Abstract
Nitrous oxide (N2O) is a potent greenhouse gas with a global warming potential > 310 times that of CO2. Owing to its rapid increase in atmospheric concentrations from industrial emissions, N2O poses increasing environmental concerns. Among the various N2O abatement technologies, catalytic decomposition can directly convert N2O into harmless N2 and O2 without generating secondary pollutants. In this study, Co3O4 spinel catalysts were synthesized using a polymer-assisted precipitation method, using polyvinyl alcohol, polyvinylpyrrolidone, or polyethylene glycol (PEG) as N2O decomposition catalysts. The PEG-mediated synthesis method yielded the most active catalyst with superior N2O decomposition efficiency. Structural and surface analyses confirmed that PEG facilitated the formation of Co2+-enriched surface sites and abundant oxygen vacancies, which are crucial active sites for N2O adsorption and activation. Moreover, these features improved the redox properties and electron transfer behavior of the resulting catalyst. In particular, the PEG-derived 5Co3O4/CeO2 catalyst exhibited enhanced N2O decomposition activity and stability even in the presence of coexisting N2O and O2, highlighting its potential for real-world applications. This study provides an effective synthetic route for Co3O4-based catalysts and potential opportunities for wide applications in industrial N2O removal.
1. Introduction
Nitrous oxide (N2O) has recently surpassed chlorofluorocarbons (CFCs) as the predominant anthropogenic ozone-depleting substance and is the third most significant greenhouse gas following CO2 and CH4 [1,2,3]. Although N2O represents a relatively small fraction of total greenhouse gas emissions, its global warming potential is approximately 310 times higher than that of CO2, and its ozone depletion potential is nearly double that of CFCs [4,5]. The continuous increase in atmospheric N2O concentrations caused by industrial activities, including nitric and adipic acid production, semiconductor manufacturing, fossil fuel combustion, co-firing power generation, and maritime emissions, has become an increasingly serious environmental concern [6,7,8]. N2O emissions are expected to double by 2050, rendering the development and implementation of effective reduction strategies urgently required. Various mitigation strategies have been developed, including thermal decomposition and catalytic reduction [9,10]. Among them, the direct catalytic decomposition of N2O into harmless N2 and O2 is a promising approach owing to the absence of secondary pollutants and its simplicity and high efficiency [11,12]. For the direct catalytic decomposition of N2O, extensive research has focused on three primary catalyst categories: supported noble metals [13,14,15], pure and composite metal oxides [11,16,17,18], and zeolite-based systems [19,20,21]. Among these, metal oxide catalysts with excellent thermal stability at high temperatures are being researched as an alternative to overcome disadvantages such as the high cost of noble metal catalysts, low hydrothermal stability, and catalyst activity degradation by other gas components of zeolite catalysts [22,23]. In particular, spinel-type Co3O4 (AB2O4), in which Co2+ occupies tetrahedral (A) sites and Co3+ occupies octahedral (B) sites, has been widely investigated for N2O decomposition. Spinel-type Co3O4 possesses the favorable redox properties of Co2+ and the high density of oxygen vacancies [24,25]. Although Co3O4-based catalysts offer advantages for N2O decomposition, they often exhibit reduced activity in the presence of interfering gases, such as O2. These interfering gases compete with N2O for active sites and hinder the reaction by suppressing the role of surface oxygen vacancies [26,27]. While several synthesis strategies, such as doping and defect engineering, have been explored for tuning the Co3+/Co2+ ratio, these methods often entail complex procedures or harsh, high-temperature post-treatments [28,29]. In contrast, the polymer-assisted synthesis offers a facile, energy-efficient, and cost-effective route to concurrently modulate the surface electronic structure and active sites [30]. This is achieved through the coordination between polymer functional groups (e.g., PVA, PVP, PEG) and cobalt precursors, which enables the precise control of the Co3+/Co2+ ratio and oxygen-vacancy formation under mild reaction conditions, without requiring additional doping or post-treatment [31,32].
To address these limitations, polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP), and polyethylene glycol (PEG) have been employed as polymeric additives owing to their distinct physicochemical properties. For example, PVA exhibits mild reducing properties [33,34], and PVP stabilizes particle dispersion [34,35]. PEG provides strong coordination capability and promotes the formation of surface-active sites and oxygen vacancies. In particular, PEG has emerged as an effective modifier, achieving the highest N2O conversion owing to its abundant hydroxyl groups and flexible molecular backbone, which facilitate enhanced interactions with metal precursors [36,37,38]. Recent density functional theory calculations have demonstrated that the polymer-induced electronic modulation of the surface cobalt oxidation states, coupled with the generation of oxygen vacancies, can effectively decrease the activation energy for N2O decomposition [39,40]. Although polymer-assisted synthesis is generally used for morphological control or particle stabilization, its potential role in modulating the surface electronic properties, particularly in oxidative environments, has not been extensively investigated.
In this study, PVA, PVP, and PEG were employed as polymeric additives to modulate the surface properties and electronic structures of Co3O4 catalysts. To evaluate their catalytic performance, Co3O4 catalysts, which were synthesized using polymer additives, were supported on CeO2 to form 5Co3O4/CeO2 composite catalysts. CeO2 was selected because it is widely known for its high oxygen storage capacity and strong redox ability, which promote the formation of surface oxygen vacancies and enhance oxygen mobility [36]. By varying the polymer additives used during synthesis, the Co2+/Co3+ ratio was successfully tuned and abundant surface oxygen vacancies were introduced, enhancing the N2O decomposition activity. The polymer-assisted strategy enabled the formation of Co3O4 surfaces rich in Co2+ and oxygen vacancies, achieving enhanced catalytic activity even under high gas hourly space velocity (GHSV) conditions. Notably, the optimized PEG catalyst maintained over 98% N2O conversion above 550 °C even under oxygen-rich conditions, further indicating its potential applicability in realistic industrial environments.
2. Materials and Methods
2.1. Materials
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) and cerium(IV) oxide (CeO2) were purchased from Sigma-Aldrich (St. Louis, MO, USA). PVA [CH2CH(OH)n], PVP [(C6H9NO)n], PEG [H((CH2CH2O)n)OH], and sodium hydroxide (NaOH) were obtained from Daejung Chemicals and Metals Co., Ltd. (Siheung-si, Gyeonggi-do, Republic of Korea).
2.2. Preparation of Polymer-Assisted Co3O4 Spinel Catalysts Using PVA, PVP, and PEG
Polymer-assisted Co3O4 spinel catalysts were synthesized using a precipitation method. Briefly, 1.0 and 10.0 g of polymer and NaOH, respectively, were dissolved in 100 mL of deionized (DI) water and stirred for 1 h. Subsequently, 15.0 g of Co(NO3)2·6H2O was dissolved in 100 mL of DI water and stirred for 1 h. A mixed solution of the polymer and NaOH was slowly added dropwise to the prepared cobalt solution. The pink precipitate formed from the initial blue solution was washed with ethanol and DI water and repeatedly centrifuged until the pH became neutral. Subsequently, the catalyst precursors were dried at 100 °C for 6 h in an oven and calcined at 650 °C for 4 h in a muffle furnace. The synthesized PEG-, PVP-, and PVA-based catalysts were labeled as PEG Co3O4, PVP Co3O4, and PVA Co3O4, respectively.
2.3. Synthesis of Polymer-Assisted Co3O4/CeO2 Catalysts
Using the impregnation method, the polymer-assisted (PVA, PVP, or PEG) catalysts were used as precursors to obtain CeO2-supported catalysts (5Co3O4/CeO2). Briefly, 5 wt% polymer-assisted (PVA, PVP, or PEG) Co3O4 was stirred in 100 mL of DI water for 30 min, while CeO2 was stirred in 150 mL of DI water for 30 min. The solutions were mixed together and stirred for 1 h. The catalysts were dried at 100 °C for 6 h in an oven and calcined at 650 °C for 4 h in a muffle furnace. The PEG-, PVP-, and PVA-assisted/CeO2-supported catalysts were labeled as PEG 5Co3O4/CeO2, PVP 5Co3O4/CeO2, and PVA 5Co3O4/CeO2, respectively. To enhance visual clarity and comprehensibility, the overall synthesis route has been illustrated as a schematic representation, which has been incorporated into the Figure S1.
