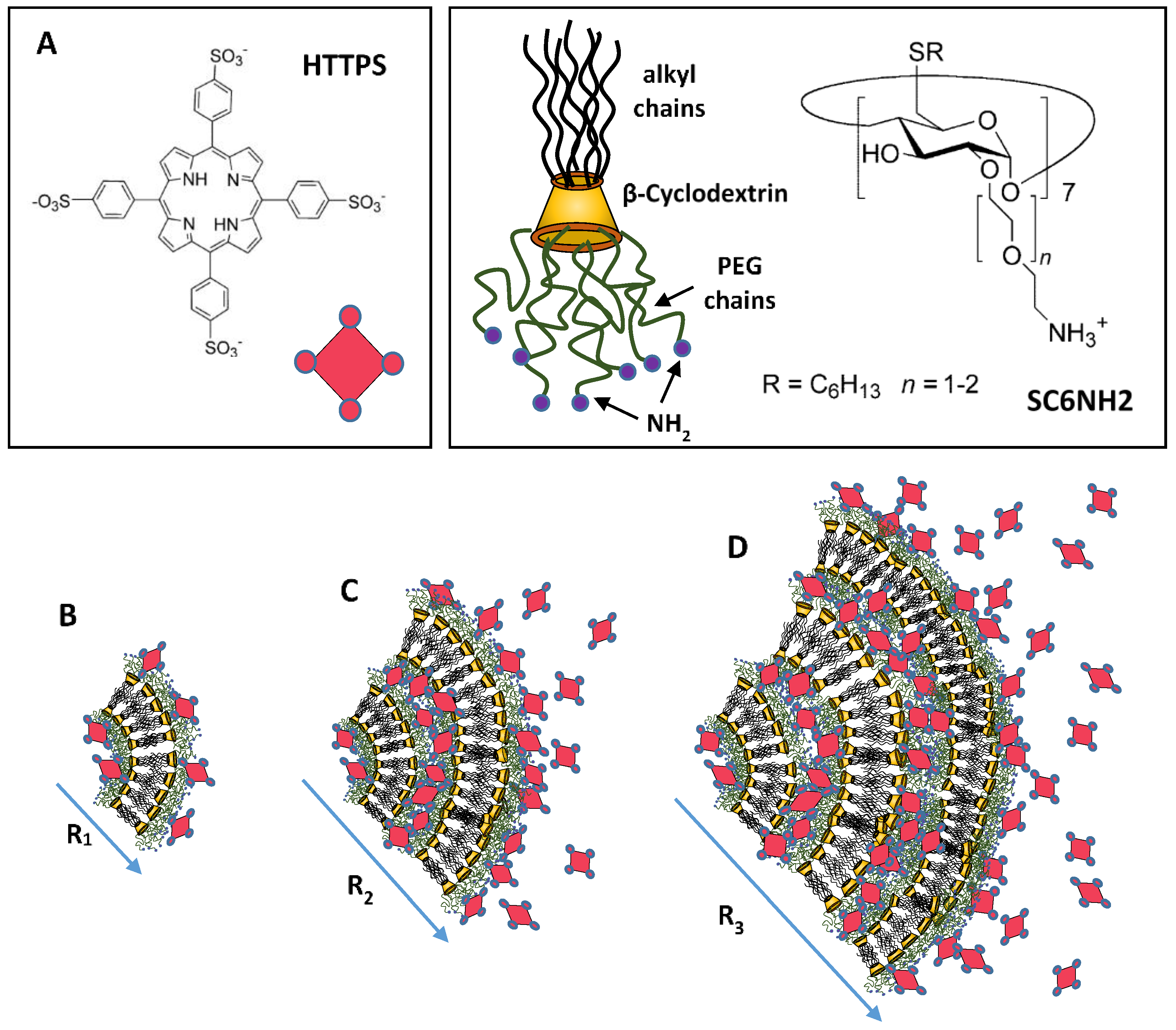
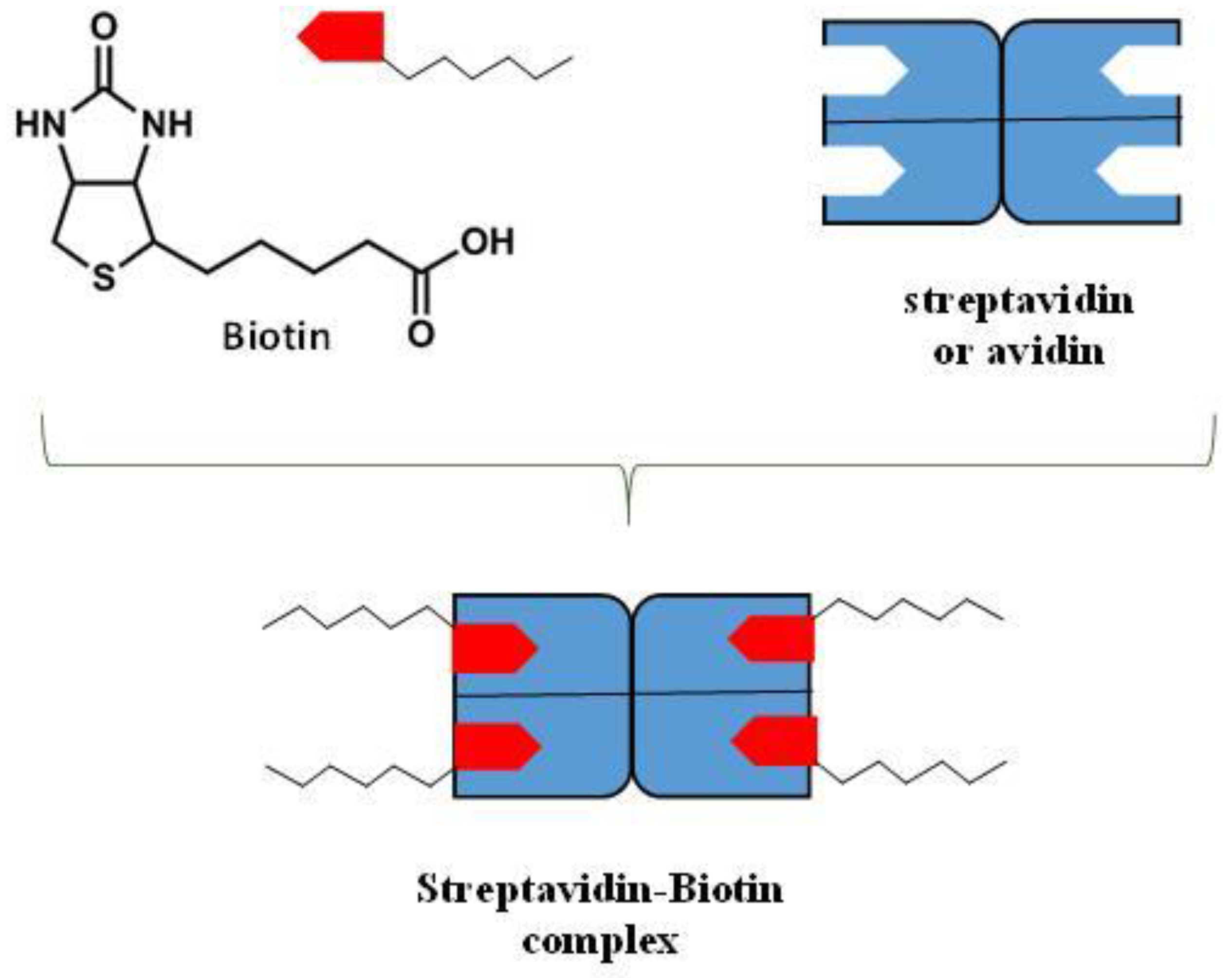
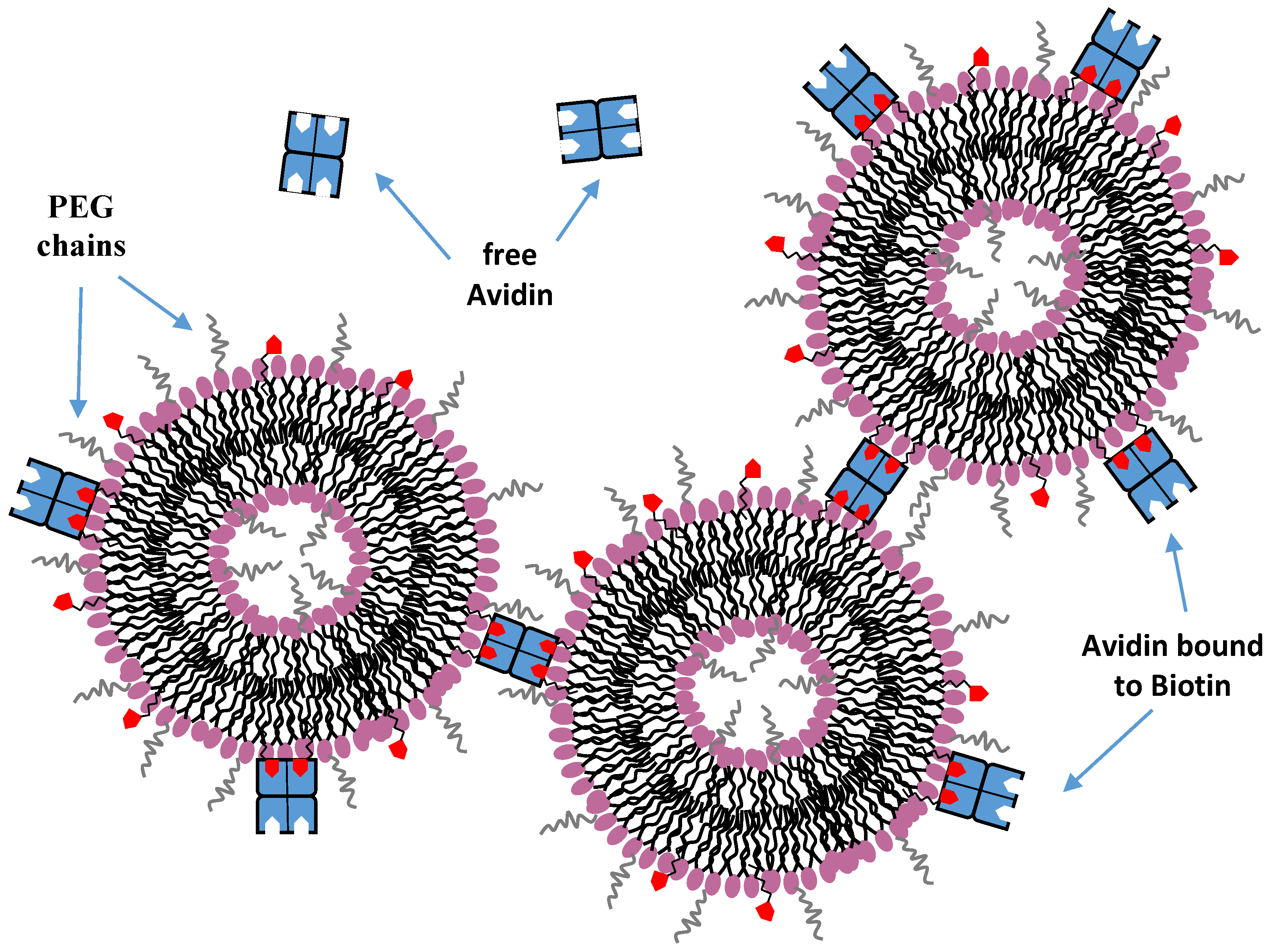
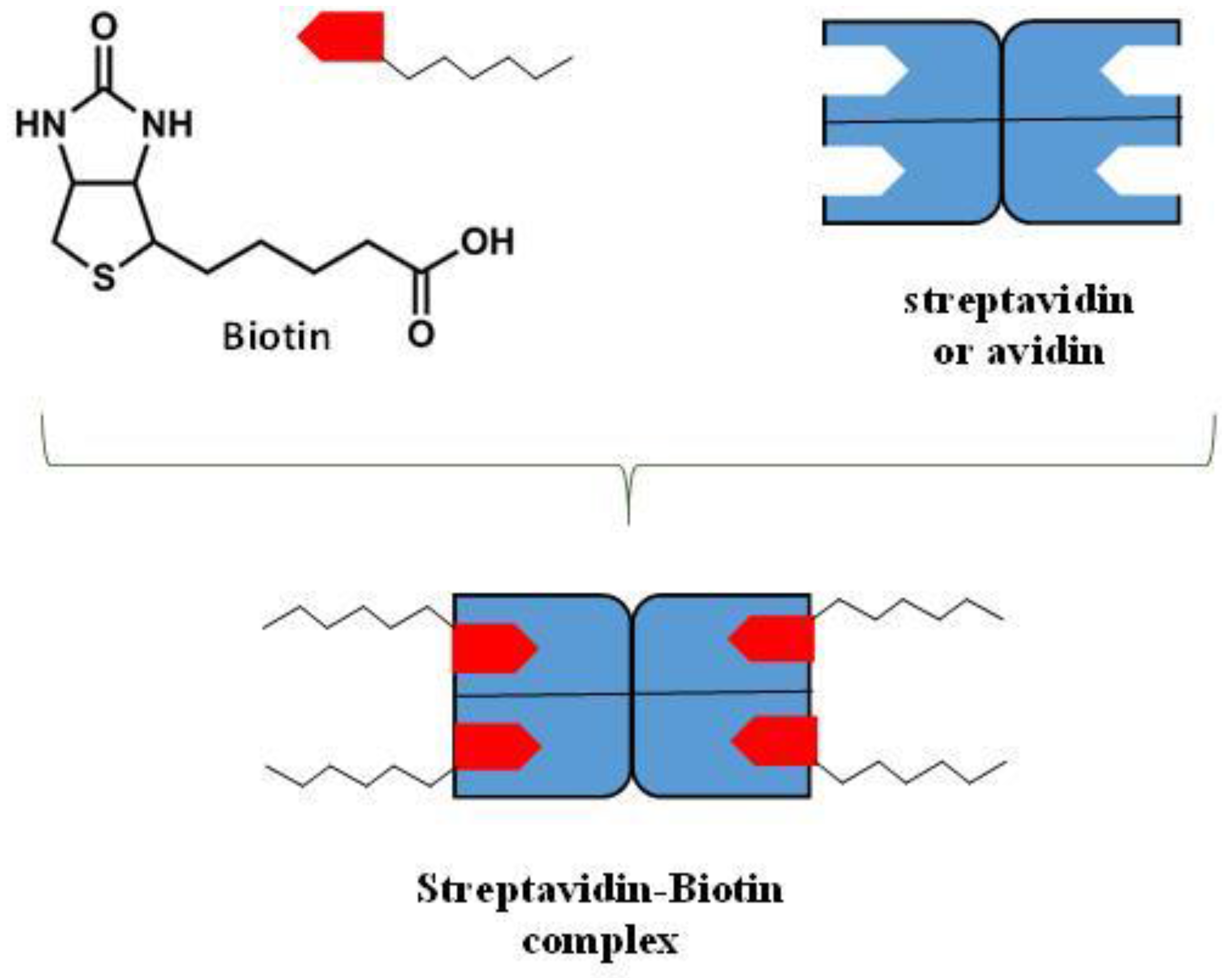
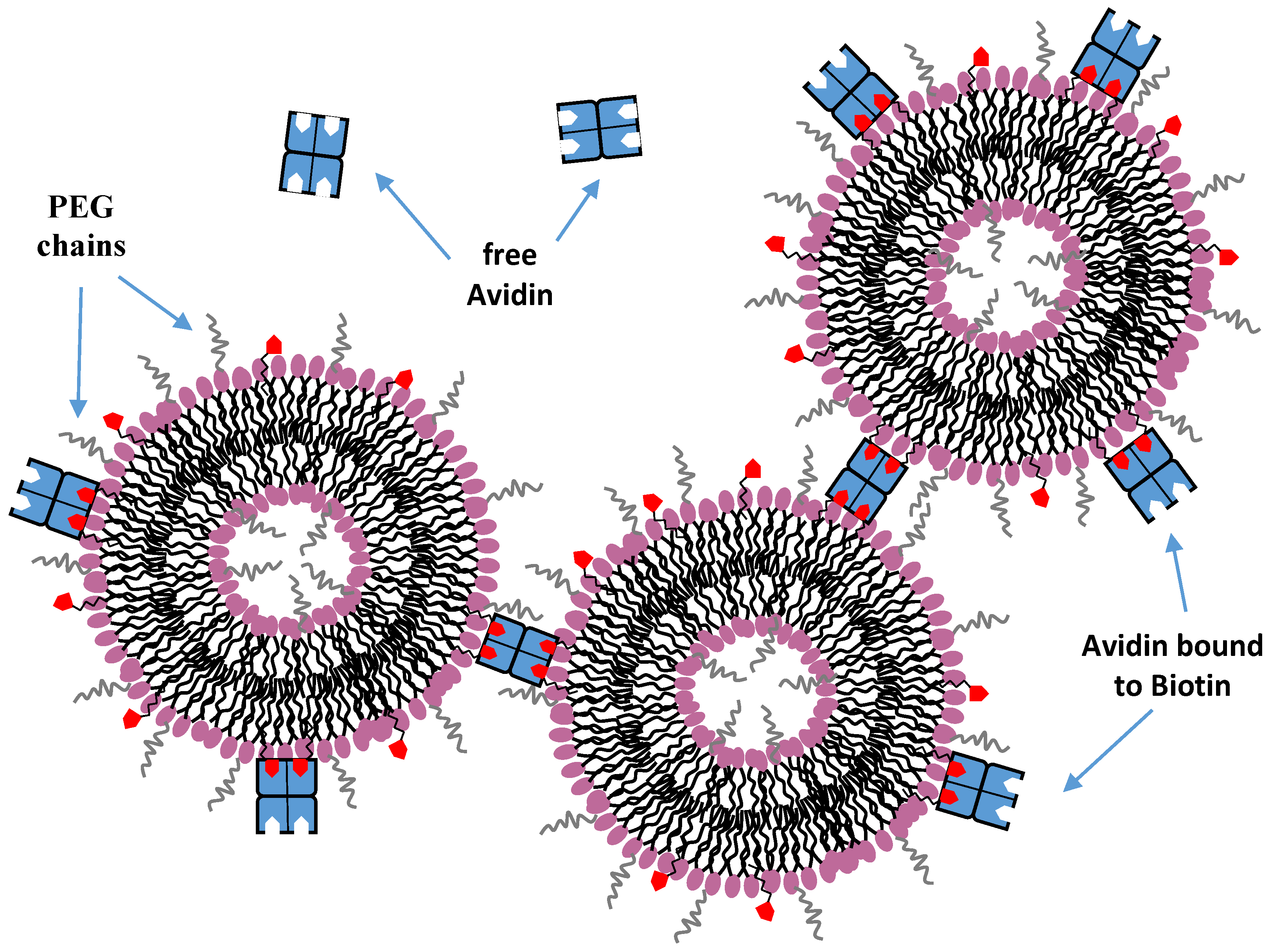
9.3. Hydrogen Bonding Mediated Interactions between Liposomes and Molecular Recognition Processes
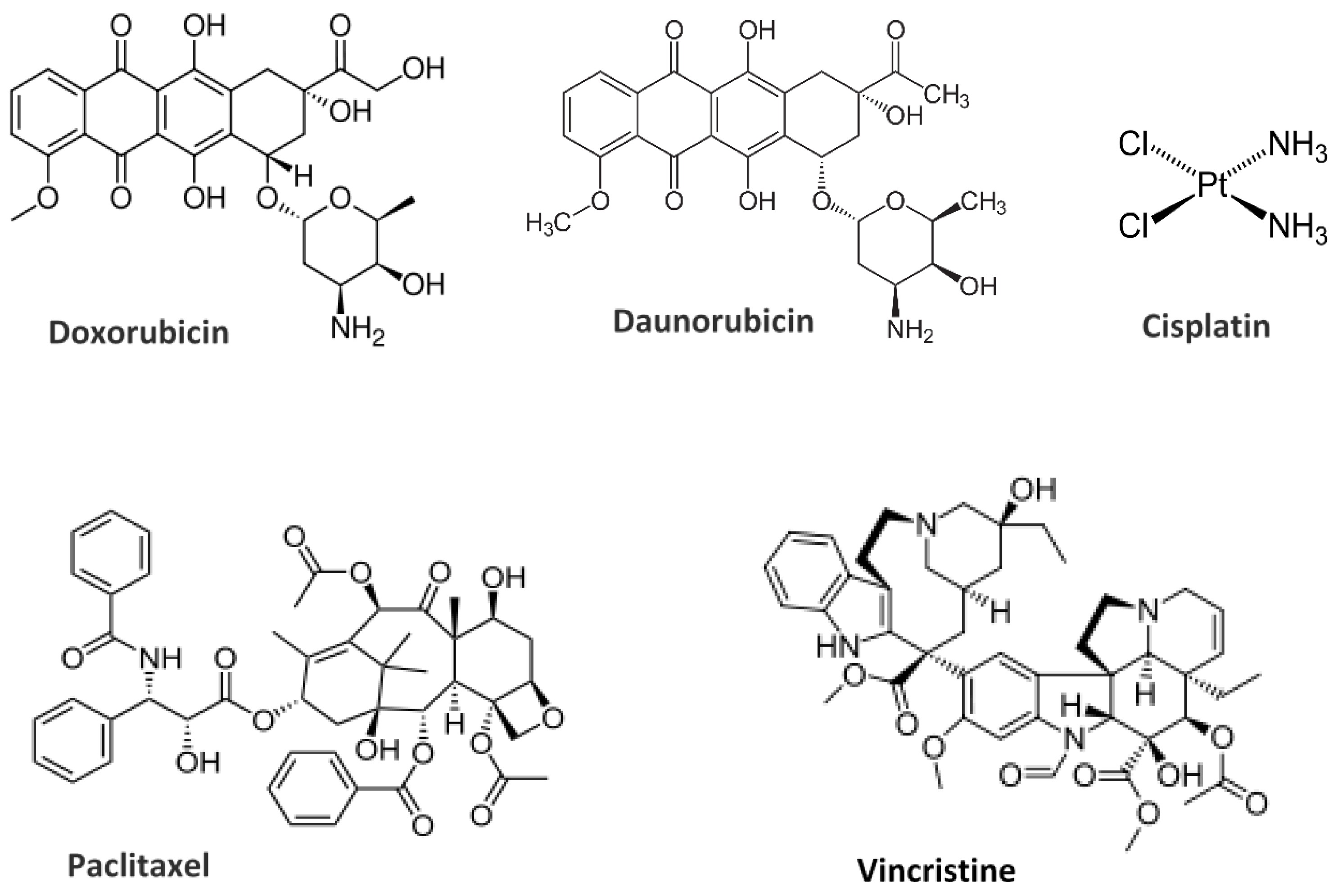
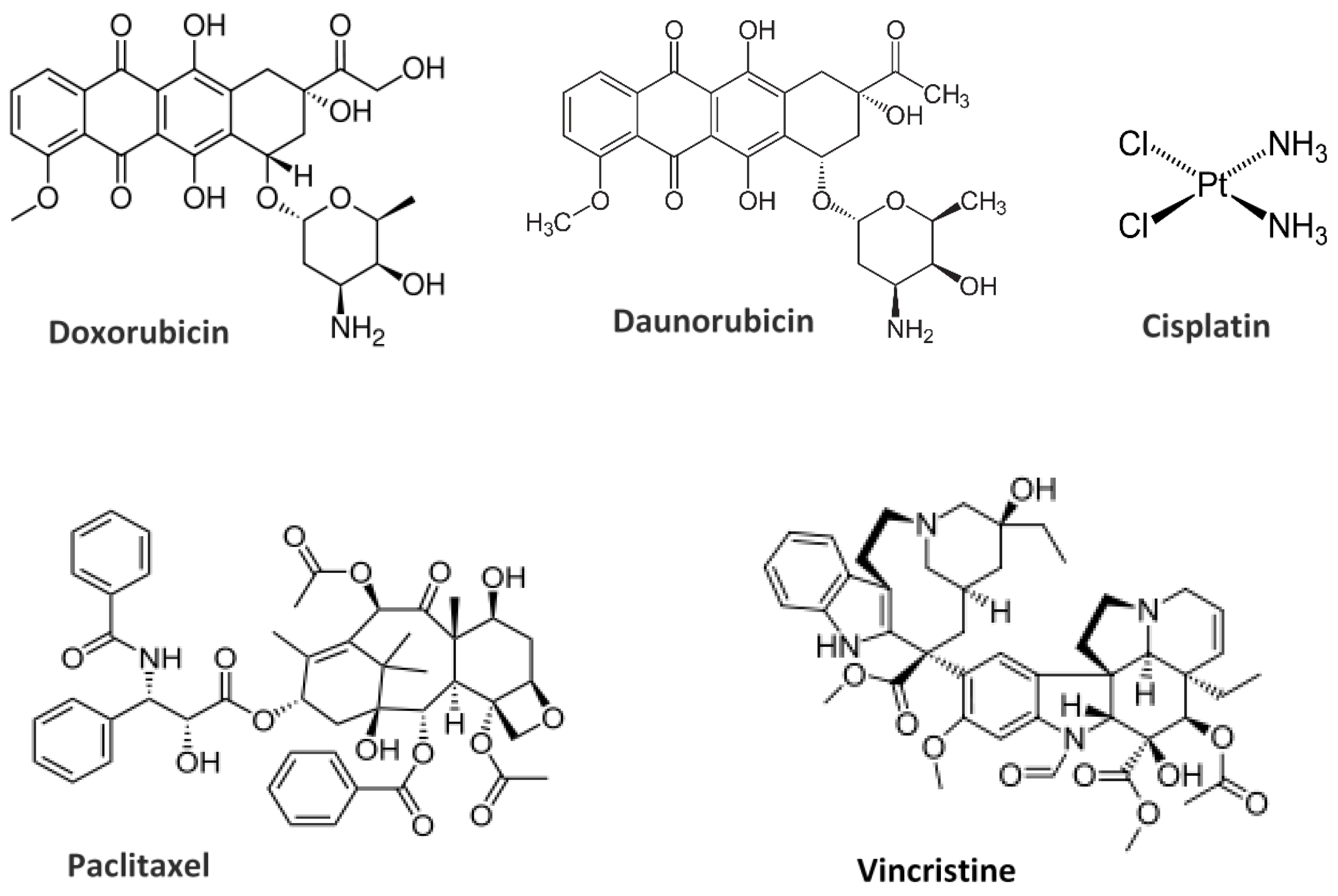
10. Liposome-Drug Interaction: Encapsulation and Delivery Approaches
10.1. Hydrophilic/Hydrophobic Drug Encapsulation and Release Properties
10.2. Triggered Drug Release from Stimuli Responsive Liposomes (SRL)
10.3. Drug Delivery with Targeted Liposome (Active Targeting)
11. Market and Clinical Development of Liposome-Based Drugs Formulations
12. Future Perspectives: The Theranostic Approach for Cancer Treatment
13. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kawasaki, E.S.; Player, A. Nanotechnology, nanomedicine, and the development of new, effective therapies for cancer. Nanomedicine 2005, 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 1, pp. 491–519. [Google Scholar]
- Sackmann, E. Physical basis of self-organization and function of membranes: Physics of vesicles. In Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 1, pp. 213–303. [Google Scholar]
- Katsaras, J.; Gutberlet, T. Lipid bilayers. In Structure and Interactions; Springer-Verlag: Berlin Heidelberg, Germany, 2000. [Google Scholar]
- Israelachvili, J.N. Physics of Amphiphiles: Micelles, Vesicles and Microemulsions; Degiorgio, V., Corti, M., Eds.; North-Holland: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Romero, E.L.; Morilla, M.J. Highly deformable and highly fluid vesicles as potential drug delivery systems: Theoretical and practical considerations. Int. J. Nanomed. 2013, 8, 3171–3186. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar]
- Aharon, D.; Weitman, H.; Ehrenberg, B. The effect of liposomes’ surface electric potential on the uptake of hematoporphyrin. Biochim. Biophys. Acta. 2011, 1808, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Tajima, S.; Takeoka, S. Evaluation of pH-responsive liposomes containing amino acid-based zwitterionic lipids for improving intracellular drug delivery in vitro and in vivo. J. Control. Release 2010, 142, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Kiselev, M.A.; Magazù, S.; Calandra, P. Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches. Adv. Condens. Matter Phys. 2015, 2015. [Google Scholar] [CrossRef]
- Helfrich, W. Elastic properties of lipid bilayers: Theory and possible experiments. Z Naturforsch C 1973, 28, 693–703. [Google Scholar] [PubMed]
