3.1. Physicochemical Properties of the Chi/PNIPAM Complexes
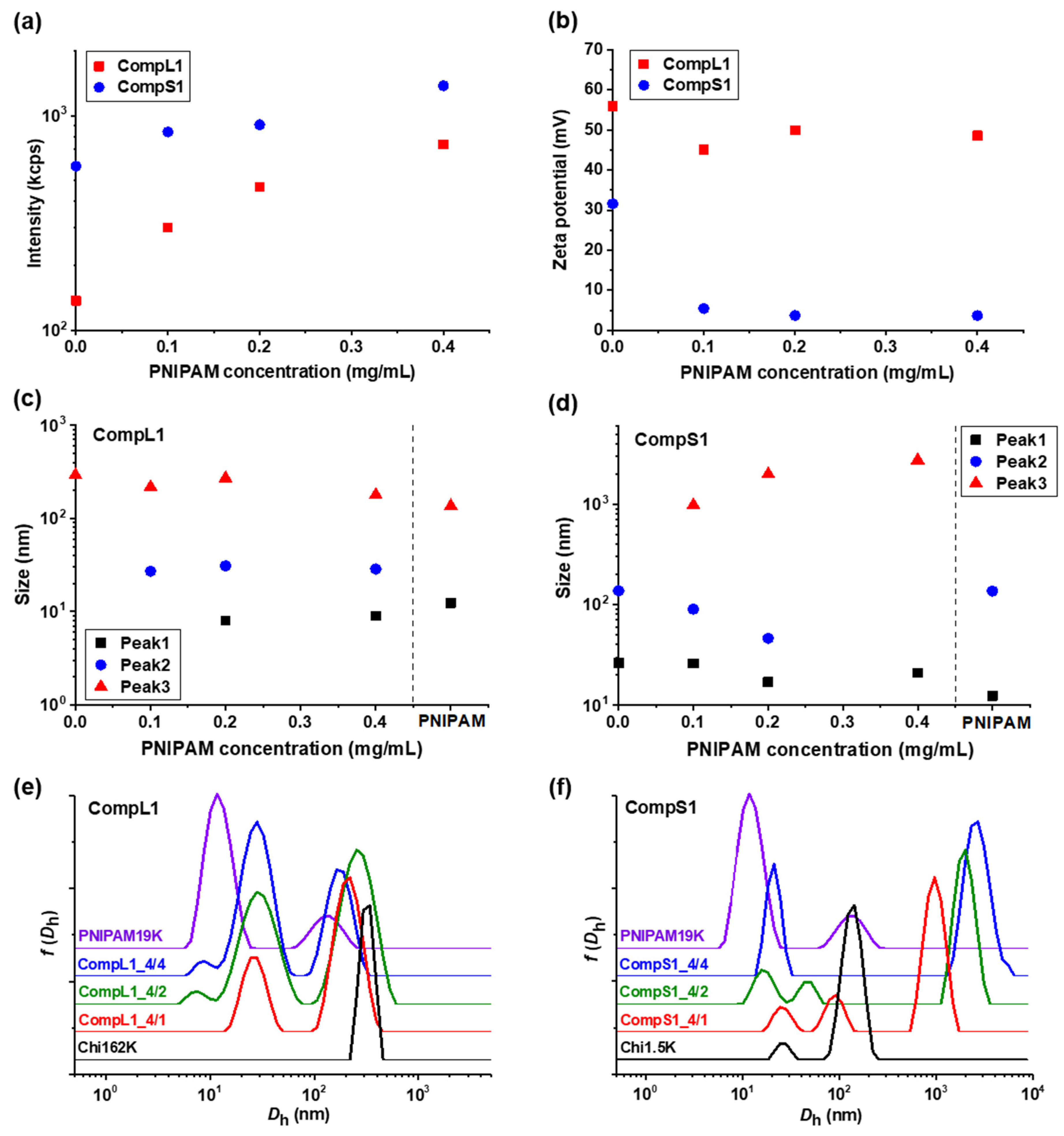
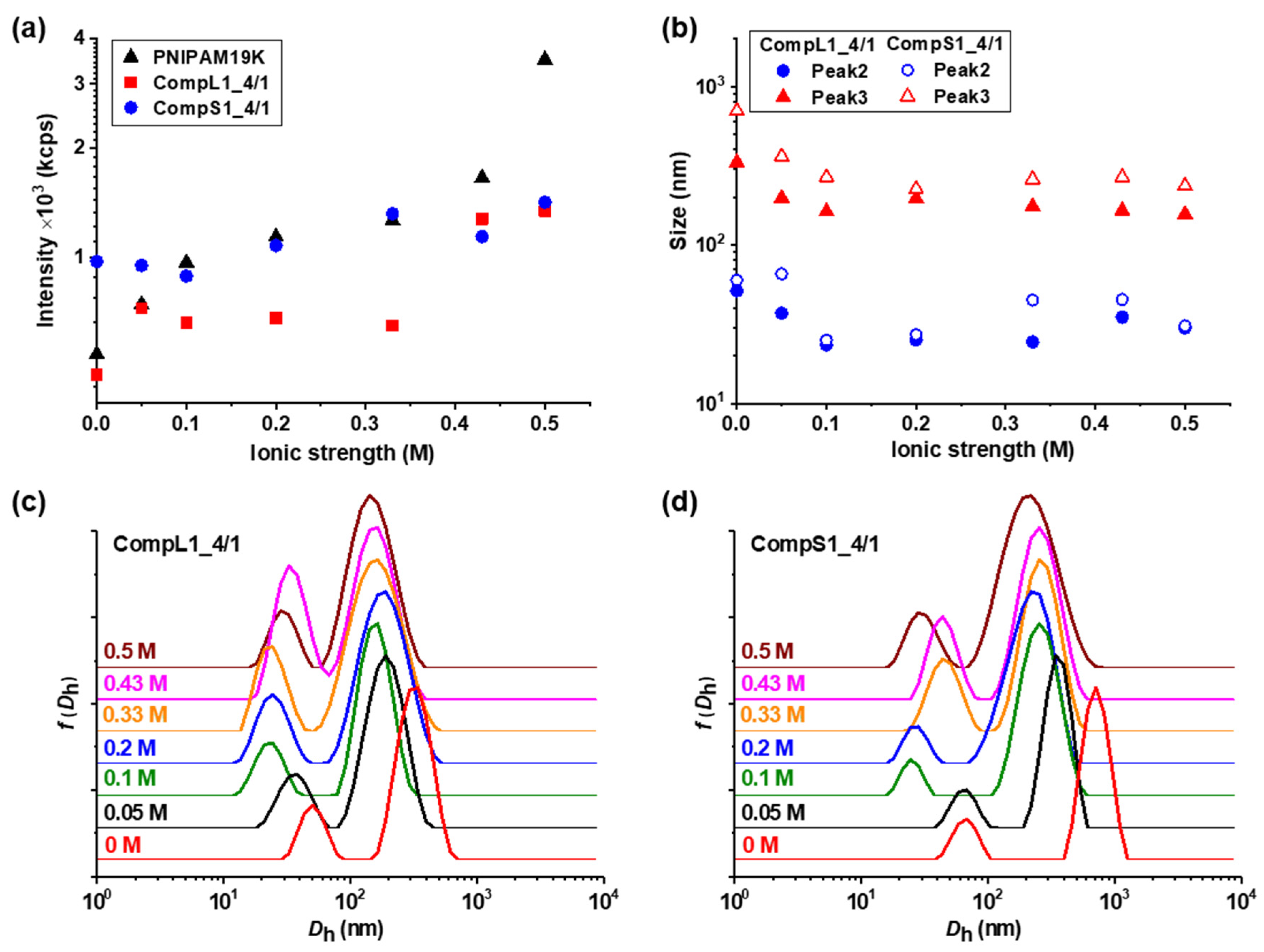
Initially, the physicochemical properties of the obtained complexes between the long or short chitosan and the PNIPAM19K sample (i.e., CompL1 or S1 samples) were assessed by DLS and ELS measurements. The scattered intensity, zeta potential, peak sizes, and size distributions for the two series of complexes with different PNIPAM contents were analyzed and the obtained results are presented in
Figure 1 as a function of the concentration of PNIPAM. It should be noted that the points at zero PNIPAM concentration indicate the corresponding values for the respective chitosan samples, that is Chi162K or Chi1.5K. Moreover, the sizes for the PNIPAM19K samples are also included in
Figure 1c,d, separated by a vertical dashed line, while the size distributions of the corresponding initial components are also shown in
Figure 1e,f for comparison. Finally, the corresponding polydispersity index values are shown in
Figure S3a. As can be observed in
Figure 1a, the scattered intensity for the CompL1 series gradually increased with increasing PNIPAM concentration. Since the scattered intensity was proportional to the mass of the complexes in solution, the observed increase confirms that the complexation between the two components occurred, and the mass of the complexes actually increased as more PNIPAM was added. In parallel, the scattered intensity for the CompS1 series also exhibited a similar increasing trend but with overall higher values, suggesting that the particles formed in this case had a greater mass or a denser conformation. This seems to be in direct correlation to the markedly higher initial scattered intensity of the Chi1.5K sample (point at 0 PNIPAM concentration), which implies that the short chitosan forms in solution particles of increased mass or density.