2.4. Catalyst Characterization
The structural characteristics of the catalysts were analyzed using X-Ray diffraction (XRD; D8 Advance, Bruker, Billerica, MA, USA) with Cu Kα radiation over a 2θ range of 10–80° to confirm the formation of the spinel Co3O4 phase and to evaluate the effect of polymer additives on crystallinity. The morphology and lattice structure of the catalysts were examined using field emission transmission electron microscopy (JEM-F200, JEOL, Ltd., Tokyo, Japan) operated at 200 kV. The specific surface areas and pore volumes of the catalysts were determined using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda methods with an ASAP 2020 instrument (Micromeritics, Norcross, GA, USA) to assess the influence of polymer-assisted synthesis on textural properties. Prior to obtaining measurements, the samples were pretreated at 150 °C for 4 h under vacuum with N2 gas to remove impurities and moisture. Bonding between the polymer and metal ion state (Co2+ and Co3+) was evaluated using Fourier-transform infrared (FT-IR) spectroscopy (Vertex 80v, Bruker, USA). The presence of residual polymer was confirmed using a micro-Raman spectrometer (NRS-5100, JASCO, Tokyo, Japan) with a laser wavelength of 532 nm. X-Ray photoelectron spectroscopy (XPS; K-Alpha, Thermo Fisher Scientific, Waltham, MA, USA) using an Al Kα radiation source (λ = 1.4866 Å) was conducted to investigate the chemical states of elements in the polymer-assisted Co3O4/CeO2 catalyst. The XPS binding energy was calibrated using the C 1s peak at 284.6 eV. Electron paramagnetic resonance (EPR) spectroscopy (Bruker, Billerica, MA, USA, EMXplus) was performed at X-band ~9.518 GHz with a magnetic field 1500 G to measure the unpaired electron signals of Co2+ ions for identification and quantification. Temperature-programmed reduction with H2 (H2-TPR) and temperature-programmed desorption with O2 (O2-TPD) and N2O (N2O-TPD) were performed to evaluate the redox behavior, oxygen mobility, using a chemisorption analyzer (AutoChem II 2920, Micromeritics, USA) by increasing the temperature from 100 to 700 °C at a rate of 10 °C/min.
2.5. Catalytic Performance Evaluation
The N2O decomposition activity was evaluated at 350–650 °C using a fixed-bed reactor with a reactor diameter and length of 12.7 and 400 mm, respectively. The feed-gas flow rate was regulated using a mass-flow controller (Brooks Instruments, Hatfield, PA, USA). The total flow rate of the feed gas was maintained at 500 sccm. For each test, 0.9 g of catalyst was loaded onto quartz wool 0.3 g to prevent channeling and maintain a stable fixed-bed configuration during the reaction. The evaluation gas flow conditions were as follows: 5000 ppm of N2O, 5 vol% O2 (when used), balanced N2, and GHSV of 60,000 h−1. FT-IR spectroscopy (CX-4000, Gasmet Technologies Oy, Helsinki, Finland) was employed to analyze the inlet and outlet reaction gas concentrations (except O2), while the O2 concentration was measured using a gas analysis spectrometer (DSM-XT, Hausnet S.R.L., Buenos Aires, Argentina). The N2O conversion efficiency was calculated using Equation (1), where [N2O]in and [N2O]out represent the N2O concentrations (ppm) in the feed and flue gases, respectively.
3. Results & Discussion
3.1. Catalytic Performance
The catalytic activity of the synthesized polymer-assisted 5Co3O4/CeO2 catalysts was evaluated for N2O decomposition using a fixed-bed reactor. As shown in Figure 1a, all polymer-assisted catalysts exhibited enhanced N2O conversion compared to the pristine Co3O4/CeO2. Among them, PEG 5Co3O4/CeO2 demonstrated the highest activity, achieving ˃99.5% conversion at 600 °C. In contrast, the pristine sample reached a maximum conversion of 57.6% under identical conditions. This trend highlights the promotional effect of polymer-induced surface modification, particularly by PEG, on catalytic performance for N2O decomposition. The catalytic activity trend observed in Figure S2 for polymer-assisted Co3O4 catalysts was consistent with that of the corresponding 5Co3O4/CeO2 catalysts. To ensure that the intrinsic catalytic activity was accurately evaluated and to eliminate surface area effects as shown in Figure S3, the N2O decomposition reaction was performed under kinetically controlled conditions, where mass-transfer limitations were excluded according to the Koros–Nowak criterion [41,42]. All catalytic measurements were carried out at a GHSV 1,000,000 h−1, while maintaining the N2O conversion below 20%, consistent with previously reported kinetic control conditions [43]. The apparent activation energies (Ea) for the Co3O4 catalysts were determined to be 73.94 kJ∙mol−1 for pristine Co3O4, 59.42 kJ∙mol−1 for PVA Co3O4, kJ∙mol−1 for PVP Co3O4, and kJ∙mol−1 for PEG Co3O4. Significantly, PEG Co3O4 exhibited the lowest Ea, indicating that the polymer-assisted precipitation strategy effectively increased the density of accessible active sites and enhanced surface reducibility, thereby improving the intrinsic catalytic activity toward N2O decomposition.
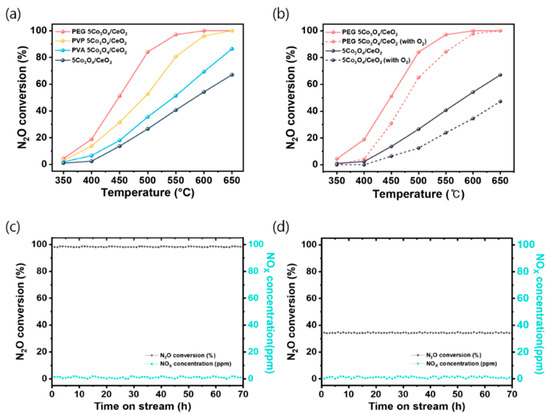
Figure 1.
(a) N2O decomposition activities of the PEG, PVP, and PVA-assisted 5Co3O4/CeO2 catalysts and pristine 5Co3O4/CeO2 under the following conditions: 5000 ppm N2O, N2 balance, GHSV of 60,000 h−1, and 350–650 °C. (b) N2O decomposition activities of PEG 5Co3O4/CeO2 and pristine 5Co3O4/CeO2 with and without 5 vol.% O2 under the following conditions: 5000 ppm N2O, N2 balance, GHSV of 60,000 h−1, and 350–650 °C. Long-term stabilities of the (c) PEG 5Co3O4/CeO2 and (d) pristine 5Co3O4/CeO2 catalysts at 600 °C for 70 h under the following conditions: 5000 ppm N2O, 5 vol.% O2, N2 balance, and GHSV of 60,000 h−1.
To investigate the effect of O2 on N2O decomposition owing to competitive adsorption on the catalyst surface, catalytic activity under O2 co-feeding conditions was evaluated for the best-performing (PEG 5Co3O4/CeO2) and least active (pristine 5Co3O4/CeO2) catalysts [31,44]. As shown in Figure 1b, the presence of 5 vol% O2 suppressed N2O conversion across all temperatures. The observed inhibitory effect is consistent with previous studies, which have reported that the coexistence of O2 suppresses N2O decomposition due to competitive adsorption on the active sites of Co3O4-based catalysts [45,46,47,48]. However, PEG 5Co3O4/CeO2 retained high activity even in the presence of O2, reaching 97.6% conversion at 600 °C. In contrast, the pristine 5Co3O4/CeO2 catalyst showed significantly reduced performance, with a maximum conversion decreasing to 35.4% under identical conditions. Thus, PEG 5Co3O4/CeO2 exhibited enhanced intrinsic activity and superior resistance to oxygen-induced inhibition. Long-term stability of the catalysts was evaluated by conducting a 70 h N2O decomposition test at 600 °C under O2 and N2O co-feeding conditions, simulating realistic oxidative environments Figure 1c,d. Because industrial N2O abatement systems typically operate under oxidizing atmospheres for extended durations, such long-term O2 durability is critical for assessing catalyst viability in commercial-scale industry.