- New, R.C. Preparation of liposomes. In Liposomes: A Practical Approach; New, R.C., Ed.; New York, Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Tao, X.; Jin, S.; Wu, D.; Ling, K.; Yuan, L.; Lin, P.; Xie, Y.; Yang, X. Effects of Particle Hydrophobicity, Surface Charge, Media pH Value and Complexation with Human Serum Albumin on Drug Release Behavior of Mitoxantrone-Loaded Pullulan Nanoparticles. Nanomaterials 2016, 6. [Google Scholar] [CrossRef]
- Israelachvili, J.; Wennerström, H. Role of hydration and water structure in biological and colloidal interactions. Nature 1996, 379, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Angel, C.A. Water, A Comprehensive Treatise; Franks, F., Ed.; Plenum: New York, NY, USA, 1972; Volume 7. [Google Scholar]
- Magazù, S.; Migliardo, F.; Telling, M.T. Study of the dynamical properties of water in disaccharide solutions. Eur. Biophys. J. 2007, 36, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Disalvo, E.A.; Lairion, F.; Martini, F.; Tymczyszyn, E.; Frías, M.; Almaleck, H.; Gordillo, G.J. Structural and functional properties of hydration and confined water in membrane interfaces. Biochim. Biophys. Acta Biomembr. 2008, 1778, 2655–2670. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.; Migliardo, F.; Takacs, E.; Vertessy, B.; Magazù, S.; Telling, M.T.F. Study of solvent-protein coupling effects by neutron scattering. J. Biol. Phys. 2010, 36, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes; Krieger: Malabar, FL, USA, 1991. [Google Scholar]
- Parsegian, V.A. Van der Waals Forces; Cambridge University Press: New York, NY, USA, 2006. [Google Scholar]
- Hunter, R.J. Foundations of Colloid Science; Oxford University Press: New York, NY, USA, 1986; Volume I−II. [Google Scholar]
- Israelachvili, J.N. Intermolecular and Surface Forces, 2nd ed.; Academic Press: San Diego, FL, USA, 1992. [Google Scholar]
- Schoenborn, B.P. Neutron Scattering for the Analysis of Membranes. Biochim. Biophys. Acta. 1976, 457, 41–55. [Google Scholar] [CrossRef]
- Zemb, T.; Lindner, P. Neutron, X-rays and Light Scattering Methods Applied to Soft Condensed Matter; North-Holland: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Fitter, J.; Gutberlet, T.; Katsaras, J. Neutron scattering in Biology. Techniques and Applications; Springer-Verlag: Berlin Heidelberg, Germany, 2006. [Google Scholar]
- Kiselev, M.A.; Lombardo, D. Structural characterization in mixed lipid membrane systems by neutron and X-ray scattering. Biochim. Biophys. Acta. 2016, (in press). [Google Scholar] [CrossRef]
- Bibi, S.; Kaur, R.; Henriksen-Lacey, M.; McNeil, S.E.; Wilkhu, J.; Lattmann, E.; Christensen, D.; Mohammed, A.R.; Perrie, Y. Microscopy imaging of liposomes: From coverslips to environmental SEM. Int. J. Pharm. 2011, 417. [Google Scholar] [CrossRef] [PubMed]
- Ruozi, B.; Belletti, D.; Tombesi, A.; Tosi, G.; Bondioli, L.; Forni, F.; Vandelli, M.A. AFM, ESEM, TEM, and CLSM in liposomal characterization: A comparative study. Int. J. Nanomed. 2011, 6, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering; John Wiley: New York, NY, USA, 1976. [Google Scholar]