Regarding the effective charge of the complexes, the measured zeta potential values are presented in
Figure 1b for both CompL1 and CompS1 series. Obviously, although no significant changes were observed upon increasing the concentration of PNIPAM for both series, the values of the zeta potential of the complexes were considerably lower than that of the corresponding chitosan samples, thus confirming the complexation between the two macromolecular components. This decrease was distinctly more pronounced in the case of the CompS1 samples, with the zeta potential values of the complexes being close to zero (which is also in agreement with the corresponding charge density values presented in
Table S2), denoting the extended neutralization of opposite charges. In other words, the effective charge of the complexes suggests a higher degree of interaction (and thus, charge neutralization) between the short chitosan and the PNIPAM19K sample, as compared to that for the long chitosan, although the nominal charge ratio (that is, analogy of total number of charges) was similar for both series of samples (CompL1 or S1, see
Table 1). This behavior can be ascribed to the possible difference in conformation of the two chitosan chains in solution, which was a direct consequence of the corresponding chain lengths of the two samples. In more detail, even in this case of semi-rigid chitosan chains, the longer one (Chi162K) was expected to adopt a somewhat coiled conformation, since its total chain length was rather high (i.e., 975 units × 0.52 nm/unit = 507 nm) in comparison to the persistence length of chitosans with high DD, which was found to be about 9 nm, thus exhibiting partial flexibility [
30]. Therefore, the charges inside the coil were less available for the subsequent electrostatic interaction with the –COO
− end groups of the PNIPAM chains, mainly after some chitosan/PNIPAM interchain interactions had occurred. On the other hand, the conformation of the short chitosan was expected to be a more extended one, as its chain length was significantly smaller (i.e., 9 units × 0.52 nm/unit = 4.7 nm). This in turn allowed the charges from the amino groups to be more accessible for interaction with the carboxyl PNIPAM chain ends. Furthermore, the fact that, in both cases, the increase in the PNIPAM concentration did not have a significant effect on the overall charge of the formed complexes can also be attributed to their different conformation/structure. It is rather likely that the assumed less compact/more loose conformation of the ones formed with the long chitosan allowed for secondary conformational rearrangements (in an attempt to maintain colloidal stability), which led to the observed preservation of the overall positive charge.
As far as the size of the complexes is concerned, from
Figure 1c,e it can be observed that for the CompL1 series, two main peaks (Peak 2 and 3) were discerned, with their size (i.e., about 30 and 220 nm, respectively) being more or less the same irrespective of the PNIPAM concentration in the complexes. These peaks denote the presence of two different scattering populations in the solutions/dispersions of the complexes, which most probably correspond to particles comprising different numbers of complexed chains. It is possible that the long chitosan exhibited some degree of self-assembly in the solution, forming multi-chain aggregates comprising a small number of chains, due to hydrophobic interactions stemming from the hydrophobicity of its backbone and pendant acetate side groups. This assumption seemed to be confirmed by the corresponding size distribution of the Chi162K sample, which showed a main peak at about 300 nm (Peak 3). Even though peaks corresponding to smaller sizes were not detected, this size fits better to the hypothesis of multi-chain aggregates rather than single chains. Therefore, the two observed populations of complexes could be the result of the interaction of either single chains or multi-chain aggregates of chitosan with the PNIPAM polymer, with the latter exhibiting some shrinking upon complexation, as evidenced by the corresponding reduction in size. Nevertheless, they seemed to adopt a rather open/loose conformation/structure (mostly dictated by that of the corresponding species of chitosan), since their sizes were not significantly affected by the interaction with an increasing number of PNIPAM chains. A small peak at about 10 nm (Peak 1) was also detected for the CompL1_4/2 and 4/4 samples and could indicate the presence of uncomplexed synthetic polymer chains, since it coincided with the corresponding peak of the PNIPAM19K sample. Another interesting remark about the PNIPAM19K sample is that it also exhibited a small degree of self-aggregation (Peak 3), likely due to hydrophobic interchain interactions (which were further enhanced by the dodecyl end groups in this case).
For the CompS1 series of complexes, the short chitosan also exhibited self-assembly in solution, since two peaks (Peak 1 and 2) with sizes about 25 and 150 nm, most likely corresponding to single chains and multi-chain aggregates, are seen in
Figure 1f. At the same time, the size distributions of the CompS1_4/1 and 4/2 samples showed three peaks, with the size of the first two being somewhat smaller than that of the corresponding peaks of the Chi1.5K sample. Apparently, both the single chains and the multi-chain aggregates of the short chitosan interacted electrostatically with the PNIPAM chains, forming complexes that were slightly smaller than the initial species of chitosan as a result of the reduction in electrostatic repulsions due to charge neutralization and/or the increase in hydrophobic interactions caused by the incorporation of PNIPAM. This combination of effects clearly weakened the colloidal stability of the formed complexes, eventually leading to extended secondary aggregation, as evidenced by the presence of the much larger population corresponding to Peak 3 with sizes almost equal or even greater than 1 μm. In fact, this secondary aggregation became more pronounced as the PNIPAM concentration increased, as the size of the largest population gradually increased, and the two smaller populations seemed to merge into one in the case of CompS1_4/4 sample. By also considering the fact that no free/uncomplexed PNIPAM chains appeared to be detected (contrary to the CompL1 series), it can be assumed that the short chitosan enabled a greater degree of interaction with the PNIPAM chains compared to the long one, as previously discussed. Of course, this could be related to the shortest polysaccharide molecule that allowed for a higher accessibility of its charges, in contrast to the longer one. One final remark concerning both CompL1 and S1 series is that the presence of the different scattering populations in solution also entailed a rather high polydispersity in all cases (see
Figure S3 and corresponding discussion).
