
Enhancing Stability of Cu/ZnO Catalysts in the CO2 Hydrogenation to Methanol by the Addition of MoO3 and ReO3 Promoters
 , ,
, ,  ,
,  ,
,  and
and
Abstract
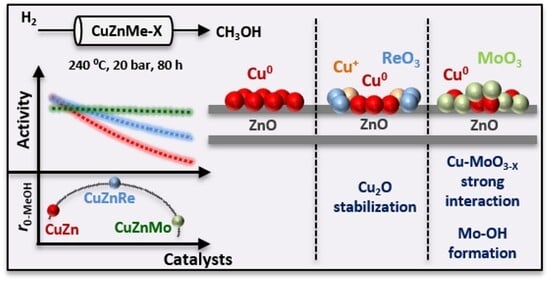
1. Introduction
2. Materials and Methods
2.1. Catalyst Synthesis
2.2. Catalyst Characterization
2.3. Catalytic Test
3. Results and Discussion
3.1. Characterization of Catalysts
3.2. Catalytic Studies
3.3. Stability Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Olah, G.A. Towards Oil Independence through Renewable Methanol Chemistry. Angew. Chem. Int. Ed. 2013, 52, 104–107. [Google Scholar] [CrossRef]
- Ebrahimzadeh Sarvestani, M.; Norouzi, O.; Di Maria, F.; Dutta, A. From Catalyst Development to Reactor Design: A Comprehensive Review of Methanol Synthesis Techniques. Energy Convers. Manag. 2024, 302, 118070. [Google Scholar] [CrossRef]
- Salahudeen, N.; Rasheed, A.A.; Babalola, A.; Moses, A.U. Review on Technologies for Conversion of Natural Gas to Methanol. J. Nat. Gas Sci. Eng. 2022, 108, 104845. [Google Scholar] [CrossRef]
- Wan, Y.; Fang, F.; Sun, R.; Zhang, J.; Chang, K. Metal Oxide Semiconductors for Photothermal Catalytic CO2 Hydrogenation Reactions: Recent Progress and Perspectives. Acta Phys.-Chim. Sin. 2023, 39, 2212042. [Google Scholar] [CrossRef]
- Pan, Z.; Han, E.; Zheng, J.; Lu, J.; Wang, X.; Yin, Y.; Waterhouse, G.I.N.; Wang, X.; Li, P. Highly Efficient Photoelectrocatalytic Reduction of CO2 to Methanol by a p–n Heterojunction CeO2/CuO/Cu Catalyst. Nano-Micro Lett. 2020, 12, 18. [Google Scholar] [CrossRef]
- Cheng, Y.; Jabeen, S.; Lei, S.; Liu, N.; Liu, Y.; Liu, Y.; Li, Y.; Wu, X.; Tong, Z.; Yu, J.; et al. N-doped carbon dots-modulated interfacial charge transfer and surface structure in FeNbO4 photocatalysts for enhanced CO2 conversion selectivity to CH4. Chem. Eng. J. 2024, 498, 155576. [Google Scholar] [CrossRef]
- Wang, Q.; Wei, H.; Liu, P.; Su, Z.; Gong, X.-Q. Recent advances in copper-based catalysts for electrocatalytic CO2 reduction toward multi-carbon products. Nano Res. Energy 2024, 3, e9120112. [Google Scholar] [CrossRef]
- Gaikwad, R.; Bansode, A.; Urakawa, A. High-Pressure Advantages in Stoichiometric Hydrogenation of Carbon Dioxide to Methanol. J. Catal. 2016, 343, 127–132. [Google Scholar] [CrossRef]
- Sha, F.; Han, Z.; Tang, S.; Wang, J.; Li, C. Hydrogenation of Carbon Dioxide to Methanol over Non−Cu-Based Heterogeneous Catalysts. ChemSusChem 2020, 13, 6160–6181. [Google Scholar] [CrossRef]
- Beck, A.; Newton, M.A.; van de Water, L.G.A.; van Bokhoven, J.A. The Enigma of Methanol Synthesis by Cu/ZnO/Al2O3-Based Catalysts. Chem. Rev. 2024, 124, 4543–4678. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Wu, Z.; Liang, B.; Huang, Y.; Zhang, T. State of the Art and Perspectives in Heterogeneous Catalysis of CO2 Hydrogenation to Methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef]
- Murthy, P.S.; Liang, W.; Jiang, Y.; Huang, J. Cu-Based Nanocatalysts for CO2 Hydrogenation to Methanol. Energy Fuels 2021, 35, 8558–8584. [Google Scholar] [CrossRef]
