Abstract
The reasonable design of low-cost, high-activity single-atom catalysts (SACs) is crucial for achieving highly efficient electrochemical CO2RR. In this study, we systematically explore, using density functional theory (DFT), the performance of transition metal (TM = Mn, Fe, Co, Ni, Cu, Zn)-doped defect-type hexagonal boron nitride (h-BN) SACs TM@B−1N (B vacancy) and TM@BN−1 (N vacancy) in both CO2RR and the hydrogen evolution reaction (HER). Integrated crystal orbital Hamiltonian population (ICOHP) analysis reveals that these catalysts weaken the sp orbital hybridization of CO2, which promotes the formation of radical-state intermediates and significantly reduces the energy barrier for the hydrogenation reaction. Therefore, these theoretical calculations indicate that the Mn, Fe, Co@B−1N, and Co@BN−1 systems demonstrate excellent CO2 chemical adsorption properties. In the CO2RR pathway, Mn@B−1N exhibits the lowest limiting potential (UL = −0.524 V), and its higher d-band center (−0.334 eV), which aligns optimally with the adsorbate orbitals, highlights its excellent catalytic activity. Notably, Co@BN−1 exhibits the highest activity in HER, while UL is −0.217 V. Furthermore, comparative analysis reveals that Mn@B−1N shows 16.4 times higher selectivity for CO2RR than for HER. This study provides a theoretical framework for designing bifunctional SACs with selective reaction pathways. Mn@B−1N shows considerable potential for selective CO2 conversion, while Co@BN−1 demonstrates promising prospects for efficient hydrogen production.
1. Introduction
The continued rise in global energy demand and the accelerated consumption of fossil fuels [1,2] have become central challenges for the sustainable development of human society in the 21st century [3,4]. Although renewable energy technologies have advanced rapidly, their inherent intermittency severely limits large-scale application [5]. In this context, electrochemical CO2RR has gained considerable attention for its potential to convert greenhouse gases into high-energy-density hydrocarbons (such as CH4, C2H4, etc.), offering both environmental benefits and energy valorization [6,7,8]. The efficiency of this technology is largely dependent on catalyst design—highly active and selective electrocatalysts can significantly lower reaction overpotentials and precisely control product distribution [9,10,11].
Traditional noble metal catalysts, such as gold (Au) [12], palladium (Pd) [13], and platinum (Pt) [14], demonstrate exceptional catalytic activity in CO2RR. However, their widespread industrial application is severely hindered by high costs, limited availability, and inadequate thermal stability [15]. Since the introduction of SACs in 2011 [16], their atomically dispersed nature has garnered significant attention. Compared to conventional catalysts, SACs achieve nearly 100% atomic utilization [17] by anchoring transition metal (TM) atoms onto a support surface [18], dramatically improving economic efficiency. Theoretical studies suggest that electron transfer between the unoccupied d orbitals of TM atoms in SACs and the antibonding orbitals of CO2 effectively weakens the C=O bond, reducing its bond energy from 806 kJ·mol−1 to less than 750 kJ·mol−1 [19]. Additionally, the highly exposed active sites enhance intermediate adsorption and lower the reaction activation energy barrier, significantly boosting catalytic efficiency [20].
Two-dimensional materials, with their high specific surface area, ultra-fast carrier mobility, and abundant active sites at the interfaces [21], have emerged as ideal support platforms for TM single-atom catalysts in CO2RR. Among these, two-dimensional h-BN [22] forms a graphene-like honeycomb structure through sp2 hybridization. The strong covalent B-N bonds impart exceptional stability to the material, making it highly resistant to extreme conditions such as high temperatures and harsh acidic or basic environments [23]. By engineering B/N vacancies, strong coordination anchoring sites can be created on the surface of h-BN, allowing for high-density loading of TM single atoms. This facilitates an efficient catalytic interface for C=O bond activation and multi-electron transfer processes [24].
In recent years, significant advancements have been made in the study of h-BN-based SACs. For instance, Okello et al. [25] successfully developed functionalized h-BN heterostructures by incorporating transition metal nanoparticles and single-atom impurities into h-BN. Through a combination of scanning transmission electron microscopy (STEM) and density functional theory (DFT) analysis, they discovered that Ta, Co, Ni, and Ir single atoms preferentially adsorb at the N-top site of h-BN. The strong agreement between experimental and theoretical results offers valuable insights for catalyst design. Chen et al. [26] loaded Pd nanoparticles (NPs) onto the surface of h-BN and found that the strong interaction between h-BN and Pd could tune the d-band center of Pd, optimizing the adsorption energy of reaction intermediates and significantly enhancing electrocatalytic performance. Li et al. [27], through DFT screening, discovered that Fe-doped h-BN (Fe@BN) exhibits the lowest barrier (0.52 eV) and the highest exothermic energy (4.37 eV) for CO oxidation, demonstrating exceptional catalytic potential. Additionally, Wang et al. [28] confirmed that Cu@O1N2-BN catalysts show a very low rate-limiting step energy barrier of 0.46 eV in the direct methane-to-methanol conversion reaction. Zhao et al. [29] found that Mo@BN monolayers exhibit an overpotential of only 0.19 V for nitrogen reduction reaction (NRR), highlighting their outstanding catalytic activity. Konopatsky A et al. [30] reported the Ag-doped layered ferrihydrite (Fh)/h-BN heterostructures synthesized via the solvent thermal synthesis method exhibited a 2.5-fold enhancement in CO2 conversion efficiency compared to the initial value, with olefin selectivity increasing from 0.31% to 0.88%.
Although h-BN-based SACs have demonstrated exceptional performance in areas such as HER [31], oxygen evolution reaction (OER) [32], and nitrogen reduction reaction (NRR) [33,34], and CO2RR, this study will conduct an in-depth theoretical investigation into the mechanism of CO2RR on h-BN-based SACs. Experimental studies have demonstrated that Fe-doped h-BN catalysts exhibit promising CO2 conversion efficiency; however, their stability remains suboptimal, necessitating further exploration of the influence of h-BN defect types (e.g., B vacancies versus N vacancies) on catalytic pathways and the extension of research to other transition metal/h-BN systems [35]. To address these gaps, we systematically explore the CO2RR performance of Fe and other transition metals within the same period (Mn, Co, Ni, Cu, and Zn) embedded in defective h-BN (TM@B−1N and TM@BN−1) through DFT calculations. The focus is on analyzing structural stability, electronic properties, and the kinetics of C=O bond activation, with the goal of providing a theoretical foundation for designing defected h-BN-based SACs and optimizing their application in CO2RR and other related reactions.
2. Materials and Methods
In this study, first-principles calculations based on DFT are performed using the Vienna Ab Initio Simulation Package (VASP 6.3.0) software [36]. The Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional [37] within the generalized gradient approximation (GGA) framework is applied, with a plane-wave cutoff energy set to 500 eV. The Brillouin zone integration is carried out using the Monkhorst–Pack method [38], generating a 3 × 3 × 1 k-point mesh. The energy convergence criterion is set to 1 × 10−5 eV/atom, and the Hellmann–Feynman force convergence criterion during structural relaxation is set to less than 0.05 eV/Å for each atom. To accurately capture interlayer van der Waals interactions, the DFT-D3 method [39] is used for van der Waals corrections. In this study, charge transfer between the catalyst and the adsorbates is analyzed based on quantum theory of atoms in molecules (QTAIM) [40] charge calculations, using the Bader charge analysis method [41]. The atomic electronic configurations for the elements involved in this study are as follows: B: 2s22p1, N: 2s22p3, Mn: 3d54s2, Fe: 3d64s2, Co: 3d74s2, Ni: 3d84s2, Cu: 3d104s1, Zn: 3d104s2.
In this study, the pristine hexagonal boron nitride (h-BN) crystal structure serves as the foundational model. It belongs to the P63/mmc space group (No. 194) with lattice parameters of a = b = 2.510 Å, c = 6.690 Å, α = β = 90°, and γ = 120° [42].