In addition, NOx concentrations in the outlet stream were monitored to assess the selectivity of the catalysts and ensure that N2O was directly decomposed into N2 and O2 without proceeding through intermediate steps involving NO or NO2 formation. Monitoring NOx is critical because N2O can follow multiple decomposition pathways, some of which involve the formation of NO or NO2 as intermediates [1]. These species may arise from incomplete reduction or side oxidation reactions. Their presence indicates both a loss of selectivity toward N2 and environmental concerns because NOx are regulated pollutants [49]. Therefore, the absence of NOx formation indicates that the reaction selectively proceeds toward N2 and O2 without generating harmful byproducts. Under N2O and O2 conditions, the PEG 5Co3O4/CeO2 catalyst exhibited only a marginal decrease in N2O conversion (from ˃99.5% to 97.2%) at 600 °C. In contrast, the pristine 5Co3O4/CeO2 catalyst showed a significant reduction in performance, with the N2O conversion decreasing from 57.6% to 35.4%. This reduction represents approximately a 20% loss in activity. Thus, the PEG additive functioned as an effective surface electronic property modulator, enhancing both redox stability and oxygen tolerance. Consequently, PEG 5Co3O4/CeO2 maintained superior catalytic activity even under oxidizing conditions.
To assess the durability of the catalysts under conditions that influence stability, time-on-stream tests were conducted at 550 and 600 °C with the stepwise introduction of O2, NO, and H2O into the N2O feed stream Figure 2a. Furthermore, Figure 2b shows the N2O decomposition of all synthesized catalysts at 600 °C under simultaneous feeding conditions. These species adversely affect catalytic performance because O2 competes with N2O for active sites while NO and H2O deactivate or block surface oxygen vacancies, resulting in decreased catalytic activity [50,51]. At 550 °C, the pristine 5Co3O4/CeO2 catalyst exhibited low baseline activity (approximately 40%), which decreased more severely to 21% (with O2), 18% (with O2 + NO), and only 6% (with O2 + NO + H2O). In contrast, the PEG 5Co3O4/CeO2 catalyst initially achieved an 84% N2O conversion under N2O-only conditions. Upon the sequential additions of O2, NO, and H2O, the conversion progressively decreased to 80%, 76%, and 59%, respectively. A similar trend was observed at 600 °C. The PEG-assisted catalyst maintained ˃99.5% N2O conversion under N2O-only conditions, with only minor reductions of 97%, 95%, and 80% following co-feeding with O2, NO, and H2O, respectively. Conversely, the pristine catalyst started at 57% conversion and decreased to 35%, 30%, and 12% under identical conditions. These results demonstrate the superior tolerance of the PEG 5Co3O4/CeO2 catalyst to the deactivation effects of oxygen, nitrogen oxides, and water vapor, which can significantly impair the N2O decomposition performance of conventional Co3O4-based catalysts. Overall, the PEG 5Co3O4/CeO2 catalyst exhibited excellent N2O removal efficiency, enhanced resistance to common inhibitors, and stable performance under various reaction conditions. Thus, the PEG 5Co3O4/CeO2 catalyst has significant potential for practical applications in industrial emission-control systems. The CeO2 support was excluded in the analysis to decouple the effect of polymers on cobalt.
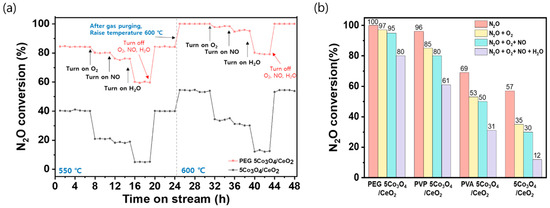
Figure 2.
N2O decomposition activities of the PEG 5Co3O4/CeO2 and pristine 5Co3O4/CeO2 catalysts under the conditions. (a) Time-on-stream stability test at 550 and 600 °C under sequential addition of O2, NO, and H2O. (b) All synthesized catalysts at 600 °C under simultaneous feeding O2, NO, and H2O. The reaction conditions were as follows: 5000 ppm N2O, 5 vol% O2, 500 ppm NO, 5 vol% H2O, N2 balance, and GHSV of 60,000 h−1.
3.2. Structural Characteristics and Surface Properties of the Catalysts
XRD was performed to examine the crystallinities and identify the crystal planes of the synthesized catalysts Figure 3a. The structure of the pristine Co3O4 catalyst was confirmed by characteristic peaks located at 19.1°, 31.3°, 36.8° and 44.6°, which corresponded to the (111), (220), (311), and (400) planes of Co3O4, respectively (JCPDS card No. 75-2480) [52]. For the polymer-assisted Co3O4 catalysts, a decrease in diffraction intensity was observed across all planes. In particular, the intensity of the main (311) crystal plane was significantly reduced in the PEG Co3O4 sample, indicating a lower crystallinity. This observation is typically associated with smaller crystallite sizes and structural disorder [53,54]. The textural properties of the catalysts were examined using BET analysis to confirm the effects of reduced crystallinity on surface properties Table 1. All polymer-assisted Co3O4 catalysts exhibited increased BET surface areas compared to the pristine Co3O4 catalyst. Among them, the PEG Co3O4 sample showed the highest increase in both surface area (m2/g) and pore volume (cm3/g). Therefore, the structural modifications induced by PEG increased the accessible surface area, which was beneficial for catalytic reactions. Based on the XRD and BET results, the reduced crystallinity and increased surface area of PEG Co3O4 likely enhanced the exposure of active sites, thereby improving catalytic activity for N2O decomposition [55]. As shown in Table 1, the crystallite sizes determined using the Scherrer equation (Equation (2)) demonstrated that the PEG Co3O4 catalyst exhibited the smallest crystallite size among the samples. Additionally, the particle size distributions were evaluated from TEM images Figure S4, and the results indicated that the PEG Co3O4 catalyst exhibited smaller particle sizes compared to the pristine sample [56]. Furthermore, the SAED patterns obtained from TEM analysis confirmed the crystalline nature of the catalysts. Specifically, the pristine Co3O4, PVA Co3O4, and PVP Co3O4 catalysts displayed relatively sharp and distinct diffraction rings, whereas the PEG Co3O4 exhibited more diffuse and blurred rings, indicating a decrease in crystallinity [57,58].
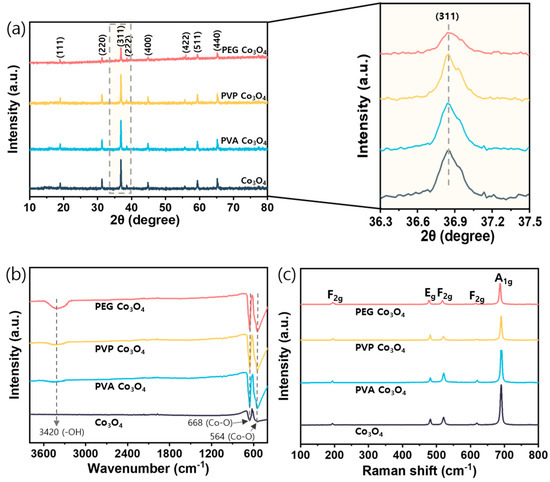
Figure 3.
Structural and vibrational characterizations of PEG Co3O4, PVP Co3O4, PVA Co3O4, and pristine Co3O4 catalysts: (a) XRD analysis (b) FT-IR spectra (c) Raman spectra.

Table 1.
BET surface areas, pore volumes and crystallite size of PEG Co3O4, PVP Co3O4, PVA Co3O4, and pristine Co3O4.