- Cored, J.; Lopes, C.W.; Liu, L.; Soriano, J.; Agostini, G.; Solsona, B.; Sánchez-Tovar, R.; Concepción, P. Cu-Ga3+-Doped Wurtzite ZnO Interface as Driving Force for Enhanced Methanol Production in Co-Precipitated Cu/ZnO/Ga2O3 Catalysts. J. Catal. 2022, 407, 149–161. [Google Scholar] [CrossRef]
- Kordus, D.; Timoshenko, J.; Divins, N.J.; Chee, S.W.; Ortega, E.; Lopez Luna, M.; Hejral, U.; Etxebarria, A.; Roldan Cuenya, B. Cu–Ga Interactions and Support Effects in CO2 Hydrogenation to Methanol Catalyzed by Size-Controlled CuGa Nanoparticles Deposited on SiO2 and ZnO. ACS Catal. 2025, 15, 17241–17254. [Google Scholar] [CrossRef]
- Sloczyński, J.; Grabowski, R.; Olszewski, P.; Kozlowska, A.; Stoch, J.; Lachowska, M.; Skrzypek, J. Effect of Metal Oxide Additives on the Activity and Stability of Cu/ZnO/ZrO2, Catalysts in the Synthesis of Methanol from CO2 and H2. Appl. Catal. A Gen. 2006, 310, 127–137. [Google Scholar] [CrossRef]
- Ma, L.; Tran, T.; Wainwright, M.S. Methanol Synthesis from CO2 Using Skeletal Copper Catalysts Containing Co-Precipitated Cr2O3 and ZnO. Top. Catal. 2003, 22, 295–304. [Google Scholar] [CrossRef]
- Calverley, E.M.; Smitht, K.J. The Effects of Carbon Dioxide, Methanol, and Alkali Promoter Concentration on the Higher Alcohol Synthesis over a Cu/ZnO/Cr2O3 Catalyst. J. Catal. 1991, 130, 616–626. [Google Scholar] [CrossRef]
- Santana, C.S.; Rasteiro, L.F.; Marcos, F.C.F.; Assaf, E.M.; Gomes, J.F.; Assaf, J.M. Influence of Al, Cr, Ga, or Zr as Promoters on the Performance of Cu/ZnO Catalyst for CO2 Hydrogenation to Methanol. Mol. Catal. 2022, 528, 112512. [Google Scholar] [CrossRef]
- Zhu, J.; Ciolca, D.; Liu, L.; Parastaev, A.; Kosinov, N.; Hensen, E.J.M. Flame Synthesis of Cu/ZnO-CeO2 Catalysts: Synergistic Metal-Support Interactions Promote CH3OH Selectivity in CO2 Hydrogenation. ACS Catal. 2021, 11, 4880–4892. [Google Scholar] [CrossRef]
- Sun, Y.; Ren, J.; Zhang, S. Breaking the H2 Pressure Dependence in Hydrogenation through Interfacial *H Reservoirs on Cu–WO3 Catalysts. ACS Catal. 2025, 15, 14331–14340. [Google Scholar] [CrossRef]
- Zhou, G.; He, Z.; Dong, X. Role of Metal Oxides in Cu-Based Catalysts with NaBH4 Reduction for the Synthesis of Methanol from CO2/H2. Catal. Lett. 2021, 151, 1091–1101. [Google Scholar] [CrossRef]
- Wang, G.; Mao, D.; Guo, X.; Yu, J. Enhanced Performance of the CuO-ZnO-ZrO2 Catalyst for CO2 Hydrogenation to Methanol by WO3 Modification. Appl. Surf. Sci. 2018, 456, 403–409. [Google Scholar] [CrossRef]
- Wang, G.; Mao, D.; Guo, X.; Yu, J. Methanol Synthesis from CO2 Hydrogenation over CuO-ZnO-ZrO2-MxOy Catalysts (M = Cr, Mo and W). Int. J. Hydrogen Energy 2019, 44, 4197–4207. [Google Scholar] [CrossRef]
- Sung Lee, J.; Ik Moon, K.; Hoon Lee, S.; Young Lee, S.; Gul Kim, Y. Modified Cu/ZnO/Al2O3 Catalysts for Methanol Synthesis from CO2/H2 and CO/H2. Catal. Lett. 1995, 34, 93–99. [Google Scholar] [CrossRef]