- Grabielle-Madelmont, C.; Lesieur, S.; Ollivon, M. Characterization of loaded liposomes by size exclusion chromatography. J. Biochem. Biophys. Methods 2003, 56, 189–217. [Google Scholar] [CrossRef]
- McNeil-Watson, F.; Tscharnuter, W.; Miller, J. A new instrument for the measurement of very small electrophoretic mobilities using phase analysis light scattering (PALS). Colloids Surf. A 1998, 140, 53–57. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C. Recognition by macrophages and liver cells of opsonized phospholipid vesicles and phospholipid headgroups. Pharm Res. 2001, 18. [Google Scholar] [CrossRef]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harashima, H.; Kiwada, H. Interactions of liposomes with cells in vitro and in vivo: Opsonins and receptors. Curr. Drug Metab. 2001, 2, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Grafmuller, A.; Shillcock, J.; Lipowsky, R. Pathway of membrane fusion with two tension-dependent energy barriers. Phys. Rev. Lett. 2007, 98. [Google Scholar] [CrossRef] [PubMed]
- Cuatrecasas, P.; Roth, T.F. Receptor-Mediated Endocytosis, Receptors and Recognition; Cuatrecasas, P., Roth, T.F., Eds.; Springer: London, UK, 1983; Volume 15. [Google Scholar]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Yang, B.; Wang, G.; Qin, G.; Wada, S.; Wang, J.Y. Two cholesterol derivative-based PEGylated liposomes as drug delivery system, study on pharmacokinetics and drug delivery to retina. Nanotechnology 2014, 25. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Marks, E.; Schneider, J.J.; Keely, S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: Selective targeting to diseased versus healthy tissue. Nanomedicine 2015, 11, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome-cell interactions in vitro: Effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient lipid-mediated DNA transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed]
- Rädler, J.O.; Koltover, I.; Salditt, T.; Safinya, C.R. Structure of DNA-cationic liposome complexes: DNA intercalation in multilamellar membranes in distinct interhelical packing regimes. Science 1997, 275. [Google Scholar] [CrossRef]
- Farago, O.; Grønbech-Jensen, N.; Pincus, P. Mesoscale computer modeling of lipid-DNA complexes for gene therapy. Phys. Rev. Lett. 2006, 96. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lee, A.; Lu, Y.; Lee, R.J. Vascular targeting of doxorubicin using cationic liposomes. Int. J. Pharm. 2007, 337, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.B.; Fukumura, D.; Brown, E.B.; Mazzola, L.M.; Izumi, Y.; Jain, R.K.; Torchilin, V.P.; Munn, L.L. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002, 62, 6831–6836. [Google Scholar] [PubMed]