FT-IR spectroscopy was performed to investigate the vibrational characteristics and chemical bonding of the synthesized catalysts. The FT-IR spectra of the polymer-assisted Co3O4 catalysts are shown in Figure 3b. These samples exhibited three characteristic absorption bands at approximately 3420, 668, and 564 cm−1 [59]. The broad band centered at 3420 cm−1 corresponded to the hydroxyl (-OH) stretching vibration. Compared to that of pristine Co3O4, the enhanced intensity of this hydroxyl band in the PEG Co3O4 sample suggests an increased surface hydroxyl content, which may be associated with higher numbers of surface defect sites and active oxygen species [60]. The presence of abundant surface hydroxyl groups can enhance catalytic activity by facilitating reactant adsorption and participating in electron transfer processes during redox reactions [61]. Two distinctive bands at 668 and 564 cm−1 corresponded to the Co-O stretching vibrations of Co2+-O and Co3+-O bonds, respectively, confirming the successful formation of a Co3O4 spinel structure in all synthesized catalysts [59]. When comparing the polymer-assisted catalysts with pristine Co3O4, significant differences were observed in the intensities of the Co3O4 stretching bands. For example, the polymer-assisted samples exhibited enhanced and sharper peaks at 668 and 564 cm−1, suggesting improved Co3O4 bonding characteristics. Moreover, the increased intensity and sharpness of these characteristic peaks indicate that the polymer additives may influence the local coordination environment during Co3O4 formation, potentially contributing to better-defined metal–oxygen bonding in the spinel structure [40,62].
Raman spectroscopy was used to investigate the structural characteristics of the synthesized Co3O4 catalysts. According to group theory, spinel Co3O4 exhibits five Raman-active modes: A1g, Eg, and three F2g modes [63]. The Raman spectra of the pristine and polymer-assisted Co3O4 catalysts are shown in Figure 3c. All samples exhibited the three characteristic Co3O4 spinel bands, thus indicating the successful formation of a Co3O4 spinel structure without impurities. For example, the A1g mode appeared at approximately 680 cm−1 (octahedral Co3+-O6 vibration), three F2g modes were located at approximately 190, 515, and 610 cm−1 (tetrahedral Co2+-O4 vibrations), and the Eg mode was observed at approximately 478 cm−1 [50]. Notably, a gradual decrease in the A1g peak intensity was observed in the polymer-assisted samples in the following order: PEG Co3O4 < PVP Co3O4 < PVA Co3O4 < pristine Co3O4. This decrease indicates a reduction from Co3+ to Co2+ during synthesis, because the A1g mode is characteristic of Co3+ species in octahedral sites [64]. The reduction process can be attributed to the thermal decomposition of the organic polymers (PVA, PVP, and PEG), which function as mild reducing agents during calcination. Their decomposition releases carbonaceous species that facilitate the partial reduction of Co3+ to Co2+, resulting in the formation of oxygen vacancies, which play a key role in enhancing catalytic activity by providing active sites for redox reactions [64,65].
3.3. Redox Behavior and Surface Oxygen State Properties
As shown in Figure 4a and Table 2, the chemical oxidation states of cobalt were investigated using XPS. The Co 2p spectrum exhibited two primary peaks corresponding to the Co2+ and Co3+ oxidation states. Particularly, the fitted peaks near 796.4 and 781.3 eV were attributed to Co2+ 2p1/2 and Co2+ 2p3/2, respectively. Moreover, the peaks located near 794.7 and 779.6 eV were assigned to Co3+ 2p1/2 and Co3+ 2p3/2, respectively [66]. The detailed peak positions and corresponding assignments are summarized in Table S1. According to previous reports, a high number of Co2+ ions on the catalyst surface is advantageous for promoting N2O decomposition because Co2+ can adsorb N2O and donate electrons to weaken and cleave N-O bonds [51,67]. The Co 2p spectra revealed that the relative ratio of the Co2+ peaks increased in the polymer-assisted Co3O4 catalysts, particularly in the PEG-assisted sample, suggesting a higher surface concentration of Co2+ species. This increased Co2+ content may contribute to improved N2O decomposition performance because Co2+ species serve as the primary active sites in the redox mechanism. In addition, the presence of surface oxygen species plays a crucial role in catalytic reactions. As shown in Figure 4b, the O 1s spectra of the catalysts comprised two main peaks. The peak located at 531.0–531.8 eV was attributed to surface-adsorbed oxygen species (Oads), while the peak at 529.1–530.0 eV corresponded to lattice oxygen species (Olatt). The ratios of Oads-to-Olatt for all samples are presented in Table 2 [44]. For the polymer-assisted Co3O4 catalysts, the Oads/Olatt ratio increased depending on the type of polymer: Co3O4 < PVA Co3O4 < PVP Co3O4 < PEG Co3O4. This increase was likely due to the formation of additional oxygen vacancies induced by adding the polymer during synthesis. The enhanced formation of surface oxygen vacancies facilitated the generation of reactive oxygen species, which are beneficial for N2O decomposition [68]. Overall, the PEG-assisted Co3O4 catalyst exhibited the highest Co2+/(Co2+ + Co3+) and Oads/(Oads + Olatt) ratios of 0.47 and 0.72, respectively, indicating enhanced oxygen vacancy formation and optimal surface properties for N2O decomposition.
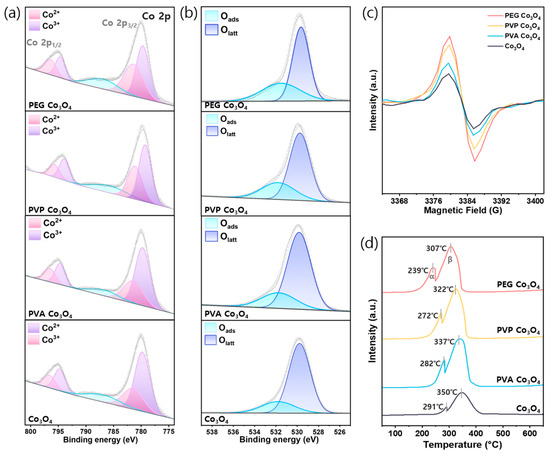
Figure 4.
Surface and redox characterizations of PEG Co3O4, PVP Co3O4, PVA Co3O4, and pristine Co3O4 catalysts: (a) Co 2p XPS spectra (b) O 1s XPS spectra. (c) EPR spectra and (d) H2-TPR profiles.
Table 2.
XPS results of the PEG Co3O4, PVP Co3O4, PVA Co3O4, and pristine Co3O4 catalysts.
EPR spectroscopy is highly sensitive to paramagnetic species, such as unpaired electrons; thus, it was employed to investigate the presence of surface Co2+ species and their associated electronic states. As shown in Figure 4c, the pristine and polymer-assisted Co3O4 catalysts exhibited a characteristic signal at g ≈ 2.003, corresponding to unpaired electrons localized on surface Co2+ ions [50]. The signal intensities followed the order: Co3O4 < PVA Co3O4 < PVP Co3O4 < PEG Co3O4, indicating an increase in the concentration of surface Co2+ species. It is likely that the thermal decomposition of polymer additives during calcination facilitated the formation of oxygen vacancies and promoted the generation of Co2+ ions on the catalyst surface. These results are consistent with the XPS analysis, further confirming the enhanced surface Co2+ species in the polymer-assisted Co3O4 catalysts [54,69].
The redox behaviors of the synthesized catalysts were investigated using H2-TPR analysis Figure 4d. All samples exhibited two distinct hydrogen reduction peaks within the range of 50–700 °C. The first peak (denoted as α, 200–300 °C) corresponded to the reduction of Co3+ to Co2+, while the second peak (denoted as β, 300–400 °C) corresponded to the reduction of Co2+ to metallic cobalt [66]. Compared to pristine Co3O4, both the α and β peaks of the polymer-assisted Co3O4 catalysts shifted to lower temperatures. Among them, the PEG-assisted Co3O4 catalyst exhibited the most pronounced shift, with α and β peaks at 239 and 307 °C, respectively, indicating enhanced surface reducibility and a more facile reduction of cobalt species. These improved redox properties, particularly in PEG-assisted Co3O4, are expected to play a key role in enhancing catalytic performance for N2O decomposition [44,70].
The TPD of O2 and N2O was performed to investigate the role of the synthesized catalysts in N2O decomposition. First, the O2-TPD profiles were recorded over 50–700 °C to examine the surface oxygen reactivity Figure 5a and Table S2. The total desorption amount, as shown in Figure 5b, exhibited the following trend: Co3O4 < PVA Co3O4 < PVP Co3O4 < PEG Co3O4. Notably, PEG Co3O4 exhibited a strong and broad desorption peak between 130 and 280 °C, attributed to active surface oxygen species [44]. Because oxygen desorption at ˂450 °C is generally associated with chemisorbed species (Oads) [66,67], these results support the XPS findings showing elevated Oads/Olatt ratios in the polymer-assisted samples. Therefore, the enhanced low-temperature oxygen desorption observed for PEG Co3O4 indicated a higher density of reactive oxygen species, which could facilitate N2O decomposition.