- Saito, M.; Fujitani, T.; Watanabe, T.; Takeuchi, M.; Kanai, Y.; Moriya, K.; Kakumoto, T. Development of Cu/ZnO-Based High Performance Catalysts for Methanol Synthesis by CO2 Hydrogenation. Energy Convers. Manag. 1995, 36, 577–580. [Google Scholar] [CrossRef]
- Han, J.; Wang, L.; Yu, J.; Fan, M.; Mao, D. CO2 Hydrogenation to Methanol over Cu-CeO2-ZrO2 Catalysts: The Significant Effect of Metal-Support Interaction. Fuel 2025, 381, 133262. [Google Scholar] [CrossRef]
- Fritsch, C.; Dornseiffer, J.; Blankenstein, J.; Noyong, M.; Groteklaes, C.; Simon, U. CO2-Hydrogenation to Methanol over CuO/ZnO Based Infiltration Composite Catalyst Spheres. ChemCatChem 2024, 16, e202400731. [Google Scholar] [CrossRef]
- Chang, S.; Na, W.; Zhang, J.; Lin, L.; Gao, W. Effect of the Zn/Ce Ratio in Cu/ZnO-CeO2 Catalysts on CO2 Hydrogenation for Methanol Synthesis. New J. Chem. 2021, 45, 22814–22823. [Google Scholar] [CrossRef]
- Gómez, D.; Vergara, T.; Ortega, M.; Diaconescu, V.M.; Simonelli, L.; Concepción, P.; Jiménez, R.; Karelovic, A. Interdependence Between the Extent of Ga Promotion, the Nature of Active Sites, and the Reaction Mechanism Over Cu Catalysts for CO2 Hydrogenation to Methanol. ACS Catal. 2024, 14, 15265–15278. [Google Scholar] [CrossRef]
- Gómez, D.; Candia, C.; Jiménez, R.; Karelovic, A. Isotopic Transient Kinetic Analysis of CO2 Hydrogenation to Methanol on Cu/SiO2 Promoted by Ga and Zn. J. Catal. 2022, 406, 96–106. [Google Scholar] [CrossRef]
- Vergara, T.; Gómez, D.; Lacerda de Oliveira Campos, B.; Herrera Delgado, K.; Concepción, P.; Jiménez, R.; Karelovic, A. Combined Role of Ce Promotion and TiO2 Support Improves CO2 Hydrogenation to Methanol on Cu Catalysts: Interplay between Structure and Kinetics. J. Catal. 2023, 426, 200–213. [Google Scholar] [CrossRef]
- Ma, L.; Yan, L.; Lu, A.-H.; Ding, Y. Effect of Re promoter on the structure and catalytic performance of Ni–Re/Al2O3 catalysts for the reductive amination of monoethanolamine. RSC Adv. 2018, 8, 8152–8163. [Google Scholar] [CrossRef]
- Tong, Q.; Zong, A.; Gong, W.; Yu, L.; Fan, Y. Rhenium-promoted Pt/WO3/ZrO2: An efficient catalyst for aqueous glycerol hydrogenolysis under reduced H2 pressure. RSC Adv. 2016, 6, 86663–86672. [Google Scholar] [CrossRef]
- Waller, D.; Stirling, D.; Stone, F.S.; Spencer, M.S. Copper-zinc oxide catalysts. Activity in relation to precursor structure and morphology. Faraday Discuss. Chem. Soc. 1989, 87, 107–120. [Google Scholar] [CrossRef]
- Li, J.; Inui, T. Characterization of precursors of methanol synthesis catalysts, copper/zinc/aluminum oxides, precipitated at different pHs and temperatures. Appl. Catal. A Gen. 1996, 137, 105–117. [Google Scholar] [CrossRef]
- Behrens, M.; Brennecke, D.; Girgsdies, F.; Kißner, S.; Trunschke, A.; Nasrudin, N.; Zakaria, S.; Idris, N.F.; Hamid, S.B.A.; Kniep, B.; et al. Understanding the complexity of a catalyst synthesis: Co-precipitation of mixed Cu,Zn,Al hydroxycarbonate precursors for Cu/ZnO/Al2O3 catalysts investigated by titration experiments. Appl. Catal. A Gen. 2011, 392, 93–102. [Google Scholar] [CrossRef]