3. Results
3.1. Subsection
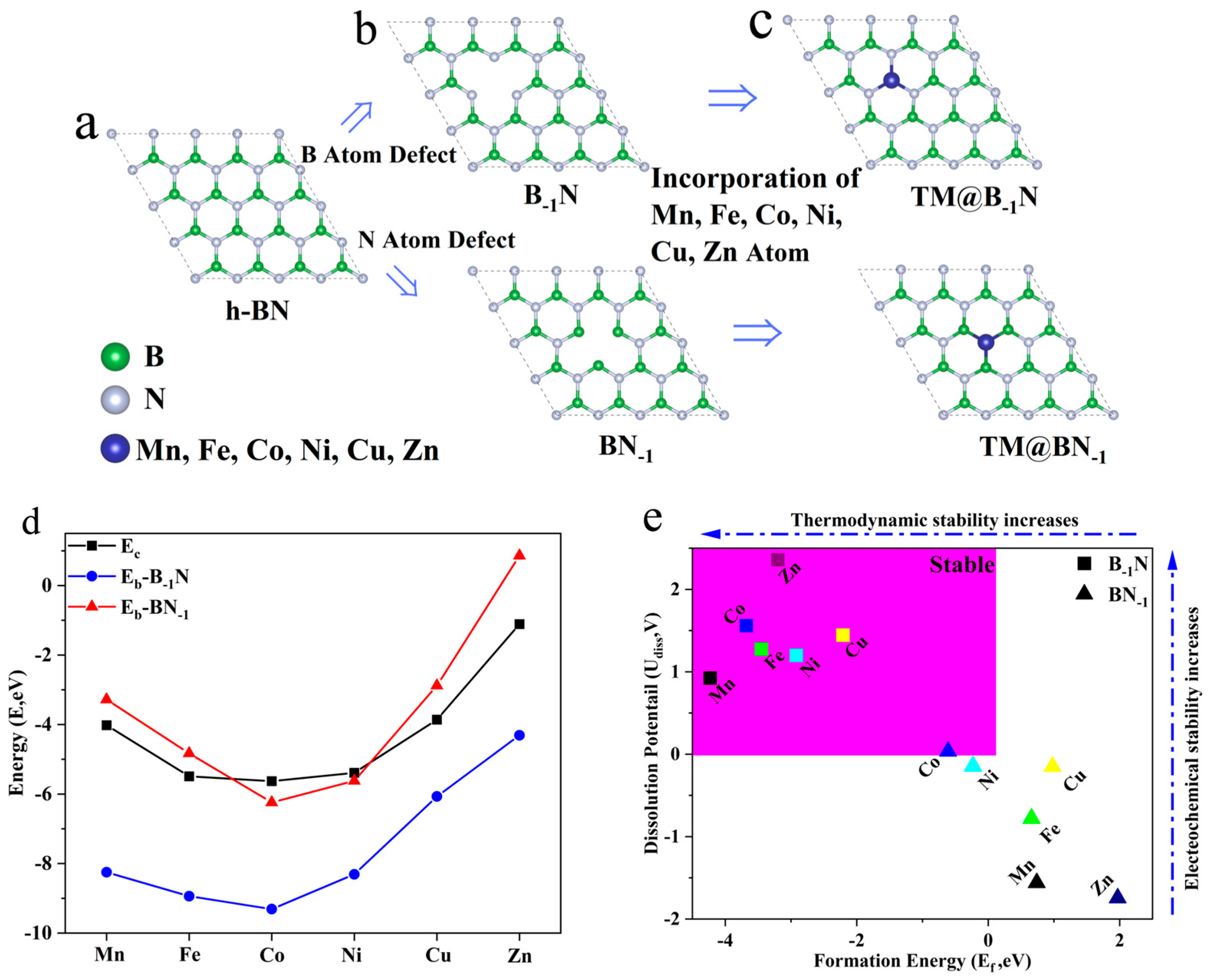
To construct the monolayer h-BN model, the structure is exfoliated along the (001) plane, and a 20 Å vacuum layer is introduced in the vertical direction to eliminate periodic interactions. After geometric optimization (Figure 1a), the equilibrium lattice parameters of the h-BN monolayer converge to a = b = 2.464 Å and c = 19.447 Å. To further reduce edge effects, a 3 × 3 × 1 supercell is constructed to establish a periodic system (Figure 1b). A single B or N atom is selectively removed from the center of the supercell, resulting in B single-vacancy (B−1N) and N single-vacancy (BN−1) defect structures, respectively [43].
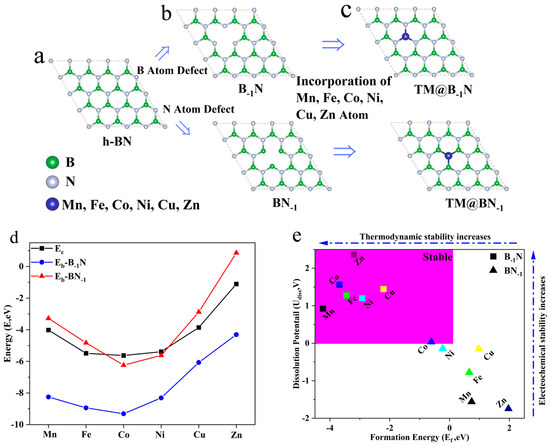
Figure 1.
(a) Structure of h-BN; (b) structure of B−1N and BN−1; (c) structure of TM@B−1N and TM@BN−1; (d) binding energy and cohesive energy in TM@B−1N and TM@BN−1; (e) formation energy and dissolution potential of metal atoms in TM@B−1N and TM@BN−1.
To construct the single-atom catalyst model (Figure 1c), transition metal atoms TM are embedded into the defect sites with sub-angstrom precision (Δz = 0.1 Å). In the B-1N system, the metal atom coordinates with three neighboring N atoms, forming an M-N3 active center (denoted as TM@B−1N). Conversely, in the BN−1 system, the metal atom bonds with three B atoms, creating an TM-B3 active site (denoted as TM@BN−1). This asymmetric embedding effectively breaks the intrinsic symmetry of the support structure, establishing an ideal model system for exploring metal–support synergistic effects in single-atom catalysts.
To assess the single-atom dispersion stability of transition metal atoms at defect sites in h-BN, this study evaluates the catalytic systems’ stability using two key criteria: binding energy (Eb) and cohesive energy (Ec). The binding energy (Eb) quantifies the strength of interaction between the metal atom and the support, calculated as [44]:
Meanwhile, the cohesive energy (Ec) represents the metal atom’s tendency to aggregate, defined as [45]:
where Etotal, Edef, EM, and Ebulk denote the total energy of the loaded system, the energy of the defective support, the energy of an isolated metal atom, and the per atom energy in the bulk metal phase, respectively. More negative values of Eb and Ec indicate stronger binding interactions.
As illustrated in Figure 1d, the condition Eb < Ec < 0 signifies that metal–support interactions outweigh metal–metal aggregation tendencies, ensuring the stability of single-atom dispersion. The results reveal that in the B-vacancy system (TM@B−1N), all TMs satisfy Eb < Ec, demonstrating the robust anchoring capability of B vacancies for transition metals. Conversely, in the N-vacancy system (TM@BN−1), only Co and Ni meet the Eb < Ec criterion, while Mn, Fe, Cu, and Zn exhibit Eb > Ec, indicating a higher propensity to aggregate into nanoclusters at N-vacancy sites.
To comprehensively assess the stability of the catalytic system, this study further evaluates the feasibility of candidate systems based on two additional criteria: thermodynamic formation energy (Ef) and electrochemical dissolution potential (Udiss). These criteria are defined as follows [46]:
where (metal, bulk) represents the standard dissolution potential of the bulk metal, and ne denotes the number of electrons involved in the dissolution process. As illustrated in Figure 1e, when Ef < 0, the metal–support composite system exhibits lower energy than its separated components, indicating potential synthetic feasibility in experiments [47]. Moreover, a positive dissolution potential (Udiss > 0) suggests that the catalyst can resist metal dissolution under reductive conditions [48]. The computational results reveal that all TM@B−1N systems satisfy Ef < 0 and Udiss > 0, confirming both synthetic feasibility and electrochemical stability. The Co@BN−1 system meets the two stability criterion which are Ef < 0 and Udiss > 0. However, the Ni@BN−1 system fails to satisfy both structural stability conditions simultaneously, particularly due to Udiss < 0, which suggests a propensity for structural reconstruction during the catalytic process. Consequently, this system will not be taken into account in subsequent studies. Therefore, all TM@B−1N and Co@BN−1 systems demonstrate both thermodynamic and electrochemical stability, making them promising candidates for catalytic applications.
According to Table S2, in TM@B−1N systems, a shorter TM-N bond length correlates with a lower Eb, indicating greater stability. For Mn@B−1N and Cu@B−1N, although the difference in TM-N bond lengths is merely 0.00113 Å, the Eb differs by as much as 2.18 eV. This suggests that nitrogen atoms exhibit stronger bonding interactions with TM atoms that possess incompletely filled penultimate electron shells. Moreover, in both Co@B−1N and Co@BN−1 configurations, comparisons of TM-B/N bond lengths and Eb consistently demonstrate a stronger affinity between TM atoms and nitrogen atoms within the substrate.