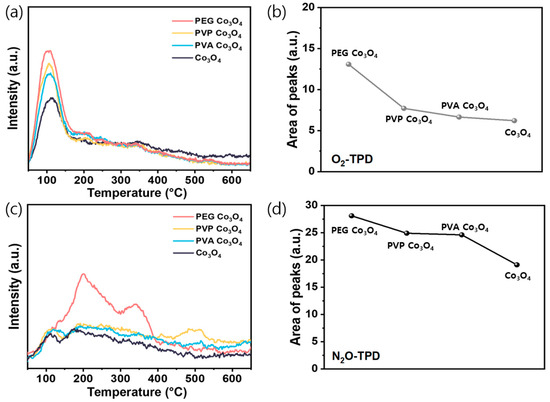
Figure 5.
O2-TPD and N2O-TPD analyses of the PEG Co3O4, PVP Co3O4, PVA Co3O4, and pristine Co3O4 catalysts: (a) O2-TPD profile (b) Peak areas of O2-TPD and (c) N2O-TPD profile (d) Peak areas of N2O-TPD.
To evaluate the N2O activation capacity, N2O-TPD analysis was performed. According to previous studies, the TPD signals in this context primarily originate from the desorption of N2O or its decomposition products adsorbed on the surface oxygen vacancies [71]. As shown in Figure 5c and Table S2, all samples displayed a primary desorption peak at <450 °C, indicating desorption of surface-adsorbed N2O or dissociation products at vacancy sites [72]. This chemisorption-related region reflects the N2O adsorption–dissociation capability of the catalysts. Among the samples, PEG Co3O4 exhibited the highest desorption intensity, followed by PVP Co3O4, PVA Co3O4, and pristine Co3O4. This order was confirmed by the temperature-programmed desorption curves and relative desorption peak areas shown in Figure 5a–d, highlighting that polymer addition enhanced the surface interactions and N2O decomposition performance.
3.4. Role of Polymer
Herein, polymer-assisted metal oxide catalysts were synthesized using polymers that can regulate the particle size and morphology and induce oxygen vacancies. All polymers coordinated with cobalt precursors during nucleation, suppressing uncontrolled hydrolysis and particle aggregation, while their subsequent thermal decomposition generated reductive intermediates that partially reduced Co3+ to Co2+ and induced oxygen vacancies [31,32,33,34]. These processes collectively enhanced redox activity and surface defect density, resulting in improved catalytic performance for N2O decomposition. Among the three polymers, PVA and PVP primarily influenced structural evolution and surface stabilization during the synthesis process. PVA, containing abundant hydroxyl groups, strongly interacted with hydrated cobalt complexes ([Co(H2O)6]2+) via hydrogen bonding, forming transient core–shell structures that inhibited grain growth and promoted uniform particle dispersion. During calcination, the decomposition of PVA released mildly reducing species, such as polyene fragments, which partially reduced Co3+ to Co2+ and generated oxygen vacancies through charge compensation associated with lattice oxygen removal. This led to a higher surface Co2+ content and enhanced exposure of active sites [34]. PVP, in contrast, contains carbonyl and pyrrolidone nitrogen groups that coordinated with cobalt ions, mitigating excessive hydrolysis and aggregation during precursor formation. Its steric hindrance further restricted crystal growth, thereby stabilizing dispersed Co3O4 nanoparticles. While PVP induced fewer oxygen vacancies than PVA, it effectively maintained particle uniformity and stabilized surface Co2+ species by suppressing excessive oxidation during calcination [73].
In particular, PEG contributed to an increased number of surface Co2+ species and the formation of oxygen vacancies during the synthesis process. These features are related to structural properties and are closely associated with redox-active surface characteristics. The mechanism responsible for the increased Co2+ ratio during the fabrication process is presented in Figure 6 and Equations (3)–(5). After Co(NO3)2·6H2O was dissolved in water to form [Co(H2O)6]2+ complexes [40], the ether oxygen groups in the PEG chain were weakly coordinated to [Co(H2O)6]2+. This resulted in the formation of PEG-[Co2+] complexes via hydrogen bonding and dipole-ion interactions. Owing to their flexible chain structure and high hydrophilicity, PEG molecules surrounded the cobalt ions, slowed hydrolysis, and facilitated uniform precipitation [74,75]. After adding NaOH, cobalt hydroxide precipitated and stabilized within the PEG matrix. During calcination, PEG decomposed particularly between 200 and 450 °C, releasing carbonaceous intermediates such as CO, CH2O, and aldehydes. These in situ formed reducing species partially reduced Co3+ to Co2+, while charge compensation was achieved via lattice oxygen removal, resulting in the formation of oxygen vacancies [76]. This trend is consistent with the XPS results, which showed a decreased Olatt signal for the PEG Co3O4 catalyst, indicating the generation of surface oxygen vacancies. These structural and electronic modifications hindered crystal grain growth and resulted in a higher surface area and number of redox-active sites, thereby enhancing the N2O adsorption and activation ability of the catalyst.
Co(NO3)2·6H2O → [Co(H2O)6]2+ + 2NO3−
[Co(H2O)6]2+ + 2OH− → Co(OH)2 + 6H2O
6Co(OH)2 + O2 → 2Co3O4 + 6H2O
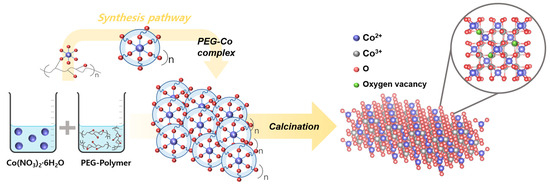
Figure 6.
Synthesis pathway of the PEG Co3O4 catalyst, which promoted Co2+ formation.
The thermal decomposition of PEG occurred in multiple stages. First, dehydration occurred at ˂180 °C, followed by the degradation of hydroxyl and ether groups between 180 and 380 °C and polyether chain scission at approximately 380–500 °C. These stages released reductive fragments that interacted with the cobalt lattice during calcination [49,76]. The resulting oxygen vacancies played a significant role in stabilizing the surface Co2+ species.
The Raman spectra showed a weakened A1g mode associated with Co3+ to Co2+, indicating partial reduction. The XPS Co 2p and O 1s spectra further confirmed the increased Co2+/Co3+ and Oads/Olatt ratios in PEG Co3O4. Moreover, EPR analysis revealed stronger signals at g ≈ 2.003, indicating increased unpaired electrons from oxygen vacancies. These findings were consistent with the improved reducibility observed in the H2-TPR results. Moreover, they were supported by the O2-TPD and N2O-TPD analyses, which indicated the enhanced desorption of surface oxygen species and N2O-derived intermediates. In summary, introducing PEG during the Co3O4 preparation process was highly beneficial for forming catalysts with enriched surface Co2+ species and oxygen vacancies, thereby enhancing the catalytic performance in N2O decomposition.