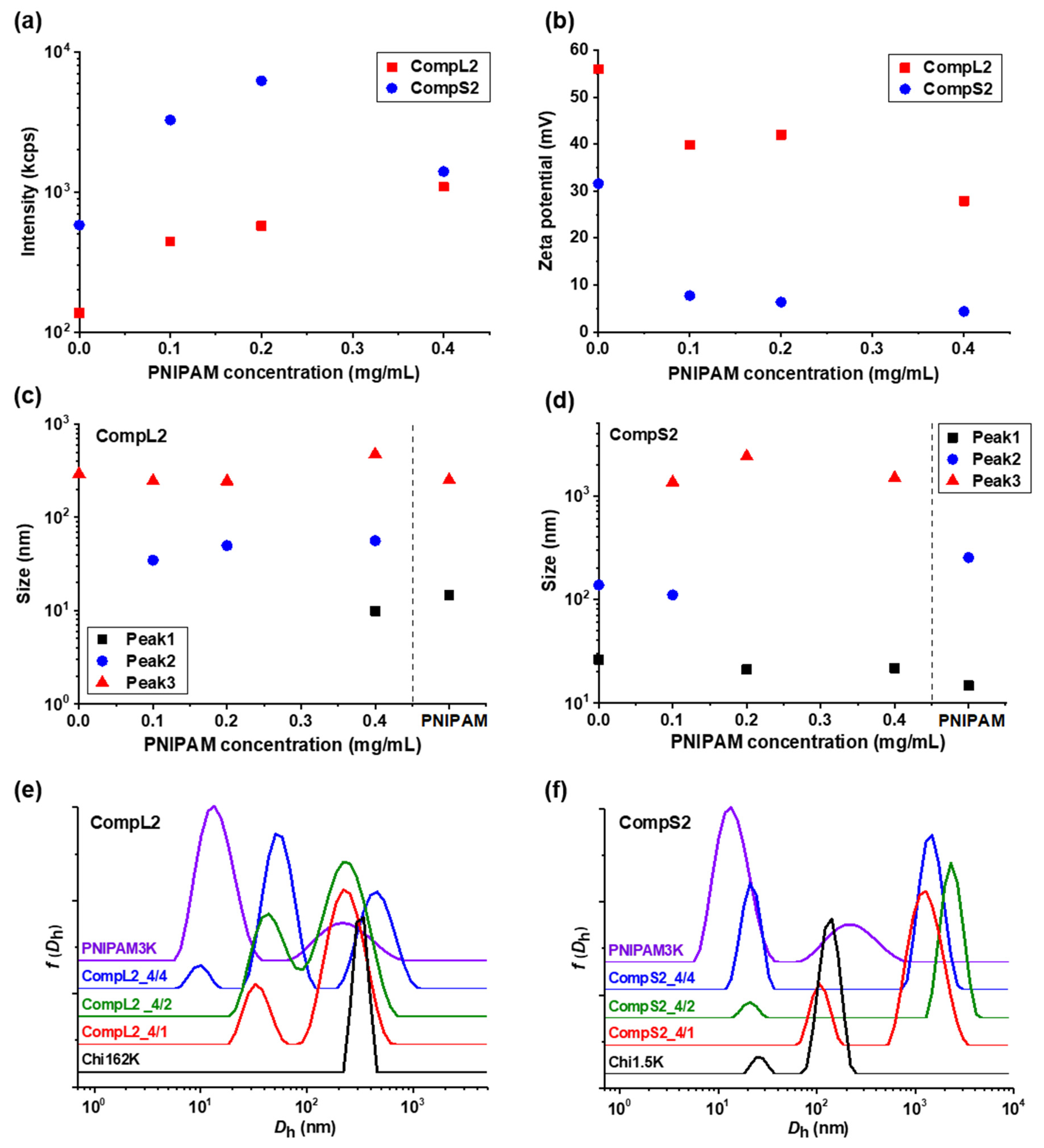
The solution behavior of the hybrid nanostructures formed between both chitosans and PNIPAM3K (i.e., CompLorS2 samples) was investigated in a similar manner by DLS and ELS measurements, and the overall physicochemical characteristics in regard to mass, charge, size, and size distribution are presented in
Figure 2, while the polydispersity index is shown in
Figure S3b. It is quite obvious that the obtained results show a similar trend to the ones already discussed for the corresponding complexes with the PNIPAM19K sample. Specifically, in
Figure 2a, an analogous increase in the scattering intensity as a function of PNIPAM concentration was observed for both series of complexes, verifying the complexation of both the long and the short polysaccharide molecules with the synthetic polymer chains. Once again, the mass of the complexes corresponding to Chi1.5K was higher than the ones for Chi162K, and, at the same time, significantly higher than that of the CompS1 series, demonstrating an increased interaction between the short chitosan and the PNIPAM3K chains. This seems to be in agreement with the fact that for the PNIPAM3K sample, the number of chains corresponding to each chitosan molecule was larger (see
Table 1), which apparently further promoted secondary aggregation. The observed decrease in intensity for the CompS2 series at the highest PNIPAM content could possibly be explained by some small amount of precipitation, also indicative of the stronger interaction. Moreover, the zeta potential measurements (
Figure 2b) show lower values for the complexes compared to the corresponding pure chitosan samples, especially for the CompS2 series. This observation is consistent with the notion that the shorter chitosan molecule was able to more easily interact with PNIPAM chains than the longer one, and hence the charge neutralization was more extended in this case, with the effective charge of these complexes being close to zero. Regarding the size of the complexes, the distributions of
Figure 2e,f reveal two main populations for the CompL2 series, corresponding to the complexes formed between the single chain and multi-chain aggregates of chitosan with the PNIPAM chains, while an additional (third) larger population indicative of secondary aggregation was observed for the CompS2 complexes. For both CompLorS2 series, the sizes of the individual peaks, as well as their variation upon increasing the concentration of PNIPAM, were quite similar to those witnessed for the CompLorS1 complexes. This fact suggests that in regard to the size of the complexes, the determining factor was mainly the conformation/self-aggregation of the chitosan molecules and, to a much lesser extent, the length of the PNIPAM chains. As in the previous case, the solutions exhibited increased polydispersity as a consequence of the coexistence of various populations (see
Figure S3b).