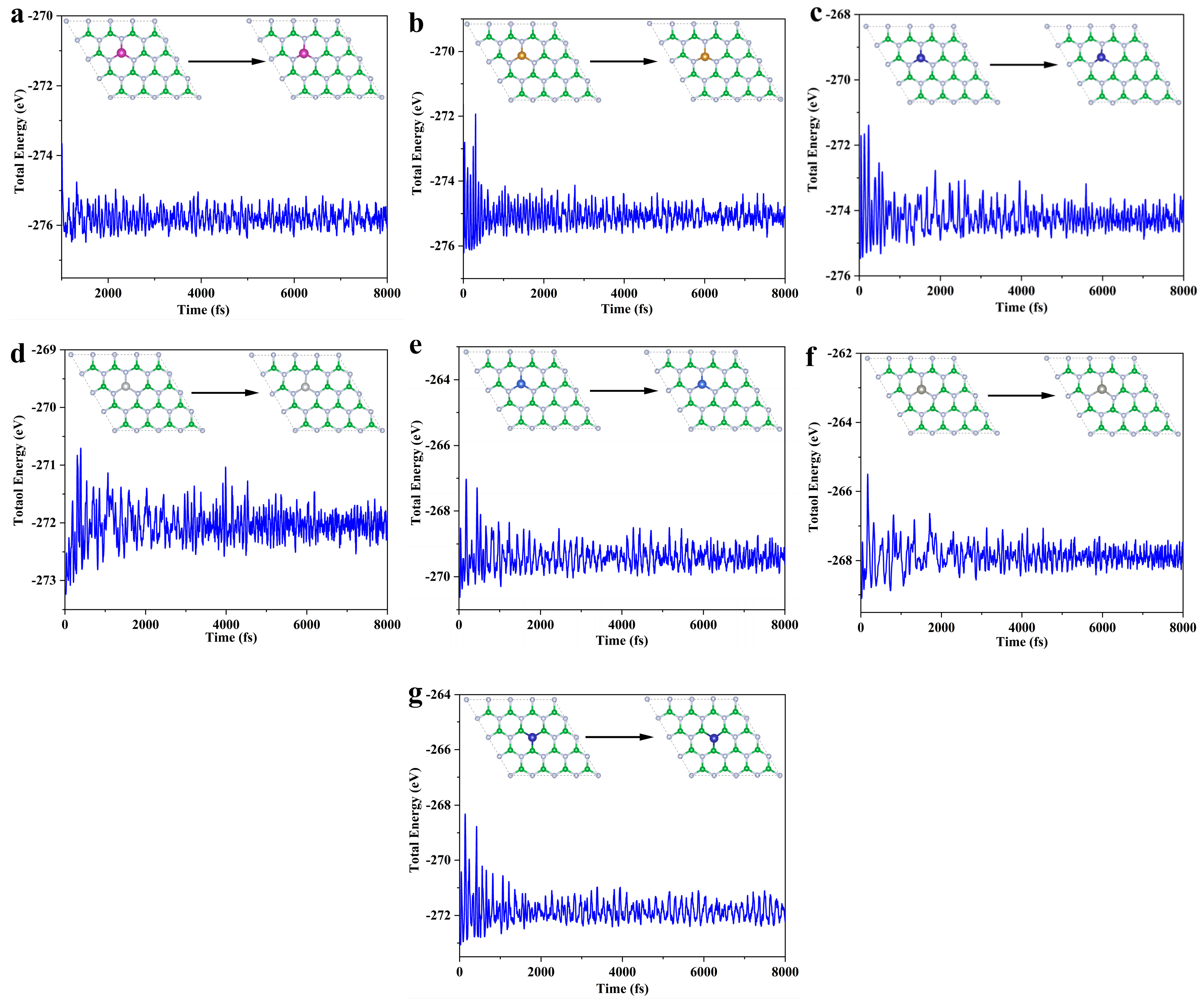
Building on the thermodynamic and electrochemical stability assessments, this study selects the TM@B−1N and Co@BN−1 systems for ab initio molecular dynamics (AIMD) [49] simulations to further evaluate their dynamic stability. The simulations are conducted at 300 K (room temperature) for a total duration of 8 ps (8000 steps, with a time step of 1 fs). As illustrated in Figure 2, the dynamic behavior analysis of all systems indicates minimal fluctuations in TM-N/B bond lengths, with no evidence of metal–support bond dissociation. Furthermore, the total energy fluctuations gradually stabilize, satisfying the criteria for dynamic stability [50]. These results confirm that the selected SACs possess both structural robustness and dynamic stability under ambient conditions, making them promising candidates for practical catalytic applications.
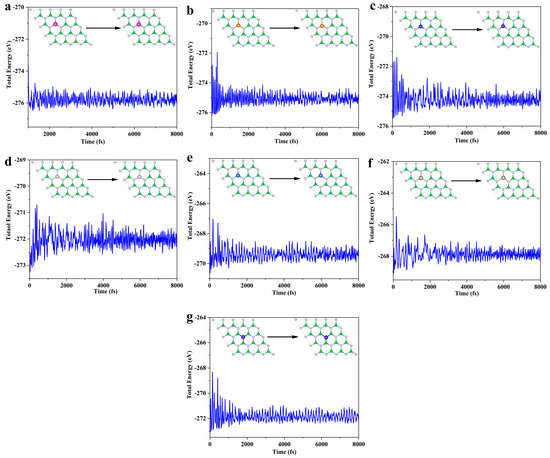
Figure 2.
SACs AIMD: (a) Mn@B−1N; (b) Fe@B−1N; (c) Co@B−1N; (d) Ni@B−1N; (e) Cu@B−1N; (f) Zn@B−1N; (g) Co@BN−1.
3.2. CO2 Adsorption and Activation
In this study, the CO2 adsorption and activation mechanisms are systematically explored through a combination of density of states (DOS) analysis and adsorption calculations. As shown in Figure S1, transition metal doping induces significant modulation of the electronic structure. Specifically, the reduced bandgap in the TM@B−1N and Co@BN−1 systems enhances their conductivity [51], thereby promoting charge transfer during the catalytic process.
The selectivity of CO2 electrochemical reduction pathways (producing CH4, CH3OH, or CO) is strongly influenced by its adsorption configuration and activation degree [52]. To quantify the interaction strength between the catalyst and reactant, the adsorption energy (Eads) is calculated as follows:
where Etotal-CO2, Etotal, and ECO2 represent the total energy of the adsorption system, the intrinsic energy of the catalyst, and the energy of a free CO2 molecule, respectively [53].