4. Conclusions
An effective synthesis strategy for Co3O4 spinel catalysts was developed using polymer (PVA, PVP, or PEG)-assisted methods to enhance the performance of N2O decomposition. The addition of these polymers facilitated the formation of Co2+-enriched surfaces and abundant oxygen vacancies, which are primary active sites for N2O activation. Among the polymers investigated, the PEG-assisted Co3O4 catalyst exhibited the most pronounced enhancement in terms of surface area, reducibility, and Oads/Olatt ratio, as demonstrated by XRD, XPS, EPR, TPD, and H2-TPR analyses. Notably, the unique characteristics of PEG, such as its abundant hydroxyl groups and flexible molecular chains, enabled strong interactions with cobalt precursors, thus significantly facilitating the preferential formation and stabilization of surface Co2+ species. The increased concentration of Co2+, which is a key redox-active site in the catalytic cycle, directly contributed to superior N2O decomposition performance. Finally, the PEG 5Co3O4/CeO2 sample achieved nearly complete N2O conversion (˃99.5%) at 600 °C. Moreover, it maintained a conversion of 97.2% under O2 co-feeding conditions, thus outperforming catalysts synthesized without polymer additives. This study demonstrates that PEG-assisted synthesis can effectively modulate the surface chemistry of Co3O4 by enriching catalytically active Co2+ sites, thereby significantly enhancing N2O decomposition activity. Thus, polymer-assisted spinel catalysts can be a practical approach for improving N2O decomposition efficiency.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/nano15211642/s1, Figure S1. A scheme of materials synthesis. Figure S2. N2O decomposition activity of PEG, PVP, and PVA-assisted Co3O4 catalysts and pristine 5Co3O4 under 5000 ppm N2O, N2 balance, GHSV 60,000 h−1, in the temperature range of 350–650 °C. Figure S3. Catalytic performance and kinetic analysis of all sample catalysts for N2O decomposition. (a) N2O conversion as function of reaction temperature (b) Reaction rates at various temperature (c) Arrhenius plot (d) Activation energy (Ea). Reaction condition: 5000 ppm N2O with N2 balance, GHSV=1,000,000 h−1. Figure S4. TEM images and SAED patterns of the catalysts. (a) and (b) Pristine Co3O4, (c) and (d) PVA Co3O4, (e) and (f) PVP Co3O4, (g) and (h) PEG Co3O4. Table S1. XPS peaks data of the PEG Co3O4, PVP Co3O4, PVA Co3O4, and pristine Co3O4 catalysts. Table S2. Peak area data from the O2-TPD and N2O-TPD analyses.
Author Contributions
N.K.: Conceptualization, investigation and writing (original draft). S.-J.K.: data curation and formal analysis. S.-H.S.: conceptualization. M.-J.L.: data curation and visualization. B.J.: software and methodology. H.-D.K.: supervision, project administration, and funding acquisition. T.W.N.: writing (review and editing) and validation. B.Y.: writing (review and editing), supervision, validation, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Ministry of Trade, Industry and Energy (MOTIE) [grant number (00432416)] and the Korea Institute of Industrial Technology (KITECH-UR-24-0035 and KITECH-JA250006).
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Zhuang, Z.; Guan, B.; Chen, J.; Zheng, C.; Zhou, J.; Su, T.; Chen, Y.; Zhu, C.; Hu, X.; Zhao, S.; et al. Review of nitrous oxide direct catalytic decomposition and selective catalytic reduction catalysts. Chem. Eng. J. 2024, 486, 150374. [Google Scholar] [CrossRef]
- Wu, X.; Du, J.; Gao, Y.; Wang, H.; Zhang, C.; Zhang, R.; He, H.; Lu, G.; Wu, Z. Progress and challenges in nitrous oxide decomposition and valorization. Chem. Soc. Rev. 2024, 53, 8379–8423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, P.; Li, Z.; Ao, C.; Zhao, X.; Lin, H. Improved activity and significant O2 resistance of Cs doped Co3O4 catalyst for N2O decomposition. J. Environ. Chem. Eng. 2024, 12, 113907. [Google Scholar] [CrossRef]
- Müller, R. The impact of the rise in atmospheric nitrous oxide on stratospheric ozone. Ambio 2021, 50, 35–39. [Google Scholar] [CrossRef]
- Li, L.; Xu, J.; Hu, J.; Han, J. Reducing Nitrous Oxide Emissions to Mitigate Climate Change and Protect the Ozone Layer. Environ. Sci. Technol. 2014, 48, 5290–5297. [Google Scholar] [CrossRef]
- Hussain, M.; Fino, D.; Russo, N. N2O decomposition by mesoporous silica supported Rh catalysts. J. Hazard. Mater. 2012, 211–212, 255–265. [Google Scholar] [CrossRef]
- Xu, X.; Xu, H.; Kapteijn, F.; Moulijn, J.A. SBA-15 based catalysts in catalytic N2O decomposition in a model tail-gas from nitric acid plants. Appl. Catal. B Environ. 2004, 53, 265–274. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Kapteijn, F.; Schöffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production: Where do we stand today? Appl. Catal. B Environ. 2003, 44, 117–151. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Y.; Wu, R.; Zhao, L.; Wei, X.; Zhao, Y. Effects of zirconia crystal phases on the catalytic decomposition of N2O over Co3O4/ZrO2 catalysts. Appl. Surf. Sci. 2020, 514, 145892. [Google Scholar] [CrossRef]
- You, Y.; Chang, H.; Ma, L.; Guo, L.; Qin, X.; Li, J.; Li, J. Enhancement of N2O decomposition performance by N2O pretreatment over Ce-Co-O catalyst. Chem. Eng. J. 2018, 347, 184–192. [Google Scholar] [CrossRef]
- Hinokuma, S.; Iwasa, T.; Kon, Y.; Taketsugu, T.; Sato, K. N2O decomposition properties of Ru catalysts supported on various oxide materials and SnO2. Sci. Rep. 2020, 10, 21605. [Google Scholar] [CrossRef]
- Li, Y.; Sundermann, A.; Gerlach, O.; Low, K.-B.; Zhang, C.C.; Zheng, X.; Zhu, H.; Axnanda, S. Catalytic decomposition of N2O on supported Rh catalysts. Catal. Today 2020, 355, 608–619. [Google Scholar] [CrossRef]
- Richards, N.; Carter, J.H.; Nowicka, E.; Parker, L.A.; Pattisson, S.; He, Q.; Dummer, N.F.; Golunski, S.; Hutchings, G.J. Structure-sensitivity of alumina supported palladium catalysts for N2O decomposition. Appl. Catal. B Environ. 2020, 264, 118501. [Google Scholar] [CrossRef]
- Centi, G.; Dall’Olio, L.; Perathoner, S. Room temperature decomposition of N2O in the presence of gaseous oxygen on prereduced Rh supported catalysts. Catal. Lett. 2000, 67, 107–112. [Google Scholar] [CrossRef]
- Marnellos, G.E.; Efthimiadis, E.A.; Vasalos, I.A. Effect of SO2 and H2O on the N2O decomposition in the presence of O2 over Ru/Al2O3. Appl. Catal. B Environ. 2003, 46, 523–539. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Djinović, P.; Tchernychova, E.; Tkachenko, O.P.; Kustov, L.M.; Pintar, A. Nanoshaped CuO/CeO2 Materials: Effect of the Exposed Ceria Surfaces on Catalytic Activity in N2O Decomposition Reaction. ACS Catal. 2015, 5, 5357–5365. [Google Scholar] [CrossRef]
- Pietrogiacomi, D.; Campa, M.C.; Carbone, L.R.; Tuti, S.; Occhiuzzi, M. N2O decomposition on CoOx, CuOx, FeOx or MnOx supported on ZrO2: The effect of zirconia doping with sulfates or K+ on catalytic activity. Appl. Catal. B Environ. 2016, 187, 218–227. [Google Scholar] [CrossRef]