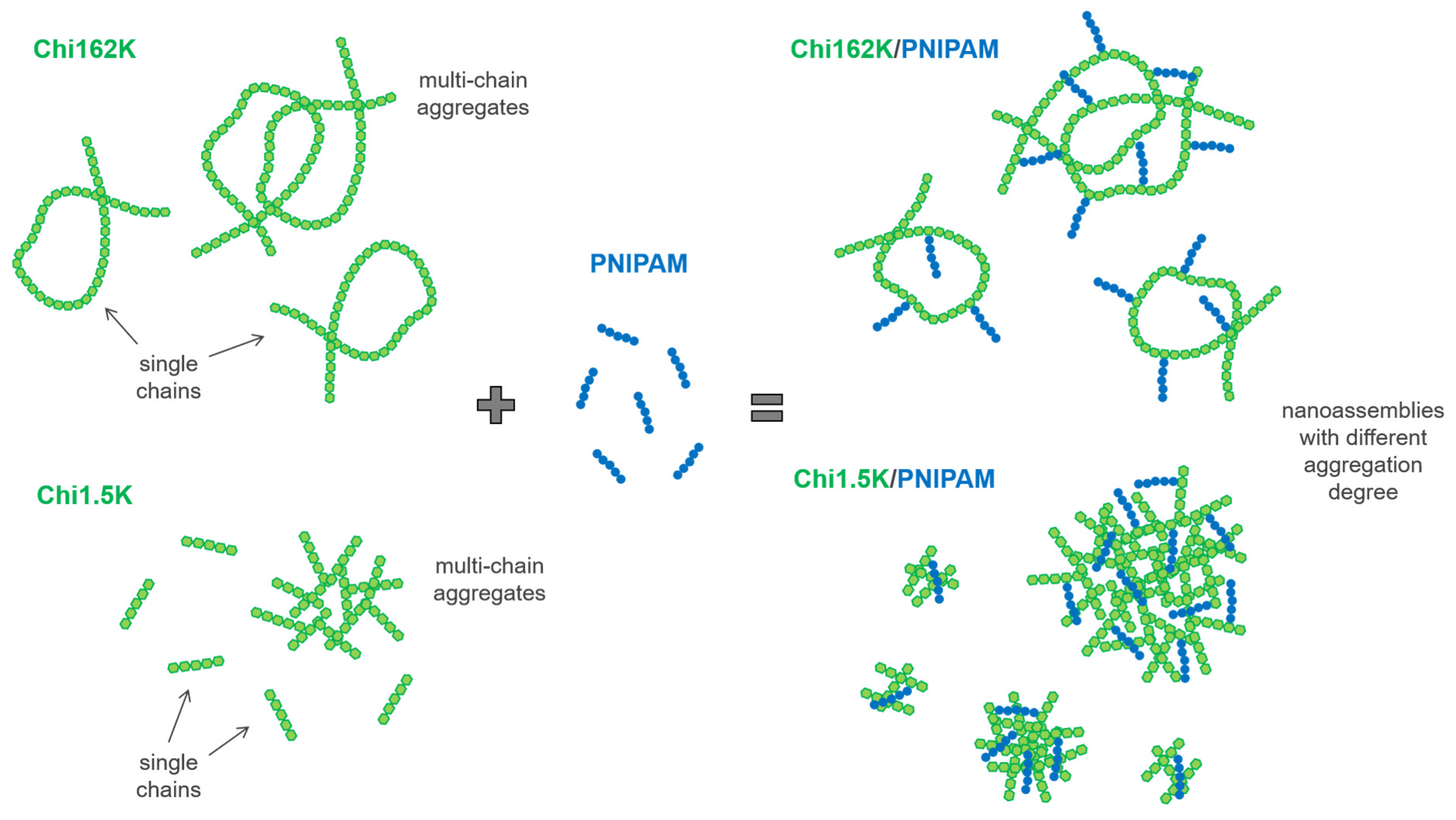
Overall, the combination of DLS and ELS results suggest that for all four investigated Chi/PNIPAM systems, different types of nanoassemblies with varying degrees of complexation/aggregation coexisted in the solution. Their respective structures seemed to be dictated mainly by the conformation of the corresponding chitosan samples, regardless of the PNIPAM chain length. A schematic representation of the complexation process between the two chitosan samples and PNIPAM chains (independent of length), along with the proposed structure of the resulting complexes/aggregates, is given in
Scheme 1.
Another interesting characteristic of the formed Chi/PNIPAM complexes is their charge density, which was indirectly determined by means of polyelectrolyte titration experiments, and the obtained values are given in
Table 2 (for the corresponding calculations see
Table S2). The polyelectrolyte titration method determined the excess charges of the complexes by neutralizing them with an oppositely charged strong polyelectrolyte. As expected, for all four series of complexes, the charge density exhibited the highest values for the samples corresponding to the 4/1 volume ratio, since these systems had the higher amounts of chitosan in excess. By increasing the PNIPAM content in the complexes, the charge density gradually decreased in all cases, as a result of the occurring charge neutralization due to the electrostatic complexation between the positive amino groups of chitosan and the negative carboxylic end groups of PNIPAM. In parallel, the complexes formed with the long chitosan (CompL1or2) had a higher charge density than the ones corresponding to the short polysaccharide molecule (CompS1or2), which exhibited a charge density close to zero (in agreement with the results obtained for zeta potential included in
Figure 1b). This finding corroborates the concept that the significantly shorter Chi1.5K molecule allows for a higher degree of interaction with the PNIPAM chains, leading to extended charge neutralization. Furthermore, as far as the effect of the length of the PNIPAM chain on the charge density of the formed complexes is concerned, the complexes corresponding to the shorter sample exhibited slightly lower values than those for the longer one. This observation seems to be in agreement with the fact that for the same volume ratio, more PNIPAM3K chains corresponded to each chitosan molecule, in comparison to the PNIPAM19K sample, as expressed by the molar ratio of the two components shown in
Table 1.
Additional information relevant to the hydrophobicity of the Chi/PNIPAM complexes was derived through fluorescence spectroscopy measurements with the aid of the pyrene probe, which has a distinctive emission spectrum indicative of the polarity of its microenvironment [
27,
28,
29]. The acquired fluorescence spectra both at 25 and 45 °C for all the series of complexes are shown in
Figure S4, while
Table 3 presents the corresponding values of the characteristic
I1/
I3 intensity ratio. Under ambient conditions, the CompS1 series exhibited
I1/
I3 ratio values of about 1.2, which indicate a fairly hydrophobic microenvironment, while the other three series of complexes showed
I1/
I3 ratio values of about 1.6, which corresponds to a rather hydrophilic one. It seems that in the case of the complexes formed between the long chitosan and either of the two PNIPAM samples, their more open/loose conformation allowed for a greater degree of hydration. On the other hand, the extended interaction and consequent secondary aggregation occurring for the CompS1 series, led to the formation of denser nanostructures with a more compact/collapsed conformation, and thus increased hydrophobicity. Quite interestingly, for the CompS2 samples, the corresponding
I1/
I3 ratio values suggest that the pyrene probe experienced a hydrophilic microenvironment, but, at the same time, excimer formation indicative of close proximity between pyrene molecules was observed, as evidenced by the relative characteristic peak (see
Figure S4). In other words, a coexistence of both isolated pyrene molecules in hydrophilic regions and hydrophobic domains/pockets, where the pyrene molecules aggregate due to increased crowding conditions, was detected. This could be correlated to the length of the PNIPAM chains, in the sense that the longer ones (i.e., PNIPAM19K sample) seemed to enable the formation of large hydrophobic regions, possibly acting as bridging elements between complexes, in contrast to the shorter ones (PNIPAM3K) that only allowed for local spatially confined hydrophobic domains.