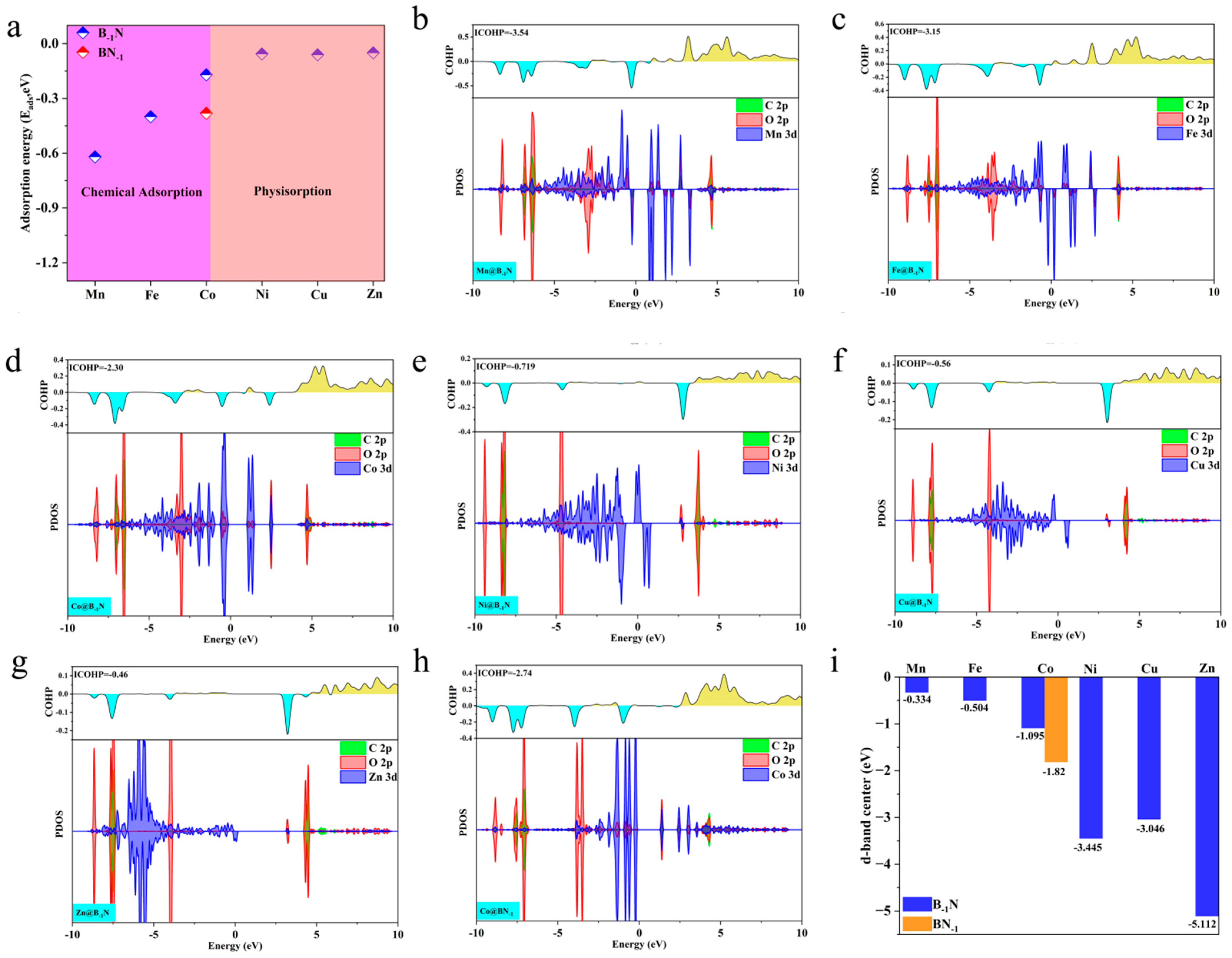
As shown in Figure 3a, Mn@B−1N, Fe@B−1N, Co@B−1N, and Co@BN−1 exhibit significantly exothermic adsorption, indicating strong chemisorption of CO2. In contrast, Ni@B−1N, Cu@B−1N, and Zn@B−1N show adsorption energies close to zero, suggesting weak physisorption. The electronic interaction mechanisms were systematically investigated through electron localization function (ELF) analysis (Figure S2). For both Mn/Fe/Co@B−1N and Co@BN−1 configurations, the ELF values spanning the full spectrum from 0 to 1 between CO2 molecules and catalytic active sites reveal significant charge redistribution patterns. This complete coverage of ELF values across the theoretical range (η = 0.4) provides conclusive evidence for the establishment of robust chemical bonding interactions, confirming the coexistence of covalent character and strong ionic bonding components in these catalyst–CO2 complexes. For the Ni/Cu/Zn@B−1N system, ELF analysis that the computed values persistently cluster near the bound (η ≈ 0), there is no exchange between the components. Electrons are localized on the components of the system. This corresponds to the interaction of closed shells. In the case of (d, e, f), there is an interaction of closed shells, i.e., van der Waals bonds, providing conclusive evidence of van der Waals-dominated interactions characterized by weak electronic coupling. These distinct adsorption modes directly impact catalytic performance. Only chemisorbed CO2 undergoes molecular bending and C=O bond elongation, facilitating effective charge transfer and molecular activation—critical prerequisites for subsequent reduction reactions.
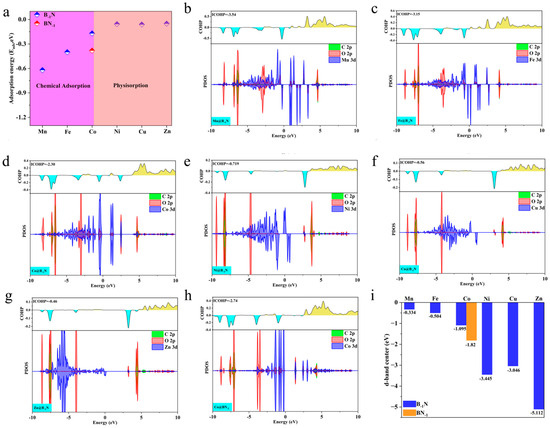
Figure 3.
(a) Adsorption energies of CO2 on TM@B−1N and Co@BN−1 surfaces; (b–h) PDOS and the COHPs of CO2 on TM@B−1N and Co@BN−1 surfaces; the bonding and antibonding states in COHP are depicted by cyan and yellow, respectively. (i) The d-band center of TM@B−1N and Co@BN−1.
To further clarify the electronic nature of CO2 activation in chemisorption and physisorption, this study employs projected density of states (PDOS) and crystal orbital Hamilton population (COHP) analyses to investigate the interaction mechanisms between metal atoms and the adsorbed CO2 molecule. As illustrated in Figure 3b–d,h, chemisorbed systems (Mn/Fe/Co@B−1N and Co@BN−1) exhibit pronounced hybridization peaks near the Fermi level, arising from the interaction between metal d orbitals and the antibonding π* orbitals of CO2. This strong orbital coupling induces molecular bending and C=O bond elongation while facilitating a dual-channel charge transfer mechanism: the unoccupied d orbitals of the metal capture electrons from the bonding orbitals of CO2, while the occupied d orbitals donate electrons into the CO2 antibonding orbitals. Consequently, the CO2 molecule acquires radical-like intermediate characteristics, enhancing its activation potential.
ICOHP analysis further substantiates these findings. As shown in Figure 3b–d,h, the chemisorbed systems exhibit strongly negative ICOHP values (Mn@B−1N: −3.54 eV, Fe@B−1N: −3.15 eV, Co@B−1N: −2.30 eV, Co@BN−1: −2.74 eV), signifying deep orbital coupling between the metal d orbitals and the CO2 π* orbitals [54]. This robust interaction induces orbital hybridization near the Fermi level with the s and p orbitals of C and O, weakening the sp hybridization strength of CO2 and lowering the C=O dissociation energy barrier. Conversely, Figure 3e–g illustrate that in the physisorbed systems (Ni/Cu/Zn@B−1N), the overlap between metal d orbitals and CO2 π* orbitals is negligible (< 0.05), and CO2 retains its linear configuration due to van der Waals interactions alone. The weak ICOHP values (Ni@B−1N: −0.72 eV, Cu@B−1N: −0.56 eV, Zn@B−1N: −0.46 eV) further confirm the feeble nature of this interaction, aligning with the minimal charge transfer (ΔQ < 0.02 e−, Table S2). These findings highlight that the strength of d-π* orbital coupling, by regulating electronic state occupancy, plays a decisive role in determining the degree of CO2 activation and ultimately dictates the selectivity of subsequent reduction reaction pathways.
The d-band center, a key descriptor of the electronic structure of transition metal surfaces, is defined as the weighted average energy of the metal d-orbital density of states. It is calculated using the following equation [55]:
A higher d-band center value indicates that the d-orbital energy levels shift closer to the Fermi level, thereby enhancing orbital hybridization with adsorbed species [56]. As shown in Figure 3i, the d-band center values of the TM@B−1N and Co@BN−1 systems follow the order: Mn@B−1N (−1.8 eV) > Fe@B−1N (−2.1 eV) > Co@BN−1 (−2.3 eV) > Co@B−1N (−2.5 eV) > Ni/Cu/Zn@B−1N (−3.2 to −3.6 eV). This trend strongly correlates with the observed decrease in Eads and ICOHP. In systems with lower d-band centers (Ni/Cu/Zn@B−1N), the d-orbital energy levels are significantly lower than the CO2 π* orbitals, preventing effective disruption of CO2 sp-hybridized bonds and resulting in weak physisorption via van der Waals interactions. In contrast, catalysts with higher d-band centers (Mn/Fe/Co-based systems) promote CO2 activation through strong d–π* antibonding orbital coupling. The predicted catalytic activity follows the order: Mn@B−1N > Fe@B−1N > Co@BN−1 > Co@B−1N, providing an electronic structure-based framework for selecting optimal reaction pathways [57].
3.3. CO2 Reduction Reaction (CO2RR) Performance
This study utilizes the computational hydrogen electrode (CHE) model proposed by Nørskov et al. [58] to assess the thermodynamic feasibility of CO2 electrochemical reduction pathways by computing the Gibbs free energy change (ΔG). The calculation is expressed as [59]:
where EH is the total energy of the system, EZPE represents the zero-point energy, and S corresponds to the entropy change, determined under a fixed-atom model for the catalyst.