- Galloni, M.G.; Campisi, S.; Gervasini, A.; Morandi, S.; Manzoli, M. How hydroxyapatite governs surface Cu(II) and Fe(III) structuring: Effects in the N2O decomposition under highly oxidant atmosphere. Appl. Catal. A Gen. 2023, 655, 119101. [Google Scholar] [CrossRef]
- Lin, F.; Andana, T.; Wu, Y.; Szanyi, J.; Wang, Y.; Gao, F. Catalytic site requirements for N2O decomposition on Cu-, Co-, and Fe-SSZ-13 zeolites. J. Catal. 2021, 401, 70–80. [Google Scholar] [CrossRef]
- Nobukawa, T.; Yoshida, M.; Okumura, K.; Tomishige, K.; Kunimori, K. Effect of reductants in N2O reduction over Fe-MFI catalysts. J. Catal. 2005, 229, 374–388. [Google Scholar] [CrossRef]
- Rutkowska, M.; Piwowarska, Z.; Micek, E.; Chmielarz, L. Hierarchical Fe-, Cu- and Co-Beta zeolites obtained by mesotemplate-free method. Part I: Synthesis and catalytic activity in N2O decomposition. Microporous Mesoporous Mater. 2015, 209, 54–65. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. Stability improvement of zeolite catalysts under hydrothermal conditions for their potential applications in biomass valorization and crude oil upgrading. Microporous Mesoporous Mater. 2017, 249, 42–54. [Google Scholar] [CrossRef]
- Rho, Y.-J.; Yoo, Y.J.; Ryu, W.-H. Research trends on minimizing the size of noble metal catalysts for Li-CO2 batteries: From nanoparticle to single atom. Korean J. Chem. Eng. 2023, 40, 461–472. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Q.; Dang, H.; Zhao, L.; Wu, R.; Li, J.; Wang, Y.; Zhao, Y. In-situ polymerization intercalation of montmorillonite to achieve Co3O4 barrier dispersion for direct catalytic decomposition of N2O. Appl. Catal. A Gen. 2023, 664, 119329. [Google Scholar] [CrossRef]
- Liu, H.; Yang, S.; Wang, G.; Liu, H.; Peng, Y.; Sun, C.; Li, J.; Chen, J. Strong Electronic Orbit Coupling between Cobalt and Single-Atom Praseodymium for Boosted Nitrous Oxide Decomposition on Co3O4 Catalyst. Environ. Sci. Technol. 2022, 56, 16325–16335. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, Z.; Li, Z.; Liu, C.; Yan, N.; Ma, L. Boosting N2O Decomposition by Fabricating the Cs–O–Co Structure over Co3O4 with Single-Layer Atoms of Cs. Environ. Sci. Technol. 2024, 58, 906–914. [Google Scholar] [CrossRef]
- Jing, Y.; He, C.; Zhang, N.; Murano, Y.; Toyoshima, R.; Kondoh, H.; Kageyama, Y.; Inomata, H.; Toyao, T.; Shimizu, K.-I. Promotional Effect of Ag on the Catalytic Decomposition of N2O in the Presence of O2 over the Al2O3-Supported Rh Catalyst. ACS Catal. 2023, 13, 12983–12993. [Google Scholar] [CrossRef]
- Hernandez Mejia, C.; van Deelen, T.W.; de Jong, K.P. Activity enhancement of cobalt catalysts by tuning metal-support interactions. Nat. Commun. 2018, 9, 4459. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Wang, Y.; Lei, L.; Zhuang, L. Controlled Aggregation of Cobalt and Platinum Atoms via Plasma Treatment for Exceptional Hydrogen Evolution Reaction Activity. Coatings 2024, 14, 1569. [Google Scholar] [CrossRef]
- Arya, R.K.; Thapliyal, D.; Pandit, A.; Gora, S.; Banerjee, C.; Verros, G.D.; Sen, P. Polymer Coated Functional Catalysts for Industrial Applications. Polymers 2023, 15, 2009. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Liu, S.; Ren, J.; Zhu, J.; Yu, Y.; Yusup, S.; Chen, D.; Zhang, S.; Huang, Y. Controllable design of phosphorus-doped cobalt sulfide for catalytic conversion of cellulose to methyl levulinate: The importance of Co3+/Co2+ ratio. Renew. Energy 2025, 250, 123322. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Ma, J.; Liu, F.; Einaga, H.; He, H. Improved and Reduced Performance of Cu- and Ni-Substituted Co3O4 Catalysts with Varying CoOh/CoTd and Co3+/Co2+ Ratios for the Complete Catalytic Oxidation of VOCs. Environ. Sci. Technol. 2022, 56, 9751–9761. [Google Scholar] [CrossRef]
- Ye, M.; Mohanty, P.; Ghosh, G. Morphology and properties of poly vinyl alcohol (PVA) scaffolds: Impact of process variables. Mater. Sci. Eng. C 2014, 42, 289–294. [Google Scholar] [CrossRef]
- Saljoughi, E.; Mohammadi, T. Cellulose acetate (CA)/polyvinylpyrrolidone (PVP) blend asymmetric membranes: Preparation, morphology and performance. Desalination 2009, 249, 850–854. [Google Scholar] [CrossRef]
- Masoudpanah, S.M. PVP-assisted hydrothermal synthesis of rod-like NiCo2O4 powders as high-performance microwave absorbers. J. Mater. Res. Technol. 2022, 20, 3264–3274. [Google Scholar] [CrossRef]
- Selli, D.; Valentin, C.D. Ab Initio Investigation of Polyethylene Glycol Coating of TiO2 Surfaces. J. Phys. Chem. C 2016, 120, 29190–29201. [Google Scholar] [CrossRef] [PubMed]
- Do, S.-B.; Lee, S.-E.; Kim, T.-O. Oxidative decomposition with PEG-MnO2 catalyst for removal of formaldehyde: Chemical aspects on HCHO oxidation mechanism. Appl. Surf. Sci. 2022, 598, 153773. [Google Scholar] [CrossRef]
- Qasem Ali, A.A.; Khan, M.U.; Siddiqui, Z.N. PEG supported Cu–Mo mixed metal oxide (CuO–MoO3@PEG): A highly efficient catalyst for the enamination of pyrimidine dione and dimedone. Curr. Res. Green Sustain. Chem. 2022, 5, 100231. [Google Scholar] [CrossRef]
- Li, S.; Zhao, J.; Song, Z.; Wang, H.; Zhang, T.; Liu, J.; Jiang, Q. New insights into the effect of polyvinyl alcohol on Co3O4 spinel oxide catalyst for N2O decomposition. Fuel 2024, 362, 130745. [Google Scholar] [CrossRef]
- Nowakowski, L.; Hudy, C.; Zasada, F.; Gryboś, J.; Piskorz, W.; Wach, A.; Kayser, Y.; Szlachetko, J.; Sojka, Z. N2O Decomposition on Singly and Doubly (K and Li)-Doped Co3O4 Nanocubes─Establishing Key Factors Governing Redox Behavior of Catalysts. J. Am. Chem. Soc. 2024, 146, 24450–24466. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.A.; Verma, A.A.; Paolucci, C.; Parekh, A.A.; Anggara, T.; Yezerets, A.; Schneider, W.F.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Identification of the active Cu site in standard selective catalytic reduction with ammonia on Cu-SSZ-13. J. Catal. 2014, 312, 87–97. [Google Scholar] [CrossRef]
- Zhang, T.; Qiu, Y.; Liu, G.; Chen, J.; Peng, Y.; Liu, B.; Li, J. Nature of active Fe species and reaction mechanism over high-efficiency Fe/CHA catalysts in catalytic decomposition of N2O. J. Catal. 2020, 392, 322–335. [Google Scholar] [CrossRef]
- Lee, M.-J.; Jeong, B.; Kim, D.; Kim, S.-J.; Choi, Y.; Ye, B.; Kim, D.-H.; Kim, H.-D.; Cho, S. Hexagonal boron nitride heterostructure to control the oxidation states and SO2 resistance of the V2O5-WO3/TiO2 catalyst for the NH3-SCR reaction across a wide temperature range. Appl. Catal. B Environ. Energy 2025, 378, 125583. [Google Scholar] [CrossRef]
- Belles, L.; Moularas, C.; Smykała, S.; Deligiannakis, Y. Flame Spray Pyrolysis Co3O4/CoO as Highly-Efficient Nanocatalyst for Oxygen Reduction Reaction. Nanomaterials 2021, 11, 925. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Shi, C. The catalytic performance of Ba-Ce-Cu catalysts for N2O decomposition. J. Environ. Chem. Eng. 2023, 11, 109970. [Google Scholar] [CrossRef]
- Smeets, P.J.; Sels, B.F.; van Teeffelen, R.M.; Leeman, H.; Hensen, E.J.M.; Schoonheydt, R.A. The catalytic performance of Cu-containing zeolites in N2O decomposition and the influence of O2, NO and H2O on recombination of oxygen. J. Catal. 2008, 256, 183–191. [Google Scholar] [CrossRef]