- Lunkenbein, T.; Schumann, J.; Behrens, M.; Schlögl, R.; Willinger, M.G. Formation of a ZnO Overlayer in Industrial Cu/ZnO/Al2O3 Catalysts Induced by Strong Metal–Support Interactions. Angew. Chem. Int. Ed. 2015, 127, 4627–4631. [Google Scholar] [CrossRef]
- Kuld, S.; Conradsen, C.; Moses, P.G.; Chorkendorff, I.; Sehested, J. Quantification of Zinc Atoms in a Surface Alloy on Copper in an Industrial-Type Methanol Synthesis Catalyst. Angew. Chem. Int. Ed. 2014, 53, 5941–5945. [Google Scholar] [CrossRef]
- Ullah Awan, S.; Hasanain, S.K.; Bertino, M.F.; Jaffari, G.H. Ferromagnetism in Li Doped ZnO Nanoparticles: The Role of Interstitial Li. J. Appl. Phys. 2012, 112, 103924. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Mendoza-Sanchez, B.; Fernandez, V.; Veenstra, R.; Dukstiene, N.; Roberts, A.; Fairley, N. Generalized Molybdenum Oxide Surface Chemical State XPS Determination via Informed Amorphous Sample Model. Appl. Surf. Sci. 2015, 326, 151–161. [Google Scholar] [CrossRef]
- Murugappan, K.; Anderson, E.M.; Teschner, D.; Jones, T.E.; Skorupska, K.; Román-Leshkov, Y. Operando NAP-XPS Unveils Differences in MoO3 and Mo2C during Hydrodeoxygenation. Nat. Catal. 2018, 1, 960–967. [Google Scholar] [CrossRef]
- Nag, N.K. A Comparative Study on the Dispersion and Carrier-Catalyst Interaction of Molybdenum Oxides Supported on Various Oxides by Electron Spectroscopy for Chemical Analysis. J. Phys. Chem. 1987, 91, 2324–2327. [Google Scholar] [CrossRef]
- Zheng, M.; Zhou, F.; Ma, H.; Songa, X.; Wu, G. Hydroxyapatite supported molybdenum oxide catalyst for selective dehydrogenation of cyclohexane to cyclohexene: Studies of dispersibility and chemical environment. RSC Adv. 2024, 14, 36461–36470. [Google Scholar] [CrossRef]
- Reddy, B.M.; Chowdhury, B.; Reddy, E.P.; Fernández, A. An XPS study of dispersion and chemical state of MoO3 on Al2O3-TiO2 binary oxide support. Appl. Catal. A Gen. 2001, 213, 279–288. [Google Scholar] [CrossRef]
- Morales-Mendoza, J.E.; Herrera-Pérez, G.; Fuentes-Cobas, L.; Hermida-Montero, L.A.; Pariona, N.; Paraguay-Delgado, F. Synthesis, Structural and Optical Properties of Cu Doped ZnO and CuO–ZnO Composite Nanoparticles. Nano-Struct. Nano-Objects 2023, 34, 100967. [Google Scholar] [CrossRef]
- Yu, T.; Zhao, X.; Shen, Z.X.; Wu, Y.H.; Su, W.H. Investigation of Individual CuO Nanorods by Polarized Micro-Raman Scattering. J. Cryst. Growth 2004, 268, 590–595. [Google Scholar] [CrossRef]
- Niaura, G. Surface-Enhanced Raman Spectroscopic Observation of Two Kinds of Adsorbed OH−Ions at Copper Electrode. Electrochim. Acta 2000, 45, 3507–3519. [Google Scholar] [CrossRef]
- Bodappa, N.; Su, M.; Zhao, Y.; Le, J.B.; Yang, W.M.; Radjenovic, P.; Dong, J.C.; Cheng, J.; Tian, Z.Q.; Li, J.F. Early Stages of Electrochemical Oxidation of Cu(111) and Polycrystalline Cu Surfaces Revealed by in Situ Raman Spectroscopy. J. Am. Chem. Soc. 2019, 141, 12192–12196. [Google Scholar] [CrossRef]
- Steimecke, M.; Araújo-Cordero, A.M.; Dieterich, E.; Bron, M. Probing Individual Cuprous Oxide Microcrystals towards Carbon Dioxide Reduction by Using In Situ Raman-Coupled Scanning Electrochemical Microscopy. ChemElectroChem 2022, 9, e202101221. [Google Scholar] [CrossRef]