- Campbell, R.B.; Ying, B.; Kuesters, G.M.; Hemphill, R. Fighting cancer: From the bench to bedside using second generation cationic liposomal therapeutics. J. Pharm. Sci. 2009, 98, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Dass, C.R. Improving anti-angiogenic therapy via selective delivery of cationic liposomes to tumour vasculature. Int. J. Pharm. 2003, 267. [Google Scholar] [CrossRef]
- Breton, M.; Berret, J.-F.; Bourgaux, C.; Kral, T.; Hof, M.; Pichon, C.; Bessodes, M.; Scherman, D.; Mignet, N. Protonation of lipids impacts the supramolecular and biological properties of their self-assembly. Langmuir 2011, 27, 12336–12345. [Google Scholar] [CrossRef] [PubMed]
- Freese, C.; Gibson, M.I.; Klok, H.-A.; Unger, R.E.; Kirkpatrick, C.J. Size- and Coating-Dependent Uptake of Polymer-Coated Gold Nanoparticles in Primary Human Dermal Microvascular Endothelial Cells. Biomacromolecules 2012, 13, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Rajabi, M.; Mousa, S.A. Multifunctional Nanomaterials and Their Applications in Drug Delivery and Cancer Therapy. Nanomaterials 2015, 5, 1690–1703. [Google Scholar] [CrossRef]
- Freese, C.; Schreiner, D.; Anspach, L.; Bantz, C.; Maskos, M.; Unger, R.E.; Kirkpatrick, C.J. In vitro investigation of silica nanoparticle uptake into human endothelial cells under physiological cyclic stretch. Part. Fibre Toxicol. 2014, 24. [Google Scholar] [CrossRef] [PubMed]
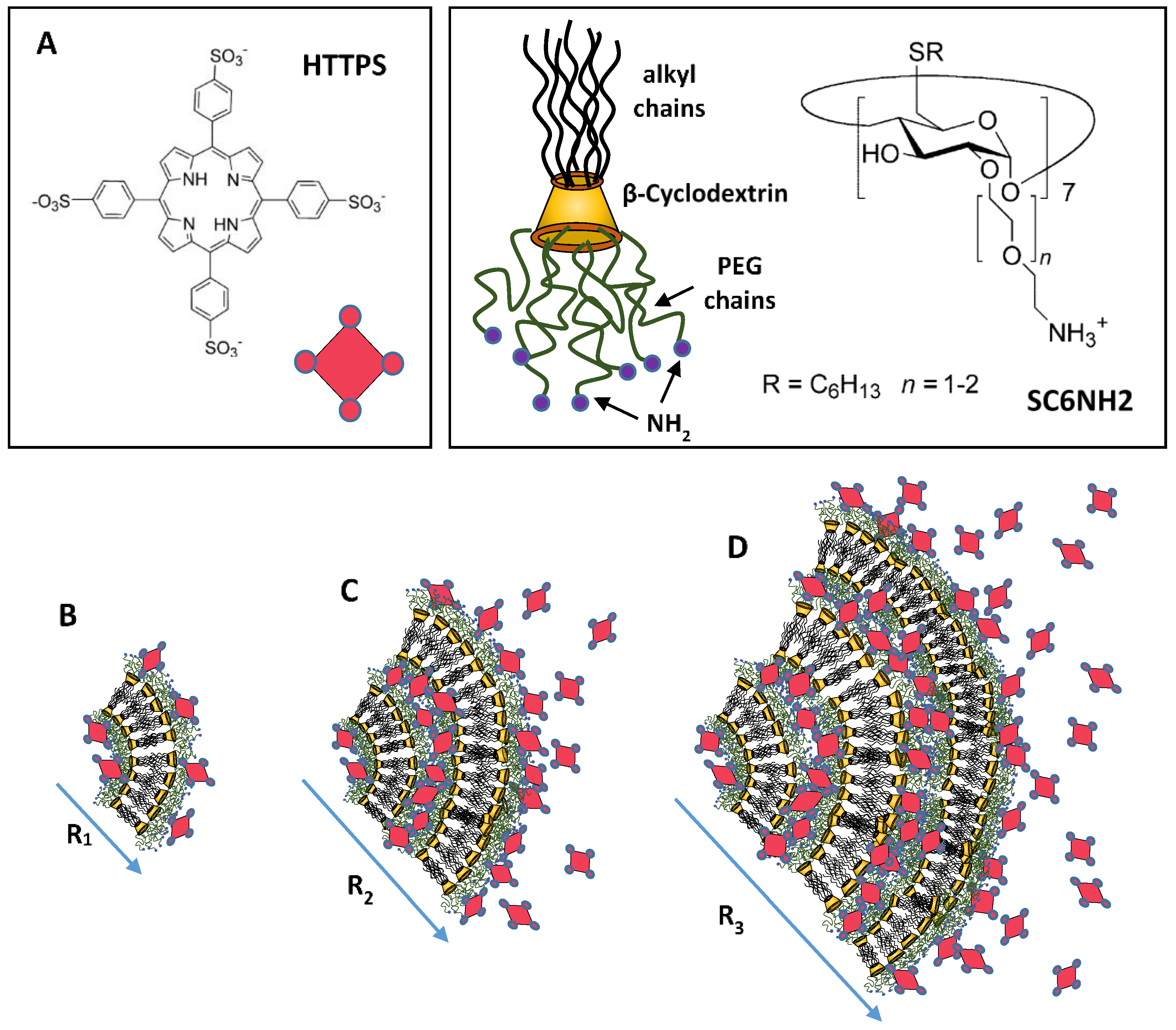
- Mazzaglia, A.; Angelini, N.; Lombardo, D.; Micali, N.; Patané, S.; Villari, V.; Scolaro, L.M. Amphiphilic Cyclodextrin Carriers Embedding Porphyrins: Charge and Size Modulation of Colloidal Stability in Heterotopic Aggregates. J. Phys. Chem. B 2005, 109, 7258–7265. [Google Scholar] [CrossRef] [PubMed]
- Mazzaglia, A.; Angelini, N.; Darcy, R.; Donohue, R.; Lombardo, D.; Micali, N.; Sciortino, M.T.; Villari, V.; Scolaro, L.M. Novel Heterotopic Colloids of Anionic Porphyrins Entangled in Cationic Amphiphilic Cyclodextrins: Spectroscopic Investigation and Intracellular Delivery. Chem. Eur. J. 2003, 9, 5762–5769. [Google Scholar] [CrossRef] [PubMed]
- Bourgaux, C.; Couvreur, P. Interactions of anticancer drugs with biomembranes: What can we learn from model membranes? J. Control Release 2014, 190, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Wanderlingh, U.; D’Angelo, G.; Branca, C.; Conti Nibali, V.; Trimarchi, A.; Rifici, S.; Finocchiaro, D.; Crupi, C.; Ollivier, J.; Middendorf, H.D. Multi-component modeling of quasielastic neutron scattering from phospholipid membranes. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, M.A.; Janich, M.; Hildebrand, A.; Strunz, P.; Neubert, R.H.H.; Lombardo, D. Structural transition in aqueous lipid/bile salt [DPPC/NaDC] supramolecular aggregates: SANS and DLS study. Chem. Phys. 2013, 424. [Google Scholar] [CrossRef]