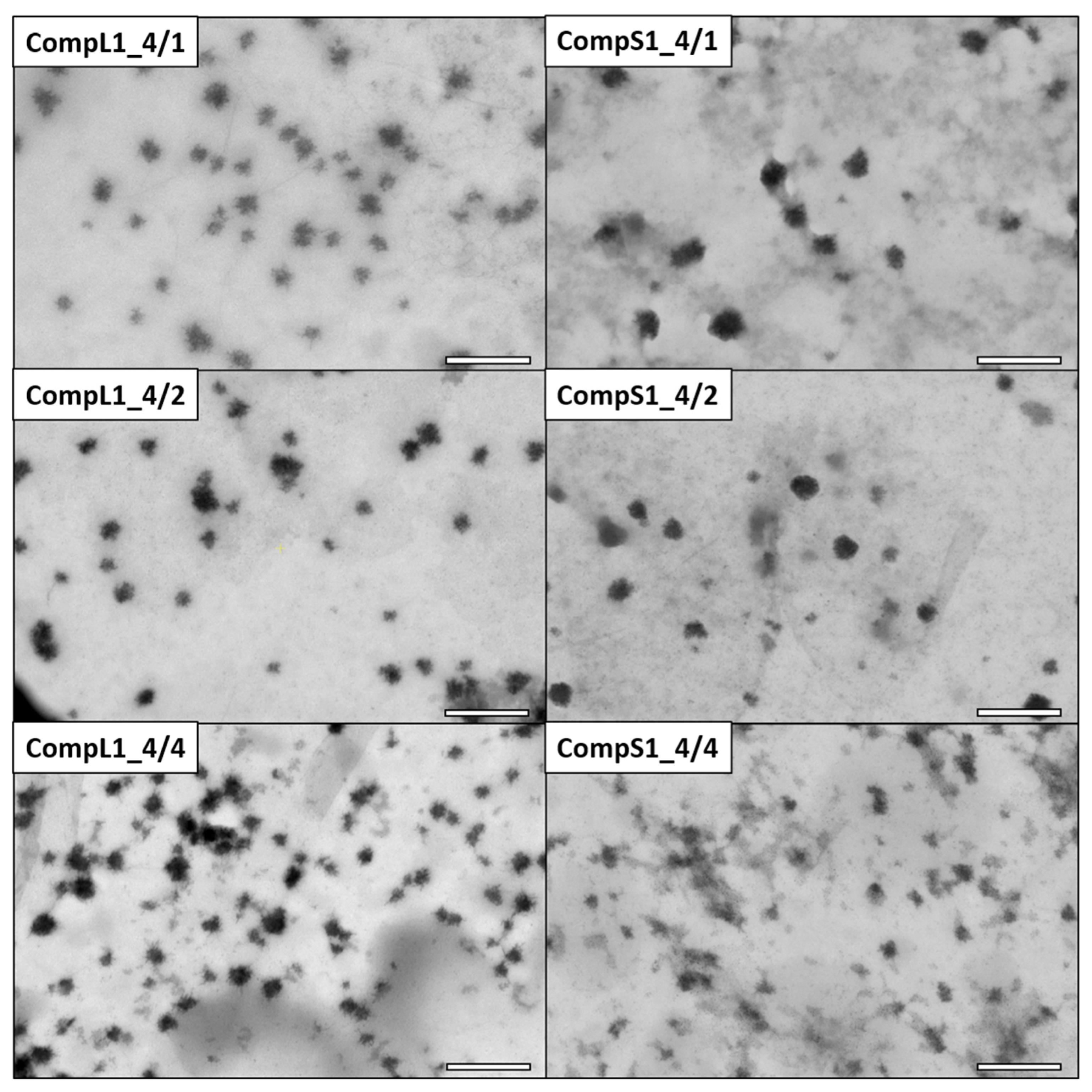
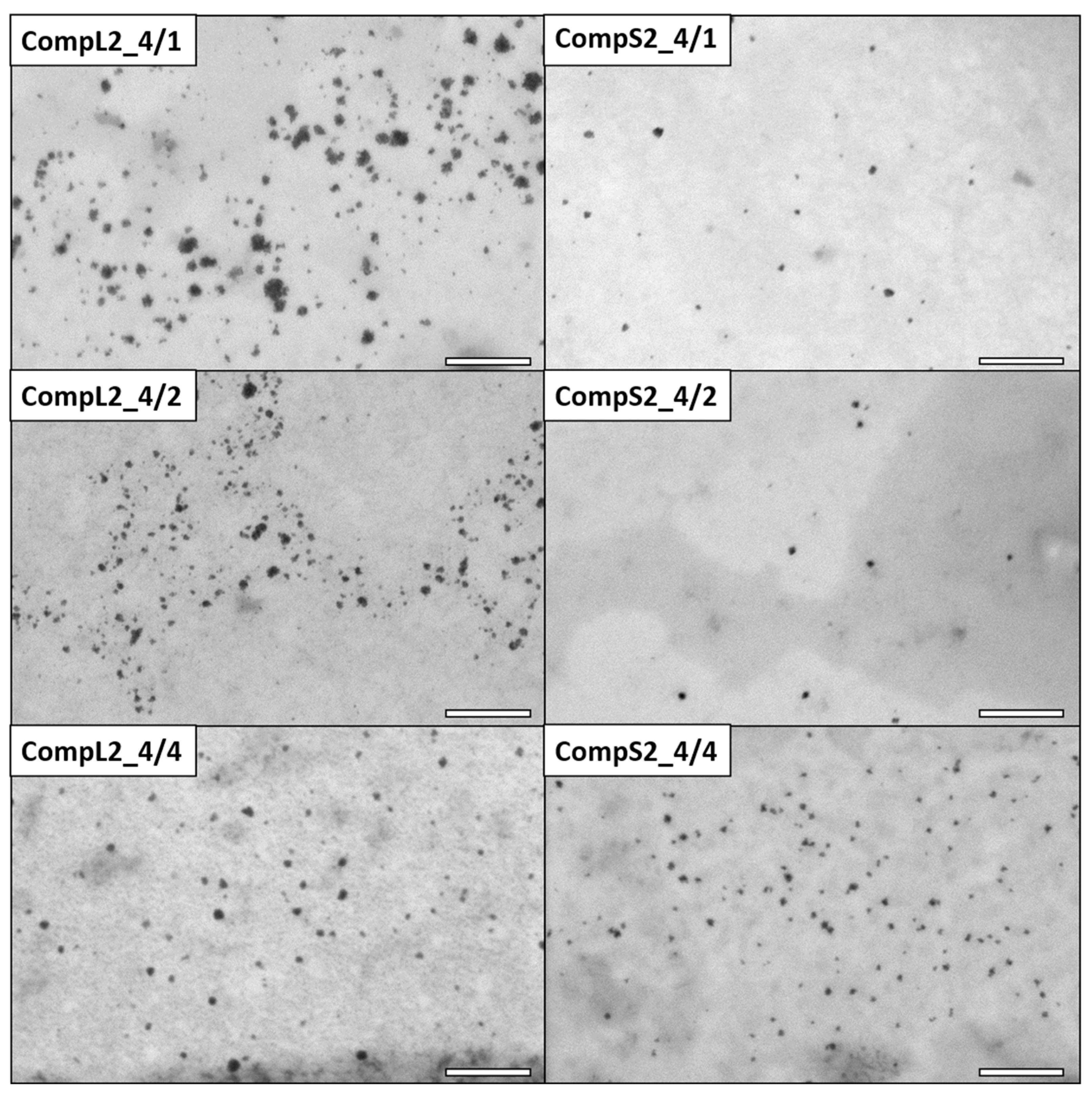
The morphology of the various Chi/PNIPAM complexes was investigated by STEM, and the obtained images for the CompLorS1 and CompLorS2 series are respectively shown in
Figure 3 and
Figure 4. In all cases, rather spherical homogeneous particles were discerned, while upon closer inspection it can be observed that the nanoparticles show an urchin-like morphology, most probably stemming from the partial rigidity of the chitosan backbone. Due to its linear structure and multitude of available functional amino groups, each chitosan chain was expected to electrostatically interact with more than one PNIPAM chain (since the synthetic polymer has only one charged end group per chain), thus creating primary complexes whose morphology would mostly be dictated by that of the conformation and aggregation state of the chitosan macromolecules. As a result of the reduction of the electrostatic repulsions due to charge neutralization that further favor the intra-chain interactions of chitosan and also reduce the overall solubility, the initial complexes aggregated together, rendering populations of clustered particles with larger sizes (see
Figure S5). The average diameter of the nanoparticles was evaluated with the aid of ImageJ software by measuring all the individual particles found in each image. In this manner, the nanoparticles’ average diameter for the CompL1 series appeared to not vary significantly with the increase in PNIPAM, in accordance with the DLS observations, with all three complexes having an average size of about 33 nm. On the other hand, the average diameter for the CompS1 samples showed a slight decrease from about 45 nm (CompS1_4/1) to 26 nm (CompS1_4/4), indicating the formation of more compact structures as the PNIPAM concentration increased.
As far as the corresponding complexes formed between the two chitosan molecules and the PNIPAM3K sample (CompL2 or S2) are concerned, the STEM images of
Figure 4 clearly show particles of smaller sizes compared with the ones containing PNIPAM19K (CompL1 or S1). In more detail, the average diameter of the nanoparticles for the CompL2 sample with the lowest amount of PNIPAM (CompL2_4/1) is about 19 nm, while the size further decreases to about 10 nm with the increase in PNIPAM concentration (CompL2_4/4). By comparison, CompS2 samples exhibited even smaller nanoparticle average diameters, ranging from about 10 nm (CompS2_4/1) to 7 nm (CompS2 4/4). Of course, it should be noted that in all cases the sizes calculated from the STEM images were expected to be smaller than the ones determined by DLS, because DLS analyzes particles in their swollen state