- Mestl, G.; Srinivasan, T.K.K. Raman Spectroscopy of Monolayer-Type Catalysts: Supported Molybdenum Oxides. Catal. Rev. Sci. Eng. 1998, 40, 451–570. [Google Scholar] [CrossRef]
- Seguin, L.; Figlarz, M.; Cavagnat, R.; Lassègues, J.-C. Infrared and Raman Spectra of MoO3, Molybdenum Trioxides and MoO3, ·XH2O Molybdenum Trioxide Hydrates. Spectrochim. Acta Part A 1995, 51, 1323–1344. [Google Scholar] [CrossRef]
- Purans, J.; Kuzmin, A.; Cazzanelli, E.; Mariotto, G. Disorder-Induced Raman Scattering in Rhenium Trioxide (ReO3). J. Phys. Condens. Matter 2007, 19, 226206. [Google Scholar] [CrossRef]
- Castriota, M.; Cazzanelli, E.; Das, G.; Kalendarev, R.; Kuzmin, A.; Marino, S.; Mariotto, G.; Purans, J.; Scaramuzza, N. Proton Presence and Motion in Rhenium-Oxide Films and Their Application to Liquid-Crystalline Cells. Mol. Cryst. Liq. Cryst. 2007, 474, 1–15. [Google Scholar] [CrossRef]
- Stencel, J.M.; Makovsky, L.E.; Sarkus, T.A.; De Vries, J.; Thomas, R.; Moulijn, J.A. Raman Spectroscopic Investigation of the Effect of H2O on the Molybdenum Surface Species in MoO3/Al2O3 Catalysts. J. Catal. 1984, 90, 314–322. [Google Scholar] [CrossRef]
- Dieterle, M.; Weinberg, G.; Mestl, G. Raman Spectroscopy of Molybdenum Oxides—Part I. Structural Characterization of Oxygen Defects in MoO3-x by DR UV/VIS, Raman Spectroscopy and X-Ray Diffraction. Phys. Chem. Chem. Phys. 2002, 4, 812–821. [Google Scholar] [CrossRef]
- Mestl, G. In Situ Raman Spectroscopy—A Valuable Tool to Understand Operating Catalysts. J. Mol. Catal. A Chem. 2000, 158, 45–65. [Google Scholar] [CrossRef]
- Payen, E.; Kasztelan, S.; Grimblot, J.; Bonnelle, J.P. Surface Chemistry of MoO3/γ-Al2O3, Catalysts Studied by Laser Raman Spectroscopy: Hydration and Dehydration Reactions and Generalization to Other Supported Systems. J. Raman Spectrosc. 1986, 17, 233–241. [Google Scholar] [CrossRef]
- Dong, X.; Li, F.; Zhao, N.; Xiao, F.; Wang, J.; Tan, Y. CO2 Hydrogenation to Methanol over Cu/ZnO/ZrO2 Catalysts Prepared by Precipitation-Reduction Method. Appl. Catal. B Environ. 2016, 191, 8–17. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.; Wang, P.; Wang, X.; Song, F.; Bai, Y.; Zhang, M.; Wu, Y.; Xie, H.; Tan, Y. Effect of Vapor-Phase-Treatment to CuZnZr Catalyst on the Reaction Behaviors in CO2 Hydrogenation into Methanol. ChemCatChem 2019, 11, 1448–1457. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhan, H.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Wang, H.; Sun, Y. Influence of Zr on the Performance of Cu/Zn/Al/Zr Catalysts via Hydrotalcite-like Precursors for CO2 Hydrogenation to Methanol. J. Catal. 2013, 298, 51–60. [Google Scholar] [CrossRef]