- Grzybek, G.; Gryboś, J.; Indyka, P.; Janas, J.; Ciura, K.; Leszczyński, B.; Zasada, F.; Kotarba, A.; Sojka, Z. Evaluation of the inhibiting effect of H2O, O2, and NO on the performance of laboratory and pilot K-ZnxCo3-xO4 catalysts supported on α-Al2O3 for low-temperature N2O decomposition. Appl. Catal. B Environ. 2021, 297, 120435. [Google Scholar] [CrossRef]
- Yu, H.; Qi, X.; Du, X.; Pan, Y.; Feng, X.; Shan, W.; Xiong, Y. The preparation of 3.0F-Co3O4 catalyst with “Yardang Landform” structure and its performance for catalyzing N2O decomposition. Mol. Catal. 2023, 537, 112960. [Google Scholar] [CrossRef]
- Ye, B.; Jeong, B.; Lee, M.-J.; Kim, T.H.; Park, S.-S.; Jung, J.; Lee, S.; Kim, H.-D. Recent trends in vanadium-based SCR catalysts for NOx reduction in industrial applications: Stationary sources. Nano Converg. 2022, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Cai, S.-C.; Xu, Z.; Chen, X.; Chen, J.; Jia, H.-P.; Chen, J. Solvothermal syntheses of Bi and Zn co-doped TiO2 with enhanced electron-hole separation and efficient photodegradation of gaseous toluene under visible-light. J. Hazard. Mater. 2017, 325, 261–270. [Google Scholar] [CrossRef]
- Long, Y.; Zhu, X.; Gao, C.; Si, W.; Li, J.; Peng, Y. Modulation of Co spin state at Co3O4 crystalline-amorphous interfaces for CO oxidation and N2O decomposition. Nat. Commun. 2025, 16, 1048. [Google Scholar] [CrossRef]
- Wójcik, S.; Grzybek, G.; Stelmachowski, P.; Sojka, Z.; Kotarba, A. Bulk, Surface and Interface Promotion of Co3O4 for the Low-Temperature N2O Decomposition Catalysis. Catalysts 2020, 10, 41. [Google Scholar] [CrossRef]
- Makhlouf, S.A.; Bakr, Z.H.; Aly, K.I.; Moustafa, M.S. Structural, electrical and optical properties of Co3O4 nanoparticles. Superlattices Microstruct. 2013, 64, 107–117. [Google Scholar] [CrossRef]
- St-Onge, V.; Cui, M.; Rochon, S.; Daigle, J.-C.; Claverie, J.P. Reducing crystallinity in solid polymer electrolytes for lithium-metal batteries via statistical copolymerization. Commun. Mater. 2021, 2, 83. [Google Scholar] [CrossRef]
- Zhou, X.J.; Shi, P.H.; Qin, Y.F.; Fan, J.C.; Min, Y.L.; Yao, W.F. Synthesis of Co3O4/graphene composite catalysts through CTAB-assisted method for Orange II degradation by activation of peroxymonosulfate. J. Mater. Sci. Mater. Electron. 2016, 27, 1020–1030. [Google Scholar] [CrossRef]
- Yu, H.; Li, Y.; Pan, Y.; Du, Y.; Feng, X.; Cui, J.; Lou, Z.; Shan, W.; Xiong, Y. Dy and K double-additive modified Co3O4 catalyst with high resistance to NOx for catalyzing N2O decomposition. Chem. Eng. J. 2025, 504, 159032. [Google Scholar] [CrossRef]
- Uvarov, V.; Popov, I. Metrological characterization of X-Ray diffraction methods for determination of crystallite size in nano-scale materials. Mater. Charact. 2007, 58, 883–891. [Google Scholar] [CrossRef]
- Kenyota, N.; Jarernboon, W.; Laokul, P. Bulk synthesis of chemically activated carbon and cobalt oxide nanocomposites as supercapacitor electrodes. J. Mater. Sci. Mater. Electron. 2024, 35, 158. [Google Scholar] [CrossRef]
- Rokicińska, A.; Łątka, P.; Olszański, B.; Żurowska, M.; Dębosz, M.; Michalik, M.; Kuśtrowski, P. Polymer template assisted construction of spherical Co3O4@meso-SiO2 yolk-shell nanoreactors for catalytic combustion of volatile organic compounds. Chem. Eng. J. 2024, 480, 148173. [Google Scholar] [CrossRef]
- Ercolino, G.; Grodzka, A.; Grzybek, G.; Stelmachowski, P.; Specchia, S.; Kotarba, A. The Effect of the Preparation Method of Pd-Doped Cobalt Spinel on the Catalytic Activity in Methane Oxidation Under Lean Fuel Conditions. Top. Catal. 2017, 60, 333–341. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, X.; Hu, X.; Zhou, W.; Zhao, Y. Effect of Formic Acid Treatment on the Structure and Catalytic Activity of Co3O4 for N2O Decomposition. Catal. Lett. 2019, 149, 1026–1036. [Google Scholar] [CrossRef]
- Jiang, Z.; Feng, X.; Deng, J.; He, C.; Douthwaite, M.; Yu, Y.; Liu, J.; Hao, Z.; Zhao, Z. Atomic-Scale Insights into the Low-Temperature Oxidation of Methanol over a Single-Atom Pt1-Co3O4 Catalyst. Adv. Funct. Mater. 2019, 29, 1902041. [Google Scholar] [CrossRef]
- Kang, B.; Guo, M.; Wu, H.; Guo, X.; Di, Z.; Wei, Y.; Jia, J.; Wang, Z.-J.; Zhang, R. Effect of alkali/alkaline-earth-metal doping on the Co3O4 spinel structure and N 2 O decomposition. Catal. Sci. Technol. 2024, 14, 2825–2837. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl. Catal. B Environ. 2007, 75, 167–174. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B Environ. 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Yi, S.; Lai, P.; Ma, G.; Pan, J.; Chen, Z.; Qin, Y.; Jiang, X. Green and facile synthesis of nanostructured Co3O4/CeO2 catalysts via a glucose-urea method for NO oxidation. Appl. Surf. Sci. 2023, 626, 157180. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, Z.; Tsubaki, N. Zeolitic Imidazolate Framework-67-Derived P-Doped Hollow Porous Co3O4 as a Photocatalyst for Hydrogen Production from Water. ACS Appl. Mater. Interfaces 2021, 13, 50996–51007. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Cai, S.; Chen, J.; Jia, H. Two-step pyrolytic engineering of carbon-doped Co3O4 with rich defects for efficient low-temperature CO oxidation. J. Mater. Chem. A 2020, 8, 6619–6630. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Zhang, L.; Jiang, D. Surface oxygen vacancies on Co3O4 mediated catalytic formaldehyde oxidation at room temperature. Catal. Sci. Technol. 2016, 6, 3845–3853. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, W.; Dang, H.; Li, L.; Wu, R.; Wang, Y.; Zhao, Y. Enhancement of N2O decomposition performance by co-doping of Ni and Y to Co3O4 catalyst. J. Environ. Chem. Eng. 2024, 12, 112463. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, D.; Li, X.; Yin, Y.; Wang, C.; Qiu, L.; Yu, J.; Chang, H. Enhancement of Cs on Co3O4 for N2O Catalytic Decomposition: N2O Activation and O2 Desorption. Ind. Eng. Chem. Res. 2022, 61, 13854–13862. [Google Scholar] [CrossRef]
- Shirayama, S.; Uda, T. Recovery of Cobalt Ion into Polyethyleneglycol (PEG) Gel Phase as Thiocyanato Complex. Mater. Trans. 2015, 56, 610–616. [Google Scholar] [CrossRef][Green Version]
- Yoo, S.H.; Kim, J.H.; Jho, J.Y.; Won, J.; Kang, Y.S. Influence of the addition of PVP on the morphology of asymmetric polyimide phase inversion membranes: Effect of PVP molecular weight. J. Membr. Sci. 2004, 236, 203–207. [Google Scholar] [CrossRef]
- Ma, S.; Bai, J.; Sun, L.; Zhao, L.; Tan, H.; Liu, L.; Peng, Z.; Zhao, X.; Xiong, D. Investigation of polyethylene glycol (PEG) assisted solvothermal synthesis of CuCoO2 nanosheets for efficient oxygen evolution reaction. Dalton Trans. 2023, 52, 13750–13757. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kim, C.; Kwon, D. Thermal degradation of poly(ethyleneglycol). Polym. Degrad. Stab. 1995, 47, 203–208. [Google Scholar] [CrossRef]
- Krause, B.; Pötschke, P. Polyethylene Glycol as Additive to Achieve N-Conductive Melt-Mixed Polymer/Carbon Nanotube Composites for Thermoelectric Application. Nanomaterials 2022, 12, 3812. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).