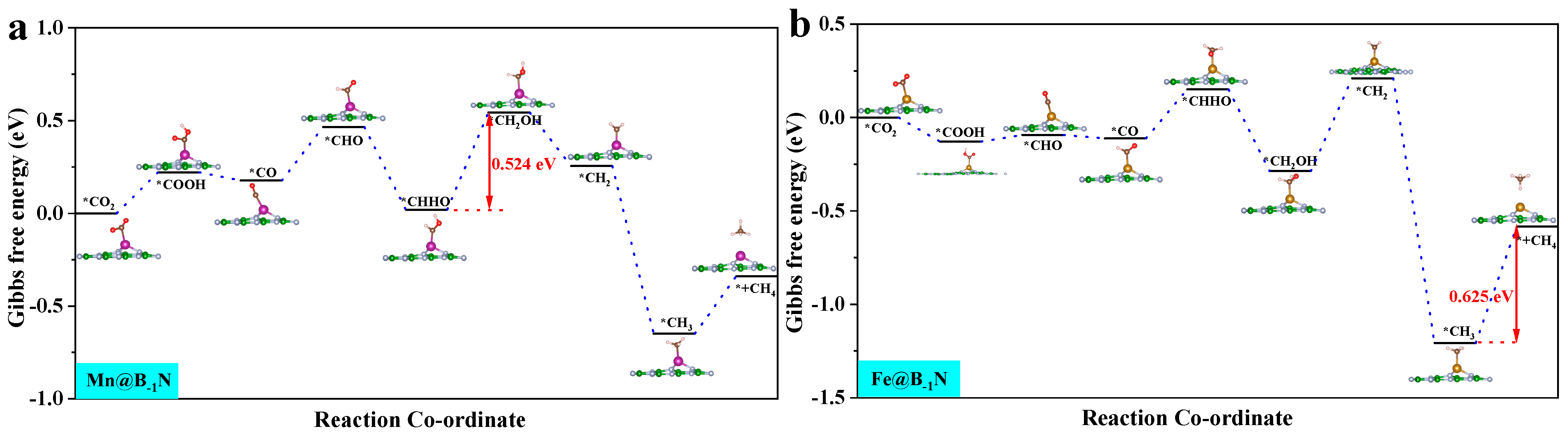
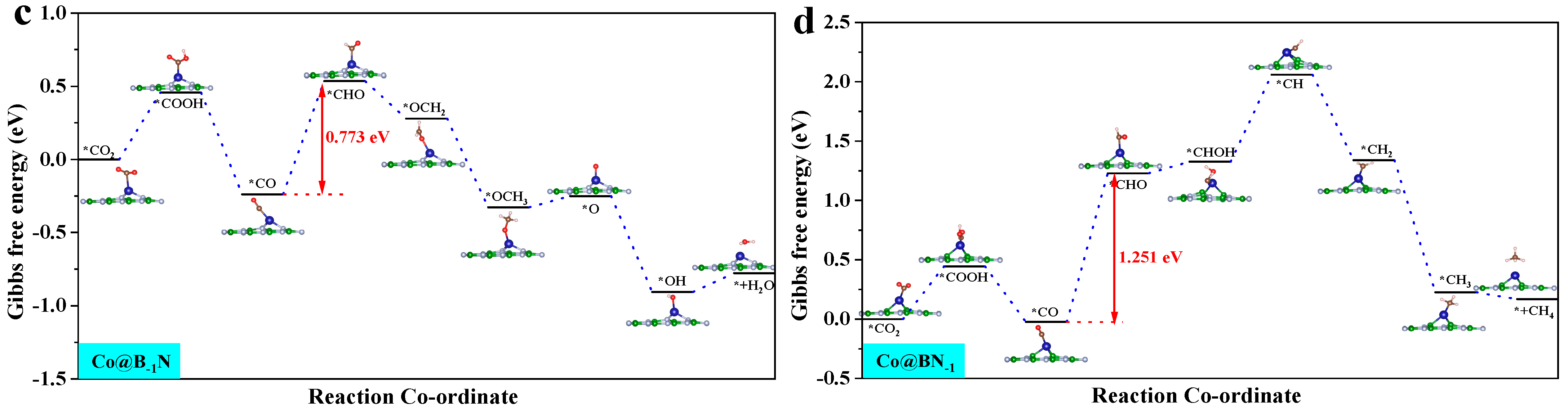
By analyzing the ΔG for the Mn@B−1N, Fe@B−1N, Co@B−1N, and Co@BN−1 catalytic systems (Figure S3), the potential determining step (PDS) for each reaction pathway is identified. According to the Brønsted–Evans–Polanyi [60] relationship, reaction steps with lower ΔG values correspond to lower activation energy barriers, thereby controlling the reaction progression. Key pathway analysis reveals the following: CO hydrogenation (*CO + H⁺ + e⁻ → *CHO/*COH): all catalysts preferentially form the *CHO intermediate. CHO hydrogenation (*CHO + H⁺ + e⁻ → *CHOH/*CH2O): Mn@B−1N and Co@BN−1 favor *CHOH formation (ΔG = 0.42 eV). In the Fe@B−1N pathway, OCHH forms first (ΔG = 0.77 eV); however, its subsequent *OH desorption barrier (1.2 eV) is significantly lower than that of the *CHOH pathway (1.8 eV), making OCHH the preferred route. Co@B−1N follows a distinct mechanism: the *COH → *CHOH step presents a prohibitively high ΔG of 3.73 eV, whereas *OCHH formation is more energetically favorable with a maximum ΔG of 2.15 eV, confirming *OCHH as the preferred intermediate.
These results indicate that Mn-/Fe-based catalysts with higher d band centers stabilize the *CHO intermediate, thereby lowering the reduction energy barrier. In contrast, Co-based systems exhibit pathway selectivity differences due to geometric steric effects, providing theoretical guidance for tuning multi-carbon product formation.
UL represents the minimum overpotential required to drive the PDS in a catalytic reaction. It is calculated as [61]:
where ΔGPDS is the Gibbs free energy change associated with the PDS, and e is the elementary charge. A more negative UL signifies a higher thermodynamic energy barrier, indicating reduced catalytic activity [62]. Figure 4 presents the optimal CO2 reduction pathways for Mn@B−1N, Fe@B−1N, Co@B−1N, and Co@BN−1 catalysts, with their UL values ranked as follows: Mn@B−1N (−0.524 V) > Fe@B−1N (−0.612 V) > Co@B−1N (−0.683 V) > Co@BN−1 (−0.718 V). For the Mn@B−1N system, the PDS is *CHHO + (H⁺ + e⁻) + H2O → *CH2OH + H2O, which corresponds to the lowest UL value, indicating the highest catalytic activity. A comprehensive comparative analysis (Table 1) reveals a monotonic decrease in UL values that exhibits strong correlation with the sequential reduction in both ICOHP parameters and d band center positions, demonstrating that the energy alignment of metal d-orbitals orchestrates catalytic performance through a dual-parameter modulation mechanism: simultaneously regulating activation kinetics through ICOHP and d band center [63].
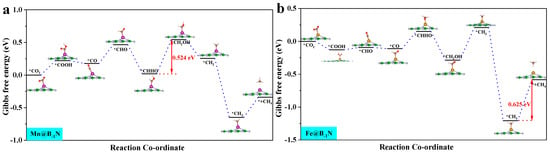
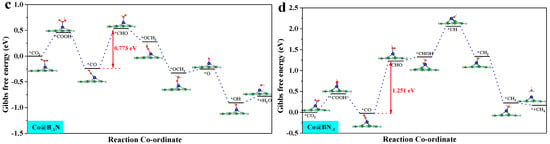
Figure 4.
Gibbs free energy step diagram of single-atom catalyst CO2RR: (a) Mn@B−1N; (b) Fe@B−1N; (c) Co@B−1N; (d) Co@BN−1. The “*” in the figure indicates the catalyst.

Table 1.
Comparative table of catalytic activity results.