in solution, while STEM technique requires the complete evaporation of the water, or, in other words, inspects their dry state. Therefore, although according to the DLS results the complexes corresponding to the two different PNIPAM samples seemed to have similar sizes, the fact that STEM images indicated a smaller size for the complexes formed with the PNIPAM3K chains is indicative of their more compact/dense structure. One final remark is that the particles seen in the STEM images of
Figure 3 and
Figure 4 are most probably primary complexes that correspond to the smaller in size populations discerned by DLS. Nevertheless, in some cases, larger clustered structures were also evidenced in STEM, as shown in
Figure S5.
Further insight into the size and structure of the formed complexes was obtained through AFM imaging.
Figure 5 and
Figure 6 present representative AFM images for the CompLorS1 and CompL2 or S2 series, respectively, while the corresponding size measurements of individual surface profiles are shown in
Figures S6–S9. From the analysis of the AFM images, it can be inferred that in all cases rather compact spherical particles, either sparsely distributed or closely packed together, were deposited on the mica substrate. For both the CompL1 and L2 series, the lowest PNIPAM content samples (CompL1 or L2_4/1) exhibited a rather interesting morphology, which could be indicative of the long chitosan conformation and the rigidity of the polysaccharide backbone. Furthermore, for all three CompL1 samples, the sizes of the individual particles were in the range of 100 to 250 nm (see
Figure S6), suggesting that they corresponded to structures formed by the aggregation of primary complexes. A narrower distribution and relatively smaller sizes were observed for the CompS1 series, as all three samples showed particles with diameters in the range of 100 to 150 nm (
Figure S7), which could be correlated with their assumed more compact/dense structure. This also stands for both the CompL2 and S2 series of samples, with the observed particles having sizes of about 100 nm (
Figures S8 and S9). Note that smaller particles were discerned in most of the obtained AFM images, indicating the coexistence of various populations of complexes comprising different numbers of chitosan molecules and PNIPAM chains. Lastly, it was also interesting that the measured heights for the complexes of the long chitosan (CompL1 or L2) were generally larger than those corresponding to the short one (CompS1 or S2). This could indicate a reduced ability to adopt more flattened conformations upon surface contact/deposition, which might be a consequence of the longer length of the polysaccharide molecule.