- Kadajji, V.G.; Betageri, G.V. Water Soluble Polymers for Pharmaceutical Applications. Polymers 2011, 3, 1972–2009. [Google Scholar] [CrossRef]
- Harris, J.M.; Martin, N.E.; Modi, M. PEGylation: A novel process for modifying pharmacokinetics. Clin. Pharmacokinet. 2001, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. Stealth' corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomedicine 2006, 1, 297–315. [Google Scholar] [PubMed]
- Morelli, C.; Maris, P.; Sisci, D.; Perrotta, E.; Brunelli, E.; Perrotta, I.; Panno, M.L.; Tagarelli, A.; Versace, C.; Casula, M.F.; et al. PEG-templated mesoporous silica nanoparticles exclusively target cancer cells. Nanoscale 2011, 3, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, K.; Tuguntaev, R.G.; Mozhi, A.; Zhang, J.; Wang, P.C.; Liang, X.J. Targeting tumor microenvironment with PEG-based amphiphilic nanoparticles to overcome chemoresistance. Nanomedicine 2016, 12, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Micali, N.; Villari, V.; Kiselev, M.A. Large structures in diblock copolymer micellar solution. Phys. Rev. E 2004, 70. [Google Scholar] [CrossRef] [PubMed]
- Mallamace, F.; Beneduci, R.; Gambadauro, P.; Lombardo, D.; Chen, S.H. Glass and percolation transitions in dense attractive micellar system. Physica A 2001, 302, 202–219. [Google Scholar] [CrossRef]
- Li, C.; Lavigueur, C.; Zhu, X.X. Aggregation and Thermoresponsive Properties of New Star Block Copolymers with a Cholic Acid Core. Langmuir 2011, 27, 11174–11179. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Mallamace, F.; Faraone, A.; Gambadauro, P.; Lombardo, D.; Chen, W.R. Observation of a re-entrant kinetic glass transition in a micellar system with temperature-dependent attractive interaction. Eur. Phys. J. E 2002, 9, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Bo, S.; Ji, X. PH/Temperature-responsive behavior of amphiphilic block copolymer micelles prepared using two different methods. Langmuir 2011, 27, 7385–7391. [Google Scholar] [CrossRef] [PubMed]
- Dzieciuch, M.; Rissanen, S.; Szydłowska, N.; Bunker, A.; Kumorek, M.; Jamróz, D.; Vattulainen, I.; Nowakowska, M.; Róg, T.; Kepczynski, M. PEGylated Liposomes as Carriers of Hydrophobic Porphyrins. J. Phys. Chem. B 2015, 119, 6646–6657. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef]
- Menger, F.M.; Gabrielson, K.D. Cytomimetic Organic Chemistry: Early Developments. Angew. Chem. 1995, 34, 2091–2096. [Google Scholar] [CrossRef]
- Cevc, G.; Richardsen, H. Lipid vesicles and membrane fusion. Adv. Drug Deliv. Rev. 1999, 38, 207–232. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Verwey, E.J.W.; Overbeek, J.T.G. Theory of the stability of lyophobic colloids; Elsevier: Amsterdam, The Netherlands, 1948. [Google Scholar]