3.4. Competition Between CO2RR and HER
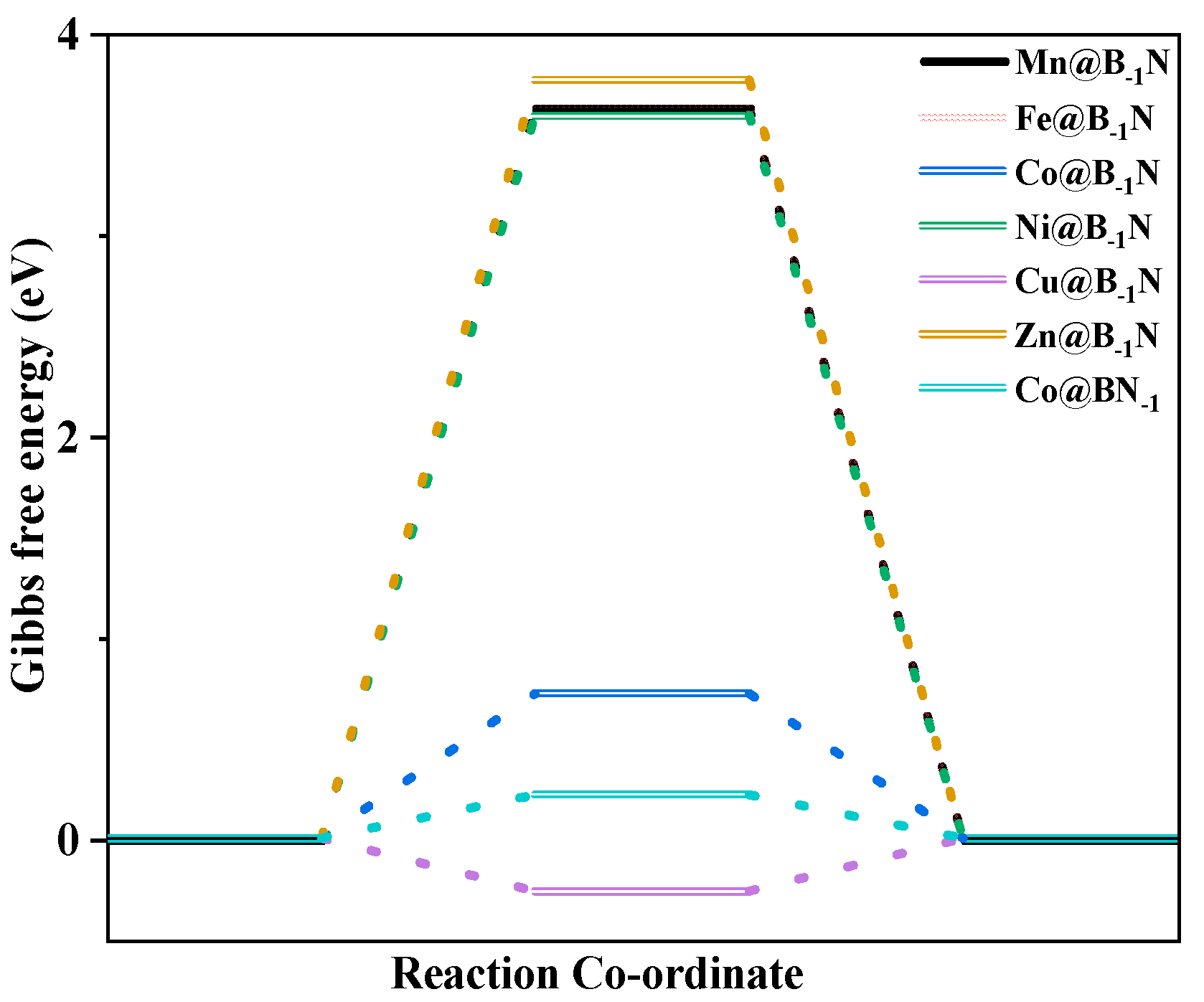
To elucidate the competitive selectivity between CO2RR and the HER on TM@B−1N and Co@BN−1 catalysts, this study systematically evaluates UL and the Gibbs free energy change (ΔG) in key intermediates to uncover the underlying mechanism [64,65]. HER performance analysis (Figure 5) reveals that Co@BN−1 demonstrates the highest hydrogen evolution activity (UL = −0.217 V), significantly surpassing other systems (Mn@B−1N: −3.631 V, Fe@B−1N: −3.635 V, Co@B−1N: −0.721 V, Cu@B−1N: −0.263 V, Zn@B−1N: −3.775 V). By comparing with Table S4, it is evident that the charge transfer between Co@BN-1 and the adsorbed H is more efficient. This superior HER activity in Co@BN−1 can be attributed to the strong hybridization between Co 3d orbitals and adjacent B 2p orbitals near the Fermi level, which markedly enhances proton (H+ + e−) adsorption and promotes the Volmer–Heyrovsky reaction pathway.
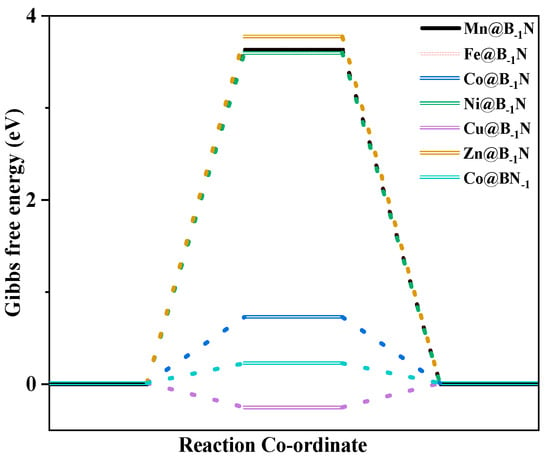
Figure 5.
Diagram of the HER Gibbs free energy step of SACs.
By analyzing Table 2 in conjunction with the Brønsted–Evans–Polanyi relationship, it is evident that the Mn/Fe/Co@B−1N systems favor CO2 reduction, as the Gibbs free energy change for CO2 activation (G*COOH) is significantly lower than that for HER (ΔG*H). In contrast, Co@BN−1 exhibits dominant HER activity due to its higher ΔG*COOH (0.28 eV) compared to ΔG*H (−0.15 eV), making hydrogen evolution energetically more favorable. This competitive mechanism underscores the critical role of the synergy between transition metal d orbitals and the coordination environment of the support in dictating CO2RR/HER pathway selectivity. By fine-tuning the relative stability of *COOH and *H intermediates, this synergy provides a precise means of steering reaction outcomes. These insights offer a dual-regulation strategy—integrating electronic structure engineering and thermodynamic modulation—for the rational design of highly selective electrocatalysts.
Table 2.
Gibbs free energy changes (ΔG) in initial protonation of CO2RR vs. HER on TM@B−1N and TM@BN−1 surfaces.
4. Conclusions
This study systematically elucidates the competitive regulation mechanisms of CO2RR and HER in transition metal-embedded defective h-BN systems (TM@B−1N and TM@BN−1). Key findings include: (1) optimal CO2RR activity: the Mn@B−1N system exhibits exceptional CO2RR performance, with its low limiting potential (UL = −0.524 V), high d-band center (−0.334 eV), and strong antibonding orbital coupling with CO2 (ICOHP = −3.54 eV) synergistically reducing the reaction energy barrier; (2) HER selectivity control: the Co@BN−1 system demonstrates HER activity (UL = −0.217 V) comparable to commercial Pt-based catalysts, driven by strong hybridization between Co-3d and B-2p orbitals near the Fermi level, which facilitates proton adsorption and Heyrovsky pathway kinetics; (3) reaction pathway competition: Mn/Fe/Co@B−1N systems activate CO2 via d-π* orbital coupling to disrupt sp hybridization, forming radical intermediates, whereas Ni/Cu/Zn@B−1N systems only adsorb linear CO2 through van der Waals interactions, resulting in ineffective activation; (4) stability and design strategy: all B-vacancy systems satisfy thermodynamic stability criteria (Eb < Ec, Udiss > 0), while only Co@BN−1 remains stable among N-vacancy configurations. Based on these insights, we propose an application-targeted design principle: Mn@B−1N with high d-band center and strong orbital coupling is optimal for CO2 conversion; Co@BN−1 is preferred for hydrogen production; and the balance between CO2RR/HER pathways can be engineered by tailoring defect types (B/N vacancies) and metal d-orbital energy levels.