3.2. Temperature Response of the Chi/PNIPAM Complexes
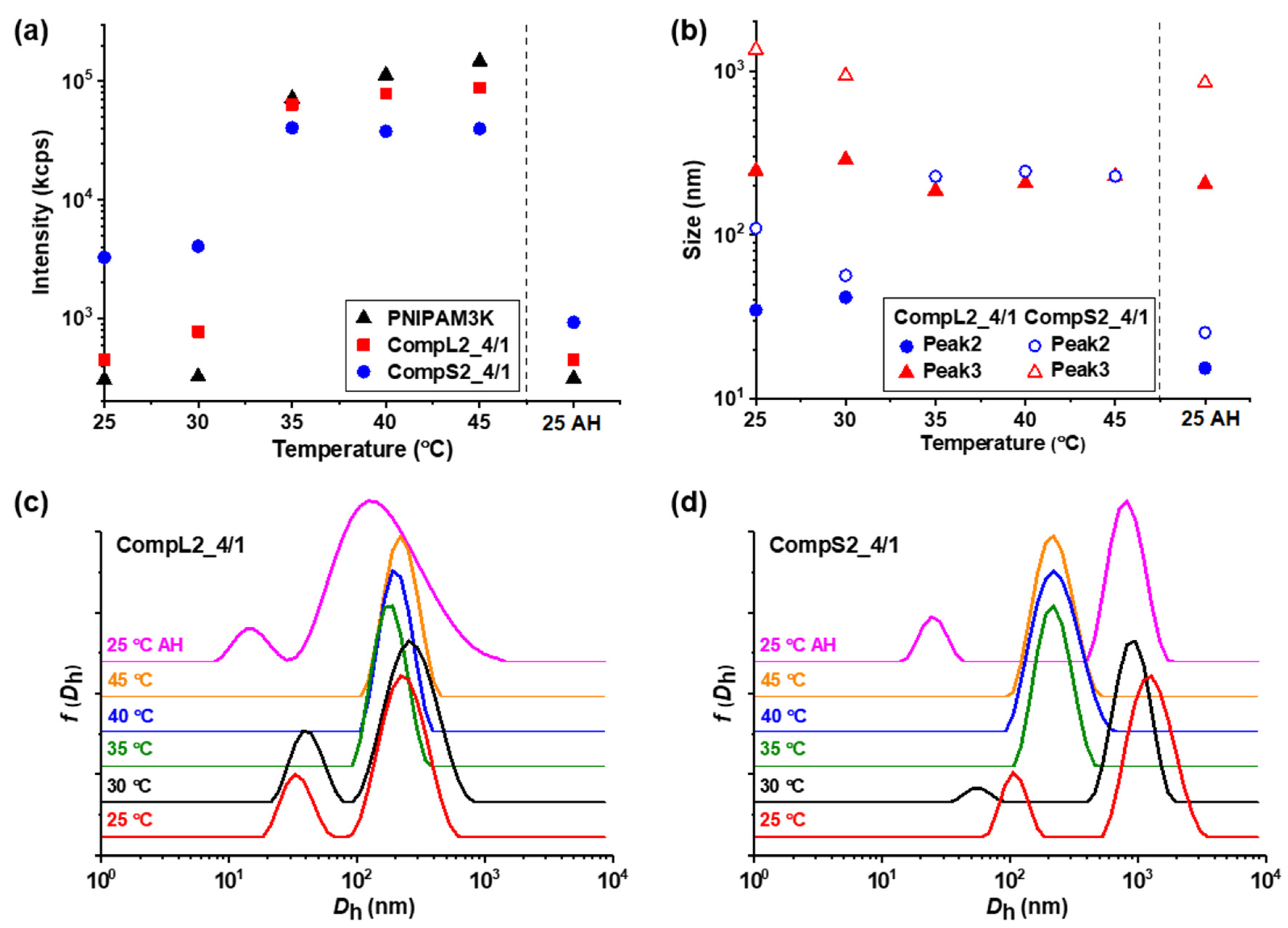
Since PNIPAM is a thermoresponsive polymer, it was expected to impart the same characteristics to the Chi/PNIPAM complexes as well, and for this reason, the influence of temperature on the formed complexes was investigated. In practice, their thermal response was examined by performing DLS measurements at different temperatures between 25 and 45 °C (with 5 °C increment steps), and the obtained results regarding the scattered intensity, peak sizes, and corresponding size distributions for the representative CompLorS1_4/1 and CompLorS2_4/1 samples are shown in
Figure 7 and
Figure 8, respectively. The influence of temperature on the rest of the Chi/PNIPAM complexes and the two PNIPAM samples was also investigated and the results can be found in the
Supplementary Materials (see Figures S10–S14). For all investigated samples, after heating up to 45 °C, the temperature was brought back to 25 °C and the sample was remeasured in order to examine the reversibility of the structures formed by the heating process. These results are denoted as “AH” (i.e., after heating) and are separated by a vertical dashed line in the graphs of scattered intensity and peak sizes.
Figure 7a and
Figure 8a reveal the influence of temperature on the scattered intensity for the representative complexes of both chitosan samples at a 4/1 volume ratio in comparison to the corresponding PNIPAM samples. At 35 °C (i.e., above the LCST of PNIPAM) an abrupt increase in intensity accompanied by a significant increase in turbidity was observed for all samples. This behavior is characteristic of the thermal phase transition of PNIPAM, and was attributed to the increase in the hydrophobic interactions that induce interchain aggregation and was common for all investigated samples of Chi/PNIPAM complexes (see also
Figures S10–S13a). Further increases in temperature up to 45 °C seemed to have no significant effect on the scattered intensity, while the initial values were fully restored upon returning to 25 °C, showcasing the reversibility of the transition. At the same time, the corresponding size distributions for the 4/1 complexes of all four systems (
Figure 7c,d and
Figure 8c,d) exhibited the same transition from two or three peaks to only one above 35 °C, which was also a characteristic trait of both PNIPAM samples (see
Figure S14). This behavior suggests the aggregation of the initial populations of complexes into single compact structures, owing to the increase in the hydrophobicity of the complexed PNIPAM chains, which in turn favors interchain interactions. The sizes of these thermally induced aggregates seems to have some correlation to the conformation of the initial complexes. In particular, for the complexes of the long chitosan molecules (CompL1or2_4/1), the species detected above 35 °C had a similar size to the corresponding initial larger populations, while in the case of the short chitosan complexes, intermediate sizes (in comparison to the initial populations) were observed for the aggregates formed as a result of the thermal transition. This fact could mean that the looser conformation of the complexes formed with the long chitosan hindered, to some degree, the inter-chain PNIPAM interactions, thus retaining more or less their initial structure upon heating. On the contrary, the complexes of the short chitosan already had a more dense/compact conformation that facilitated to a greater extent the thermally induced PNIPAM interactions, leading to further increases in their compactness. Similar thermal transitions were observed for the rest of the Chi/PNIPAM complexes (see
Figures S10–S13), with the only difference worth mentioning being those observed for the sizes of the CompLorS2_4/4 samples (
Figure S13). In this case, the long chitosan complexes seemed to become more compact upon heating, while those of the short chitosan maintained their size. Apparently, as the PNIPAM content increased, the inter-chain interactions became more dominant and even the relatively loose initial (i.e., before heating) conformation of the CompL2 complexes collapsed, while the already too-compact structure of the CompS2 ones did not allow for additional reduction in their size. Nevertheless, after the solutions were cooled back to room temperature, they all returned to their initial state, again verifying the reversibility of the systems.
The enhancement of the overall hydrophobic interactions in the systems of the formed Chi/PNIPAM complexes upon increasing the temperature above the LCST of PNIPAM, was also confirmed through the pyrene fluorescence measurements performed at 45 °C. As seen in
Table 3, the corresponding
I1/
I3 ratios exhibited more or less lower values compared to the ones at ambient conditions in almost all cases, demonstrating an increase in the hydrophobicity of the microenvironment probed by pyrene. As already discussed, the innate conformation of both the complexes and their constituent macromolecules greatly influenced their structural/conformational thermal transition, and this was also evidenced through the changes in the
I1/
I3 ratios. In particular, the complexes containing the PNIPAM19K sample exhibited a somewhat larger decrease in the
I1/
I3 ratios that the ones formed with the PNIPAM3K, which is to be expected since the longer PNIPAM chains entailed more extensive inter-chain aggregation.