- Kumar, S.; Aswal, V.K.; Kohlbrecher, J. Small-Angle Neutron Scattering Study of Interplay of Attractive and Repulsive Interactions in Nanoparticle–Polymer System. Langmuir 2016, 32, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.P.; Mc Donald, I.A. Theory of Simple Liquids; Academic Press: New York, NY, USA, 1986. [Google Scholar]
- Belloni, L. Neutron X-ray and Light Scattering; Lindner, P., Zemb, T., Eds.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Cantù, L.; Corti, M.; Zemb, T.; Williams, C. Small angle X-ray and neutron scattering from ganglioside micellar solutions. J. Phys. IV 1993, 3, 221–227. [Google Scholar] [CrossRef]
- Abramo, M.C.; Caccamo, C.; Costa, D.; Pellicane, G.; Ruberto, R.; Wanderlingh, U. Effective interactions in lysozyme aqueous solutions: A small-angle neutron scattering and computer simulation study. J. Chem. Phys. 2012, 136. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D. Modeling dendrimers charge interaction in solution: Relevance in biosystems. Biochem. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Paleos, C.M.; Tsiourvas, D.; Sideratou, Z. Hydrogen bonding interactions of liposomes simulating cell-cell recognition: A review. Orig. Life Evol. Biosph. 2004, 34, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Chiruvolu, S.; Walker, S.; Israelachvili, J.; Schmitt, F.J.; Leckband, D.; Zasadzinski, J.A. Higher order self-assembly of vesicles by site-specific binding. Science 1994, 264. [Google Scholar] [CrossRef]
- Noppl-Simson, D.A.; Needham, D. Avidin-Biotin Interactions at Vesicles Surfaces: Adsorption and Binding, Cross-Bridge Formation, and Lateral Interactions. Biophys. J. 1996, 70, 1391–1401. [Google Scholar] [CrossRef]
- Ma, M.; Gong, Y.; Bong, D. Lipid membrane adhesion and fusion driven by designed, minimally multivalent hydrogen-bonding lipids. J. Am. Chem. Soc. 2009, 131, 16919–16926. [Google Scholar] [CrossRef] [PubMed]
- Marques, B.F.; Schneider, J.W. Sequence-Specific Binding of DNA to Liposomes Containing Di-Alkyl Peptide Nucleic Acid (PNA) Amphiphiles. Langmuir 2005, 21, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Stengel, G.; Zahn, R.; Höök, F. DNA-Induced Programmable Fusion of Phospholipid Vesicles. J. Am. Chem. Soc. 2007, 129, 9584–9585. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Zhao, H.; Lee, J.I.; Reynolds, K.; Zhang, L.; Temple, R.; Lesko, L.J. Therapeutic protein-drug interactions and implications for drug development. Clin. Pharmacol. Ther. 2010, 87, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Ceresa, C.; Nicolini, G.; Rigolio, R.; Bossi, M.; Pasqua, L.; Cavaletti, G. Functionalized mesoporous silica nanoparticles: A possible strategy to target cancer cells reducing peripheral nervous system uptake. Curr. Med. Chem. 2013, 20, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Pogodin, S.; Werner, M.; Sommer, J.-U.; Baulin, V.A. Nanoparticle-Induced Permeability of Lipid Membranes. ACS Nano 2012, 6, 10555–10561. [Google Scholar] [CrossRef] [PubMed]