The DFT calculations in this study reveal that Mn@B−1N is the most suitable catalyst for CO2 reduction in h-BN-based single-atom catalysts, with a limiting potential of −0.524 V, while Co@BN−1 promotes hydrogen evolution with a limiting potential of −0.217 V, providing structural selection criteria for experimental synthesis. By controlling the type of metal (such as Mn or Co) and the type of coordinating defects (B or N vacancies), we can directionally optimize the hybridization strength between the d orbitals and the substrate, balancing the competition between CO2 activation and hydrogen adsorption, thereby guiding the preparation of highly selective bifunctional catalysts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/nano15080628/s1, Table S1: Eb, EC, Ebulk, Udiss of metals, Ne during the dissolution and Ef; Table S2: The bond length of TM-N/B in TM@B−1N and Co@B−1N. Figure S1: Density of States (DOS) of TM (Mn, Fe, Co, Ni, Cu, Zn)@B−1N and Co@BN−1. Figure S2: ELF values on the (–1, 1, –1) isosurface of the SCAs: (a) Mn@B−1N; (b) Fe@B−1N; (c) Co@B−1N; (d) Ni@B−1N; (e) Cu@B−1N; (f) Zn@B−1N; (g) Co@BN−1. Table S3: The number of charges transferred by a monatomic catalyst to CO2. Table S4: The number of charges transferred by SACs to H. Figure S3: Seven specific reaction pathways for the production of CH4 through the CO2RR (Carbon Dioxide Reduction Reaction), Gibbs free energy step diagram of single-atom catalyst CO2RR: (a) Mn@B−1N; (b) Fe@B−1N; (c) Co@B−1N; (d) Co@BN−1.
Author Contributions
Conceptualization, L.W. (Lai Wei), L.Z., L.W. (Liqian Wu) and Y.H.; data curation, L.Z.; formal analysis, C.Z., Q.C., L.W. (Liqian Wu) and Y.H.; funding acquisition, L.W. (Lai Wei) and L.Z.; investigation, X.Y., X.Z. and Y.H.; methodology, L.W. (Lai Wei) and Y.H.; project administration, L.W. (Lai Wei) and L.Z.; resources, X.Z., L.Z. and Y.H.; software, L.Z.; supervision, L.Z.; validation, X.Y., Q.C., X.Z. and Y.H.; visualization, C.Z.; writing—original draft, X.Y.; writing—review and editing, X.Y., L.Z. and Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Xinjiang Yili Normal University Discipline Comprehensive Strength Enhancement Special Project (22XKZZ24); and Science and Technology Project of Xinjiang Yili Kazak Autonomous Prefecture (YZ2022B021).
Data Availability Statement
The original contributions presented in this study are included in the article, and further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shui, J.; Jia, Y.; Zhang, H.; Cen, W.; Tang, S.; Han, Y. Design principles of two-dimentional transition metal carbides based CO2RR catalysts. Chem. Eng. J. 2025, 507, 160716. [Google Scholar] [CrossRef]
- Tu, Q.; Parvatker, A.; Garedew, M.; Harris, C.; Eckelman, M.; Zimmerman, J.B.; Anastas, P.T.; Lam, C.H. Electrocatalysis for chemical and fuel production: Investigating climate change mitigation potential and economic feasibility. Environ. Sci. Technol. 2021, 55, 3240–3249. [Google Scholar] [CrossRef]
- Jiang, W.; Tang, T.; Zhang, Y.; Hu, J. Synergistic modulation of non-precious-metal electrocatalysts for advanced water splitting. Acc. Chem. Res. 2020, 53, 1111–1123. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef]
- Hu, B.; Li, Z.; Wang, B.; Chen, L.; Wang, X.; Hu, X.; Bai, Z.; Li, Y.; Chen, G.; Luo, X. Construction of Co-In dual single-atom catalysts for photocatalytic CO2 reduction into CH4. Appl. Catal. B Environ. Energy 2025, 371, 125196. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Z.; Dai, L.; Li, B.; Li, Z. High-curvature single-atom catalysts for electrocatalysis: A review. Appl. Catal. A: General. 2025, 694, 120160. [Google Scholar] [CrossRef]
- Wang, W.; Gao, Y.; Li, H.; Tian, F.; Li, D.; Cui, T. Unraveling electrochemical CO reduction of the single-atom transition metals supported on N-doped phosphorene. Appl. Surf. Sci. 2021, 545, 148953. [Google Scholar] [CrossRef]
- Fan, J.; Yang, L.; Zhu, W. Transition metal-anchored BN tubes as single-atom catalysts for NO reduction reaction: A study of DFT and deep learning. Fuel 2025, 386, 134302. [Google Scholar] [CrossRef]
- Tang, T.; Bai, X.; Wang, Z.; Guan, J. Structural engineering of atomic catalysts for electrocatalysis. Chem. Sci. 2024, 15, 5082–5112. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, Z. Platinum single-atom catalysts: A comparative review towards effective characterization. Catal. Sci. Technol. 2019, 9, 4821–4834. [Google Scholar] [CrossRef]
- Liu, X.; Xu, M.; Wan, L.; Zhu, H.; Yao, K.; Linguerri, R.; Chambaud, G.; Han, Y.; Meng, C. Superior catalytic performance of atomically dispersed palladium on graphene in CO oxidation. Acs Catal. 2020, 10, 3084–3093. [Google Scholar] [CrossRef]
- Tanna, H.P.; Baraiya, B.A.; Jha, P.K. Co Implanted Ψ-graphene: A Non-Noble Metal Single-Atom Catalyst for Proficient CO Oxidation Reaction. Mol. Catal. 2024, 556, 113907. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Chang, B.; Wu, S.; Wang, Y.; Sun, T.; Cheng, Z. Emerging single-atom iron catalysts for advanced catalytic systems. Nanoscale Horiz. 2022, 7, 1340–1387. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, Q.; Fu, Y.; Lei, C.; Yang, B.; Li, Z.; Lei, L.; Wu, G.; Hou, Y. Single Atom Electrocatalysts: Carbon-Rich Nonprecious Metal Single Atom Electrocatalysts for CO2 Reduction and Hydrogen Evolution. Small Methods 2019, 3, 1970033. [Google Scholar] [CrossRef]
- Ju, L.; Tan, X.; Mao, X.; Gu, Y.; Smith, S.; Du, A.; Chen, Z.; Chen, C.; Kou, L. Controllable CO2 electrocatalytic reduction via ferroelectric switching on single atom anchored In2Se3 monolayer. Nat. Commun. 2021, 12, 5128. [Google Scholar] [CrossRef]
- Liu, J.; Kong, X.; Zheng, L.; Guo, X.; Liu, X.; Shui, J. Rare earth single-atom catalysts for nitrogen and carbon dioxide reduction. Acs Nano 2020, 14, 1093–1101. [Google Scholar] [CrossRef]
- Han, Q.; Chen, N.; Zhang, J.; Qu, L. Graphene/graphitic carbon nitride hybrids for catalysis. Mater. Horiz. 2017, 4, 832–850. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, H.; Abbas, Q.; Liu, J.; Shi, J.; Xia, X.; Zhang, H.; Du, G. Growth and characterization of porous sp2-BN films with hollow spheres under hydrogen etching effect via borazane thermal CVD. Appl. Surf. Sci. 2018, 452, 314–321. [Google Scholar] [CrossRef]
- Kawrani, S.; Nada, A.A.; Bekheet, M.F.; Boulos, M.; Viter, R.; Roualdes, S.; Miele, P.; Cornu, D.; Bechelany, M. Enhancement of calcium copper titanium oxide photoelectrochemical performance using boron nitride nanosheets. Chem. Eng. J. 2020, 389, 124326. [Google Scholar] [CrossRef]
- Zhou, X.; Meng, X.; Wang, J.; Shang, N.; Feng, T.; Gao, Z.; Zhang, H.; Ding, X.; Gao, S.; Feng, C. Boron nitride supported NiCoP nanoparticles as noble metal-free catalyst for highly efficient hydrogen generation from ammonia borane. Int. J. Hydrogen Energ. 2019, 44, 4764–4770. [Google Scholar] [CrossRef]
- Ngome Okello, O.F.; Doh, K.; Kang, H.S.; Song, K.; Kim, Y.; Kim, K.H.; Lee, D.; Choi, S. Visualization of Transition Metal Decoration on h-BN surface. Nano Lett. 2021, 21, 10562–10569. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, J.; Li, P.; Zhao, G.; Wang, G.; Jiang, Y.; Chen, J.; Dou, S.X.; Pan, H.; Sun, W. Hexagonal boron nitride as a multifunctional support for engineering efficient electrocatalysts toward the oxygen reduction reaction. Nano Lett. 2020, 20, 6807–6814. [Google Scholar] [CrossRef]
- Li, D.; Li, W.; Zhang, J. Fe doped BN monolayer: A promising low-cost single atom catalyst for promoted CO oxidation activity. Appl. Surf. Sci. 2020, 525, 146567. [Google Scholar] [CrossRef]