3.4. Drug Loading Capacity of the Chi/PNIPAM Complexes
The ability of the specific chitosan–PNIPAM hybrid nanostructures to serve as potential drug delivery systems was explored by examining their capacity to encapsulate a model hydrophobic drug, namely curcumin (CUR). Curcumin is widely known for its antioxidant, anti-inflammatory, antimicrobial, and anticancer properties [
34], and, at the same time, it is a fluorescent molecule that can be used for bioimaging purposes [
35,
36]. Unfortunately, the low solubility of curcumin in water (below 6 μg/mL) restricts its broad applicability since it results in low cellular uptake, poor oral bioavailability, and low chemical stability [
34]. One possible solution is the use of suitable polymeric carriers in an attempt to improve the bioavailability of curcumin and thus improve its effectiveness. Among the various Chi/PNIPAM complexes that were previously investigated, CompL1_4/4 was chosen for the encapsulation of CUR, as the combination of the long chitosan (Chi162K) molecule with the long PNIPAM19K chains is considered to be the most stable one, while the high PNIPAM content was expected to further enhance the interaction with the hydrophobic drug. Three different drug loading ratios were investigated by adding 2.5, 5, and 10%
w/
w of CUR in regard to the total mass of the complex, and the mixing was performed at 40 °C in order to benefit from the phase transition of the PNIPAM chains, as described in detail in
Section 2.3.
The physicochemical properties of the resulting CUR-loaded CompL1_4/4 samples were investigated by performing DLS measurements on 1:10 diluted solutions, and
Figure 11 presents the obtained scattered intensity values, and the peak sizes from the corresponding size distributions. Moreover, the morphology of the CUR-loaded nanoparticles was evaluated by STEM for the representative CompL1_4/4 + 5%CUR sample and a characteristic image is shown in
Figure 11d. As can be seen, the DLS results reveal that the CUR-loaded complexes exhibited a drastic increase in scattered intensity, signifying an increase in the mass of the structures (
Figure 11a). Meanwhile, as far as the size of the drug-loaded complexes is concerned, a transition from the two peaks discerned for the initial complex to one peak with an intermediate size was detected, which was accordingly accompanied by a parallel significant decrease in the polydispersity index (i.e., from about 0.6 to 0.1). The combination of the observed changes confirmed the successful incorporation of CUR and further suggested the shrinking of the initial complexes and the formation of more compact/dense structures, apparently as a result of the increase in hydrophobic interactions due to the presence of the CUR molecules. Still, the overall positive effective charge of the primary Chi/PNIPAM complexes was preserved, as the sample with the highest CUR loading content (i.e., CompL1_4/4 + 10%CUR) exhibited a zeta potential value of about +30 mV. Finally, the morphology of the drug-loaded nanostructures remained spherical and in close correlation to that of the initial complex, while their corresponding average size was about 40 nm. This was slightly higher than that derived from STEM for the initial complex (i.e., about 33 nm), which could also be indicative of the more dense/compact structures formed after CUR loading.
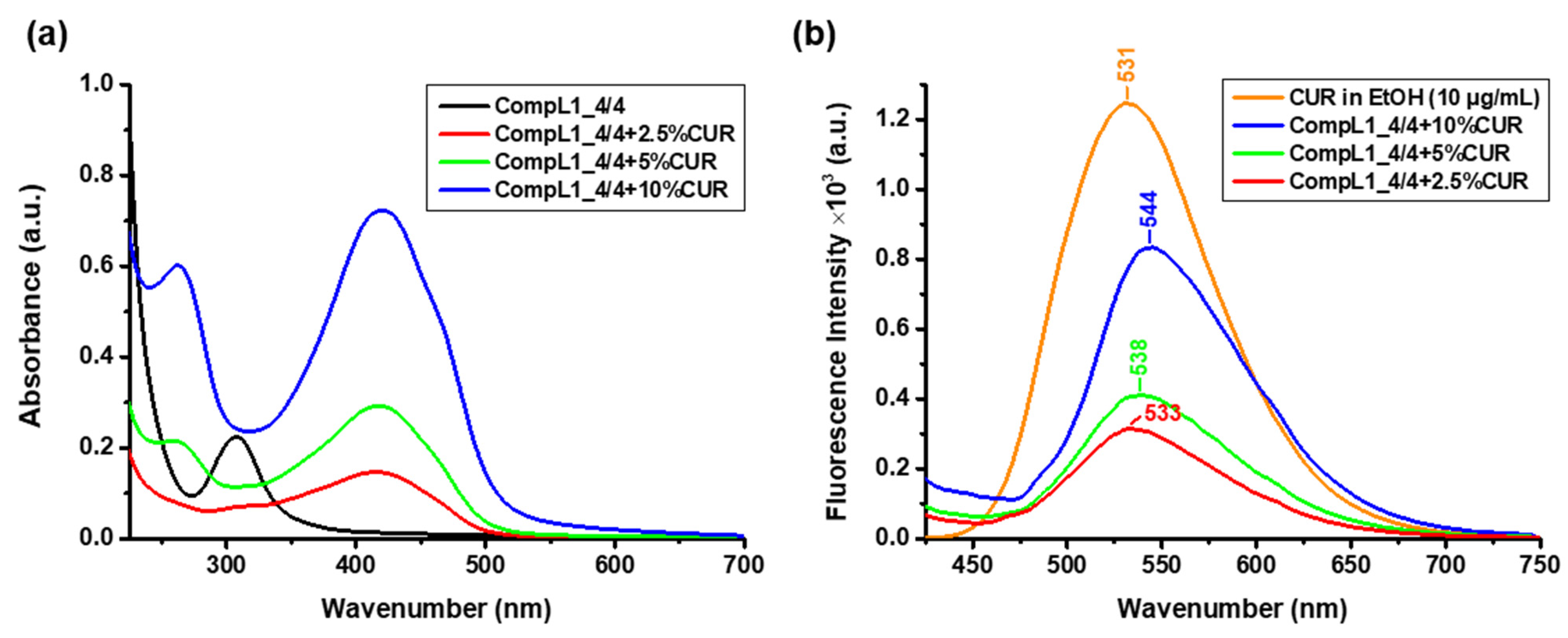
In order to determine the concentration of the encapsulated CUR, UV–Vis spectroscopic measurements were performed for the same diluted solutions and the acquired spectra are shown in
Figure 12a, along with the corresponding spectrum of the CompL1_4/4 for comparison. From the obtained values of the absorbance at 427 nm and the constructed calibration curve of CUR in ethanol (
Figure S2), the actual concentration of encapsulated CUR in the solutions/dispersions of the complexes was derived. The corresponding values for each loading ratio are given in
Table 4, along with the concentration of the added CUR (i.e., nominal concentration) and the encapsulation efficiency EE% was calculated according to Equation (1). The estimated EE% values were in the range of 43 to 61% for the three CUR contents investigated (i.e., 2.5, 5, and 10%
w/
w), indicating a sufficient drug loading capacity for the initial Chi162K + PNIPAM19K complex, which was undoubtedly correlated to the overall hydrophobicity of the system. Finally, fluorescence spectroscopy was employed to verify the fluorescent properties of the CUR-loaded complexes and thus their potential use in relevant bioimaging applications.
Figure 12b presents the obtained spectra for the three different investigated CUR contents, along with the corresponding spectrum of pure CUR in ethanol (with a concentration of 10 μg/mL). In comparison to pure CUR, an apparent gradual red shift of the characteristic CUR peak from 531 to 544 nm can be seen for the drug-loaded complexes. This shift is attributed to the hydrophobic interactions of CUR with the PNIPAM chains (especially since the loading procedure was conducted above the LCST) [
37,
38], while it is possible that the hydrophobic regions of the chitosan backbone also facilitate CUR encapsulation. Nevertheless, all three formulations exhibited strong fluorescent properties.



 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}