- Choi, Y.; Dong Sim, G.; Jung, U.; Park, Y.; Youn, M.H.; Chun, D.H.; Rhim, G.B.; Kim, K.Y.; Koo, K.Y. Copper catalysts for CO2 hydrogenation to CO through reverse water–gas shift reaction for e-fuel production: Fundamentals, recent advances, and prospects. Chem. Eng. J. 2024, 492, 152283. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Ratio Cu/Me a | BET Surface Area (m2/g) b | Crystallite Size, nm c | H2-TPR | N2O-TPR e | XPS f | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| (2 0 0) Facet of Cu0 | (1 1 0) Facet of ZnO | Maximum Temperature (°C) d | µmol CuS/gcat | µmol CuS/m2 | Cu/Zn Ratio | Cu/Me Ratio | Cu+/Cu0 Ratio f1 | |||
| CuZn | -- | 10.1 | 23.1 | 43.8 | 210.4 | 1006 | 99.6 | 0.87 | -- | 0.149 |
| CuZnRe-0.06 | 3281 | 9.2 | 17.0 | 40.1 | 220.2 | 1730 | 189.1 | 0.90 | -- | 0.237 |
| CuZnRe-0.50 | 409 | 8.4 | 16.4 | 38.7 | 238.6 | 2560 | 303.9 | 0.67 | 32.8 | 0.210 |
| CuZnRe-3.50 | 57 | 8.6 | 16.4 | 40.8 | 202.3 | 1168 | 135.8 | 0.96 | 29.6 | 0.590 |
| CuZnMo-0.06 | 1690 | 13.7 | 13.7 | 41.0 | 219.1 | 1668 | 122.1 | 0.78 | 2.07 | 0.385 |
| CuZnMo-0.44 | 239 | 28.4 | 8.3 | 48.8 | 287.7 | 1666 | 58.6 | 1.03 | 5.02 | 0.163 |
| CuZnMo-3.50 | 29 | 25.0 | 8.4 | 18.6 | 350.8 | 1815 | 72.5 | 0.77 | 0.80 | 0.157 |
| Catalyst a | K b 20 h | Ratio (KCuZn/KCuZnM-X) 20 h | % c 20 h | K 80 h | Ratio (KCuZn/KCuZnM-X) 80 h | % c 80 h | Crystallite Size, nm d | Crystallite Size, nm a | ||
|---|---|---|---|---|---|---|---|---|---|---|
| (2 0 0) Facet of Cu0 | (1 1 0) Facet of ZnO | (2 0 0) Facet of Cu0 | (1 1 0) Facet of ZnO | |||||||
| CuZn | 0.164 | 1.0 | -- | 0.168 | 1.0 | -- | 23.1 | 43.8 | 42.8 | 46.3 |
| CuZnRe0.50 | 0.087 | 1.9 | 47.0 | 0.092 | 1.8 | 45.2 | 16.4 | 38.7 | 27.9 | 44.9 |
| CuZnMo0.06 | 0.066 | 2.5 | 59.8 | 0.100 | 1.7 | 40.5 | 13.7 | 41.0 | 33.1 | 47.3 |
| CuZnMo0.44 | 0.030 | 5.5 | 81.7 | 0.044 | 3.8 | 73.8 | 8.3 | 48.8 | 13.5 | 50.1 |
| CuZnMo3.50 | 0.016 | 10.3 | 90.3 | 0.030 | 5.6 | 82.1 | 8.4 | 18.6 | 10.6 | 16.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soriano Rodríguez, J.; Nieto, J.M.L.; Rodriguez-Castellón, E.; Infantes, A.; Gómez, D.; Concepción, P. Enhancing Stability of Cu/ZnO Catalysts in the CO2 Hydrogenation to Methanol by the Addition of MoO3 and ReO3 Promoters. Nanomaterials 2025, 15, 1730. https://doi.org/10.3390/nano15221730
Soriano Rodríguez J, Nieto JML, Rodriguez-Castellón E, Infantes A, Gómez D, Concepción P. Enhancing Stability of Cu/ZnO Catalysts in the CO2 Hydrogenation to Methanol by the Addition of MoO3 and ReO3 Promoters. Nanomaterials. 2025; 15(22):1730. https://doi.org/10.3390/nano15221730
Chicago/Turabian StyleSoriano Rodríguez, Jose, José Manuel López Nieto, Enrique Rodriguez-Castellón, Antonia Infantes, Daviel Gómez, and Patricia Concepción. 2025. "Enhancing Stability of Cu/ZnO Catalysts in the CO2 Hydrogenation to Methanol by the Addition of MoO3 and ReO3 Promoters" Nanomaterials 15, no. 22: 1730. https://doi.org/10.3390/nano15221730
APA StyleSoriano Rodríguez, J., Nieto, J. M. L., Rodriguez-Castellón, E., Infantes, A., Gómez, D., & Concepción, P. (2025). Enhancing Stability of Cu/ZnO Catalysts in the CO2 Hydrogenation to Methanol by the Addition of MoO3 and ReO3 Promoters. Nanomaterials, 15(22), 1730. https://doi.org/10.3390/nano15221730