- Wang, S.; Xin, Y.; Yuan, J.; Wang, L.; Zhang, W. Direct conversion of methane to methanol on boron nitride-supported copper single atoms. Nanoscale 2022, 14, 5447–5453. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef]
- Konopatsky, A.; Kustov, A.; Evdokimenko, N.; Shesterkina, A.; Varlamova, L.A.; Teplyakova, T.; Leybo, D.; Antipina, L.Y.; Sorokin, P.B.; Fang, X. Layered Ferrihydrite and BN Nanoparticle Heterostructures Doped with Ag for CO2 Hydrogenation. ACS Appl. Nano Mater. 2024, 7, 10257–10267. [Google Scholar] [CrossRef]
- Wong, H.; Tang, T.W.; Chen, H.; Xu, M.; Wang, J.; Cai, Y.; Goddard, W.A.; Luo, Z. Dual role of h-BN as an artificial solid-electrolyte interface layer for safe zinc metal anodes. J. Mater. Chem. A 2024, 12, 4195–4203. [Google Scholar] [CrossRef]
- Lu, Y.; Li, B.; Xu, N.; Zhou, Z.; Xiao, Y.; Jiang, Y.; Li, T.; Hu, S.; Gong, Y.; Cao, Y. One-atom-thick hexagonal boron nitride co-catalyst for enhanced oxygen evolution reactions. Nat. Commun. 2023, 14, 6965. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wu, D.; Su, Y. The effect of grain boundary in hexagonal boron nitride on catalytic activity of nitrogen reduction reaction. Appl. Surf. Sci. 2022, 593, 153468. [Google Scholar] [CrossRef]
- Li, Y.; Gao, D.; Zhao, S.; Xiao, Y.; Guo, Z.; Fang, Y.; Lin, J.; Liu, Z.; Huang, Y.; Guo, K. Carbon doped hexagonal boron nitride nanoribbon as efficient metal-free electrochemical nitrogen reduction catalyst. Chem. Eng. J. 2021, 410, 128419. [Google Scholar] [CrossRef]
- Leybo, D.; Firestein, K.L.; Evdokimenko, N.D.; Ryzhova, A.A.; Baidyshev, V.S.; Chepkasov, I.V.; Popov, Z.I.; Kustov, A.L.; Konopatsky, A.S.; Golberg, D.V. Ball-milled processed, selective Fe/h-BN nanocatalysts for CO2 hydrogenation. ACS Appl. Nano Mater. 2022, 5, 16475–16488. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, Y.; Kang, L.; Zhang, C.; Wang, D.; Wang, C.; Zhang, M.; Wu, X. First-principles investigation of hydrogen storage capacity of Y-decorated porous graphene. Appl. Surf. Sci. 2017, 399, 463–468. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Bader, R.F.; Nguyen-Dang, T.T. Quantum theory of atoms in molecules–Dalton revisited. Adv. Quantum Chem. 1981, 14, 63–124. [Google Scholar]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Wyckoff, R.W. Structure of Crystals; The Chemical Catalog Company, Inc.: New York, NY, USA, 1931. [Google Scholar]
- Sun, M.; Wang, S.; Du, Y.; Yu, J.; Tang, W. Transition metal doped arsenene: A first-principles study. Appl. Surf. Sci. 2016, 389, 594–600. [Google Scholar] [CrossRef]
- Kropp, T.; Lu, Z.; Li, Z.; Chin, Y.C.; Mavrikakis, M. Anionic single-atom catalysts for CO oxidation: Support-independent activity at low temperatures. Acs Catal. 2019, 9, 1595–1604. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, L.; Wang, Y.; Liu, J.; Muravev, V.; Alexopoulos, K.; Filot, I.A.; Vlachos, D.G.; Hensen, E.J. Stability of heterogeneous single-atom catalysts: A scaling law mapping thermodynamics to kinetics. Npj Comput. Mater. 2020, 6, 144. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K. Electrochemical dissolution of surface alloys in acids: Thermodynamic trends from first-principles calculations. Electrochim. Acta 2007, 52, 5829–5836. [Google Scholar] [CrossRef]
- Karmodak, N.; Vijay, S.; Kastlunger, G.; Chan, K. Computational screening of single and di-atom catalysts for electrochemical CO2 reduction. Acs Catal. 2022, 12, 4818–4824. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the activity and selectivity challenges of electrocatalysts toward the nitrogen reduction reaction via atomically dispersed biatom catalysts. J. Am. Chem. Soc. 2020, 142, 5709–5721. [Google Scholar] [CrossRef]
- Ma, J.; Shang, S.; Kim, H.; Liu, Z. An ab initio molecular dynamics exploration of associates in Ba-Bi liquid with strong ordering trends. Acta Mater. 2020, 190, 81–92. [Google Scholar] [CrossRef]
- Lv, L.; Shen, Y.; Liu, J.; Meng, X.; Gao, X.; Zhou, M.; Zhang, Y.; Gong, D.; Zheng, Y.; Zhou, Z. Computational screening of high activity and selectivity TM/g-C3N4 single-atom catalysts for electrocatalytic reduction of nitrates to ammonia. J. Phys. Chem. Lett. 2021, 12, 11143–11150. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, S.; Shao, Y.; Zhou, S.; Wu, X.; Zhu, J.; Tang, D. Relationship between the electric structures calculated by the first principles calculation method and the photoelectrocatalysis degradation of Ir-doped SnO2 electrodes. Appl. Surf. Sci. 2017, 422, 891–899. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Y.; Lou, F.; Yu, Z. Theoretical study of single transition metal atom catalysts supported on two-dimensional Nb2NO2 for efficient electrochemical CO2 reduction to CH4. J. CO2 Util. 2022, 62, 102069. [Google Scholar] [CrossRef]
- Araujo, R.B.; Rodrigues, G.L.; Dos Santos, E.C.; Pettersson, L.G. Adsorption energies on transition metal surfaces: Towards an accurate and balanced description. Nat. Commun. 2022, 13, 6853. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef]
- Gao, Z.; Li, A.; Liu, X.; Ma, C.; Li, X.; Yang, W.; Ding, X. Density functional study of the adsorption of NO on Nin (n= 1, 2, 3 and 4) clusters doped functionalized graphene support. Appl. Surf. Sci. 2019, 481, 940–950. [Google Scholar] [CrossRef]
- Zhou, J.; Yuan, D.; Huang, B.; Xie, T.; Cai, L.; Hu, W. Highly effective Ru-based Heusler alloy catalysts for N2 activation: A theoretical study. Appl. Surf. Sci. 2022, 575, 151658. [Google Scholar] [CrossRef]
- Yu, J.; Li, J.; Xu, C.; Li, Q.; Liu, Q.; Liu, J.; Chen, R.; Zhu, J.; Wang, J. Modulating the d-band centers by coordination environment regulation of single-atom Ni on porous carbon fibers for overall water splitting. Nano Energy 2022, 98, 107266. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Dong, S.; Li, Y.; Zhang, X.; Hu, H.; Yan, B.; Ren, J.; Wan, L.; Zhao, H.; Sun, F. Pt single-atom loaded on nonmetallic elements (C, N, P, S) doped ZrO2 in photocatalytic hydrogen evolution: First principles. Mater. Des. 2023, 231, 112068. [Google Scholar] [CrossRef]
- Bronsted, J.N. Acid and Basic Catalysis. Chem. Rev. 1928, 5, 231–338. [Google Scholar] [CrossRef]
- Wang, M.; Shi, H.; Tian, M.; Chen, R.; Shu, J.; Zhang, Q.; Wang, Y.; Li, C.; Wan, N.; Lei, S. Single nickel atom-modified phosphorene nanosheets for electrocatalytic CO2 reduction. ACS Appl. Nano Mater. 2021, 4, 11017–11030. [Google Scholar] [CrossRef]
- Lv, P.; Wu, D.; He, B.; Li, X.; Zhu, R.; Tang, G.; Lu, Z.; Ma, D.; Jia, Y. An efficient screening strategy towards multifunctional catalysts for the simultaneous electroreduction of NO3−, NO2− and NO to NH3. J. Mater. Chem. A 2022, 10, 9707–9716. [Google Scholar] [CrossRef]
- Xie, L.; Huang, W.; Chen, J.; Chen, H.; Hou, C.; Ni, Q.; Huang, T.; Gui, L.; Wang, X. Selective oxidation of β-keto ester modulated by the d-band centers in DA conjugated microporous metallaphotoredox catalysts containing M− salen (MZn, Cu and Co) and triazine monomers. J. Colloid. Interf. Sci. 2024, 665, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Su, H.; Chan, S.H.; Sun, Q. CO2 electroreduction performance of transition metal dimers supported on graphene: A theoretical study. Acs Catal. 2015, 5, 6658–6664. [Google Scholar] [CrossRef]
- Back, S.; Lim, J.; Kim, N.; Kim, Y.; Jung, Y. Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 2017, 8, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).