- Barreca, D.; Laganà, G.; Toscano, G.; Calandra, P.; Kiselev, M.A.; Lombardo, D.; Bellocco, E. The interaction and binding of flavonoids to human serum albumin modify its conformation, stability and resistance against aggregation and oxidative injuries. Biochim. Biophys. Acta. Gen. Subj. 2014, (in press). [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Moghaddam, B.; Kirby, D.J.; Mohammed, A.R.; Perrie, Y. The role of lipid geometry in designing liposomes for the solubilisation of poorly water soluble drugs. Int. J. Pharm. 2013, 453, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.O.; Claro de Souza, M.; Petrilli, R.; Barcellos, J.P.; Lee, R.J.; Marchetti, J.M. Liposomes as carriers of hydrophilic small molecule drugs: Strategies to enhance encapsulation and delivery. Colloids. Surf. B 2014, 123, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J. Control Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, M.A.; Lombardo, D.; Lesieur, P.; Kisselev, A.M.; Borbely, S.; Simonova, T.N.; Barsukov, L.I. Membrane self-assembly in mixed DMPC/NaC systems by SANS. Chem. Phys. 2008, 345, 173–180. [Google Scholar] [CrossRef]
- Wang, Y.; Shim, M.S.; Levinson, N.S.; Sung, H.W.; Xia, Y. Stimuli-Responsive Materials for Controlled Release of Theranostic Agents. Adv. Funct. Mater. 2014, 24, 4206–4220. [Google Scholar] [CrossRef] [PubMed]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: state of the art review. Int. J. Nanomed. 2014, 9, 4387–4398. [Google Scholar]
- Poon, R.T.P.; Borys, N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin. Pharmacother. 2009, 10, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; He, C.Q.; Lin, A.H.; Gu, W.; Chen, Z.P.; Li, W.; Cai, B.C. Thermosensitive liposomes with higher phase transition temperature for targeted drug delivery to tumor. Int. J. Pharm. 2014, 475, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-targeted liposome design: Challenges and fundamental considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updat. 2014, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.; Amitay, Y.; Ohana, P.; Shmeeda, H.; Gabizon, A. Targeting of pegylated liposomal mitomycin-C prodrug to the folate receptor of cancer cells: Intracellular activation and enhanced cytotoxicity. J. Control Release 2016, 225, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Minh le, V.; Liu, J.; Angelov, B.; Drechsler, M.; Garamus, V.M.; Willumeit-Römer, R.; Zou, A. Baicalin loaded in folate-PEG modified liposomes for enhanced stability and tumor targeting. Colloids Surf. B 2016, 140, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Kékicheff, P.; Iss, J.; Fajolles, C.; Charitat, T.; Daillant, J.; Marques, C.M. Sliding tethered ligands add topological interactions to the toolbox of ligand–receptor design. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Roy, I.; Yang, C.; Prasad, P.N. Nanochemistry and Nanomedicine for Nanoparticle-based Diagnostics and Therapy. Chem. Rev. 2016, 11, 2826–2885. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-L.; Xu, J.; Jin, Y.-J.; Zhao, D.-X.; Xie, H.-Y. Ru(II) polypyridyl complex-incorporated and folate-conjugated vehicle for cancer cell imaging and photoinduced inactivation. Analyst 2016, 141, 2948–2954. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-I.; Yeh, M.-K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar]
- Fan, Y.; Zhang, Q. Development of liposomal formulations: From concept to clinical investigations. Asian J. Pharm. Sci. 2013, 8, 81–87. [Google Scholar] [CrossRef]
- Yu, G.; Jie, K.; Huang, F. Supramolecular Amphiphiles Based on Host-Guest Molecular Recognition Motifs. Chem. Rev. 2015, 115, 7240–7303. [Google Scholar] [CrossRef] [PubMed]
- Calandra, P.; Caschera, D.; Liveri, V.T.; Lombardo, D. How self-assembly of amphiphilic molecules can generate complexity in the nanoscale. Colloids. Surf. A 2015, 484, 164–183. [Google Scholar] [CrossRef]
- Khaja, F.; Jayawardena, D.; Kuzmis, A.; Önyüksel, H. Targeted Sterically Stabilized Phospholipid siRNA Nanomedicine for Hepatic and Renal Fibrosis. Nanomaterials 2016, 6. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
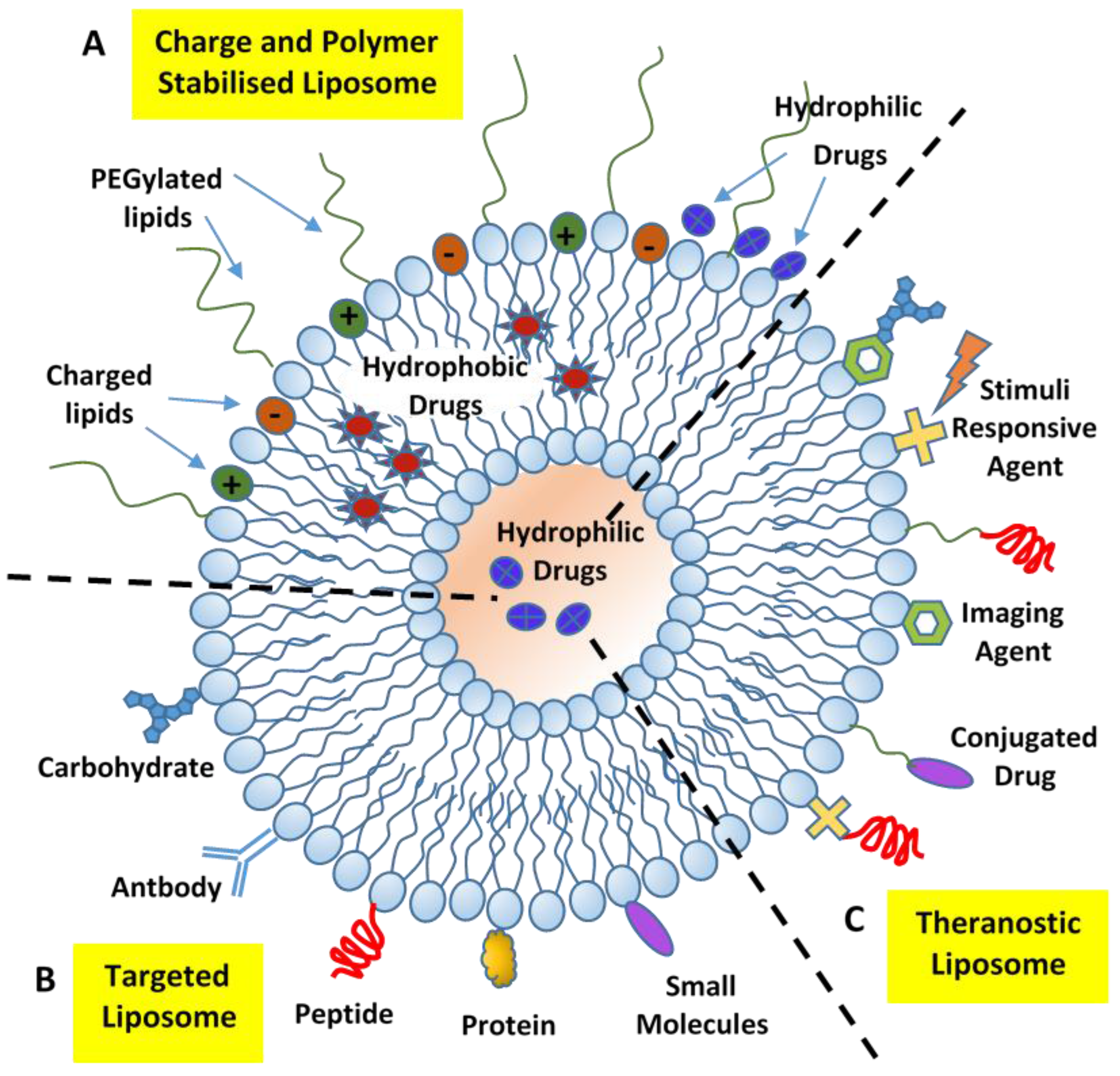{kind=link}
| Interaction Type | kJ/mol | Distance range (nm) |
|---|---|---|
| Covalent bond | 100–400 | 0.07–0.50 |
| Hydrogen bond | 4–120 | 0.3 |
| Hydrophobic interaction | <40 | variable |
| Electrostatic/ionic interaction | 20 | 0.25 |
| Van-der-Waals interaction | 0.4–5 | 0.3–0.6 |
| Steric stabilization | variable | variable |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardo, D.; Calandra, P.; Barreca, D.; Magazù, S.; Kiselev, M.A. Soft Interaction in Liposome Nanocarriers for Therapeutic Drug Delivery. Nanomaterials 2016, 6, 125. https://doi.org/10.3390/nano6070125
Lombardo D, Calandra P, Barreca D, Magazù S, Kiselev MA. Soft Interaction in Liposome Nanocarriers for Therapeutic Drug Delivery. Nanomaterials. 2016; 6(7):125. https://doi.org/10.3390/nano6070125
Chicago/Turabian StyleLombardo, Domenico, Pietro Calandra, Davide Barreca, Salvatore Magazù, and Mikhail A. Kiselev. 2016. "Soft Interaction in Liposome Nanocarriers for Therapeutic Drug Delivery" Nanomaterials 6, no. 7: 125. https://doi.org/10.3390/nano6070125
APA StyleLombardo, D., Calandra, P., Barreca, D., Magazù, S., & Kiselev, M. A. (2016). Soft Interaction in Liposome Nanocarriers for Therapeutic Drug Delivery. Nanomaterials, 6(7), 125. https://doi.org/10.3390/nano6070125