
Quantum Mechanics MP2 and CASSCF Study of Coordinate Quasi-Double Bonds in Cobalt(II) Complexes as Single Molecule Magnets


 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussions
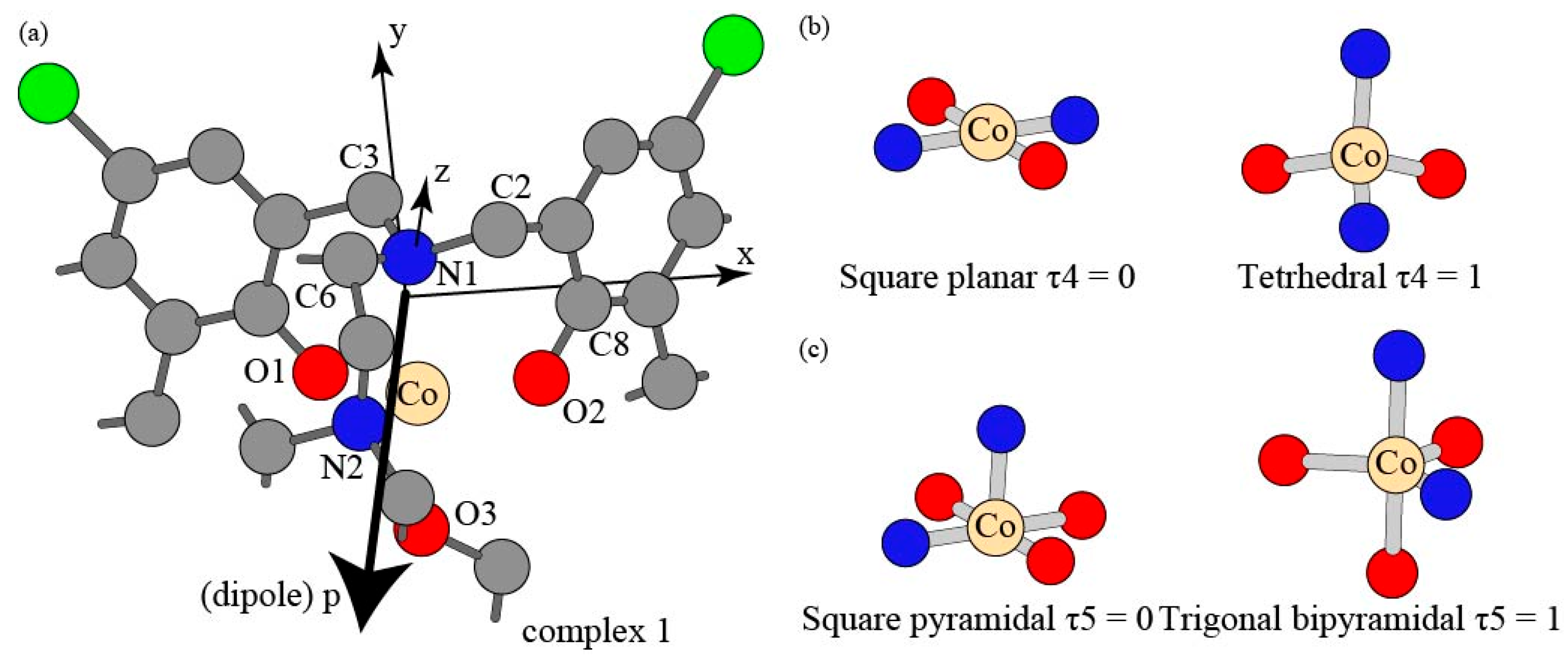
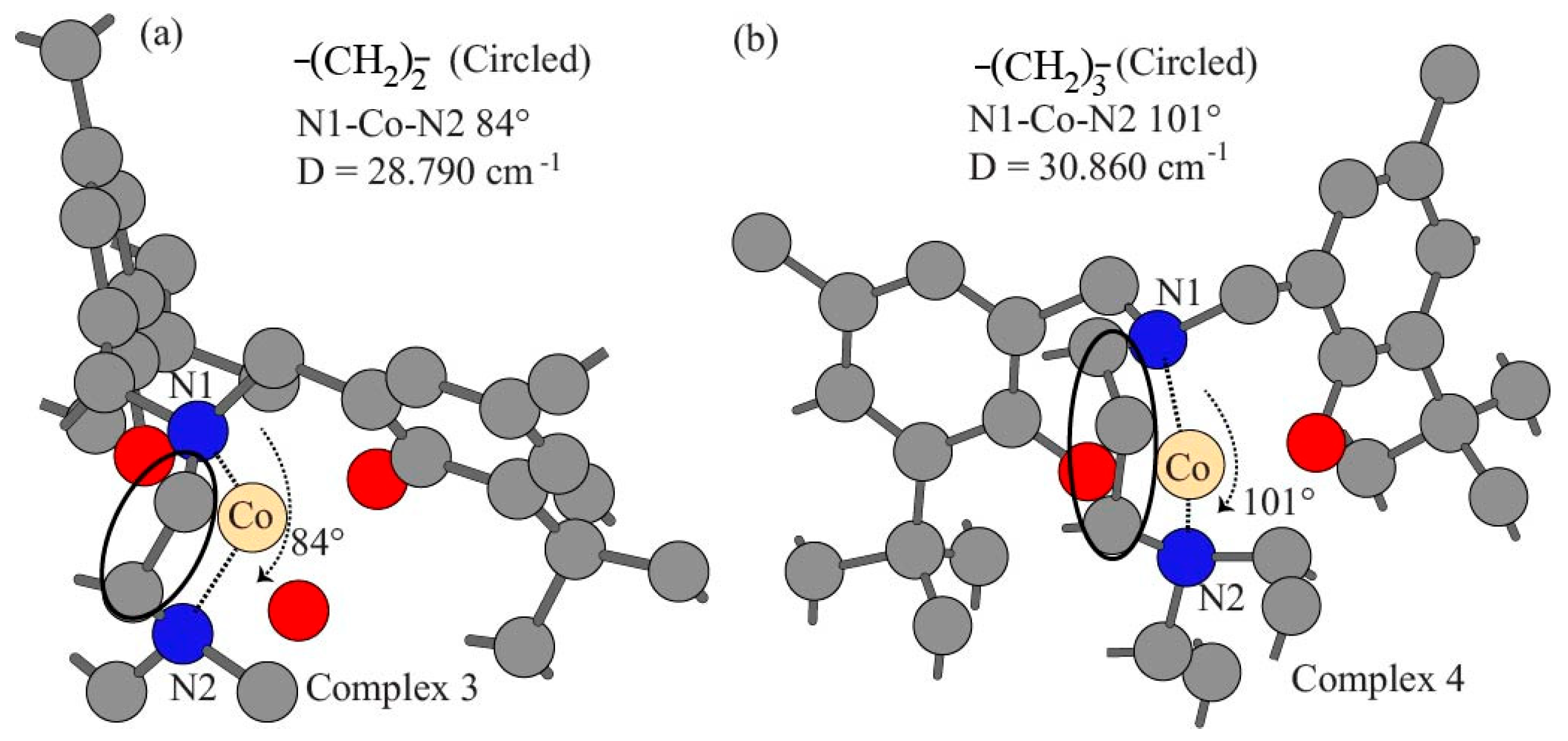
3.1. Structural Parameter and Molecular Geometry
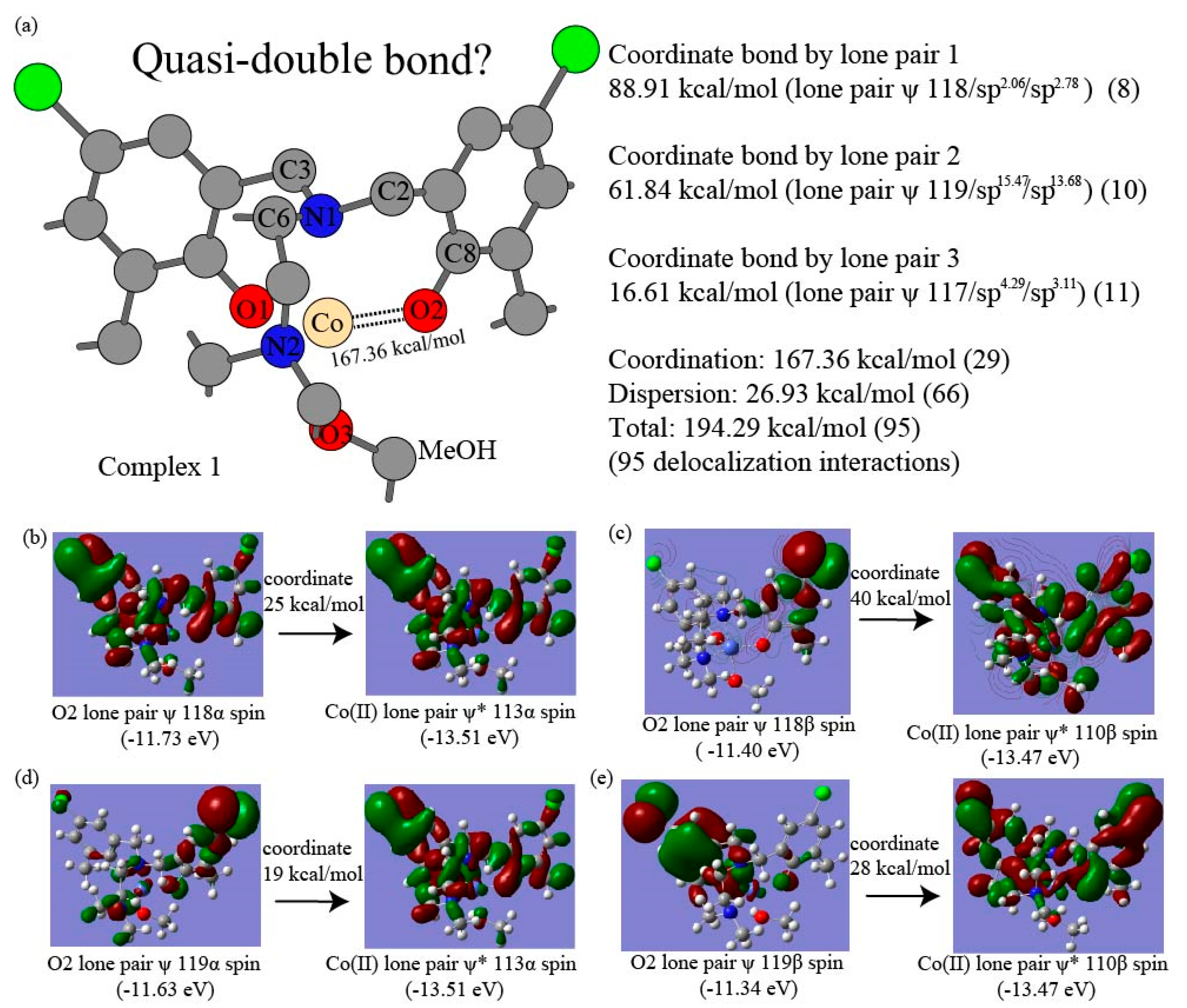
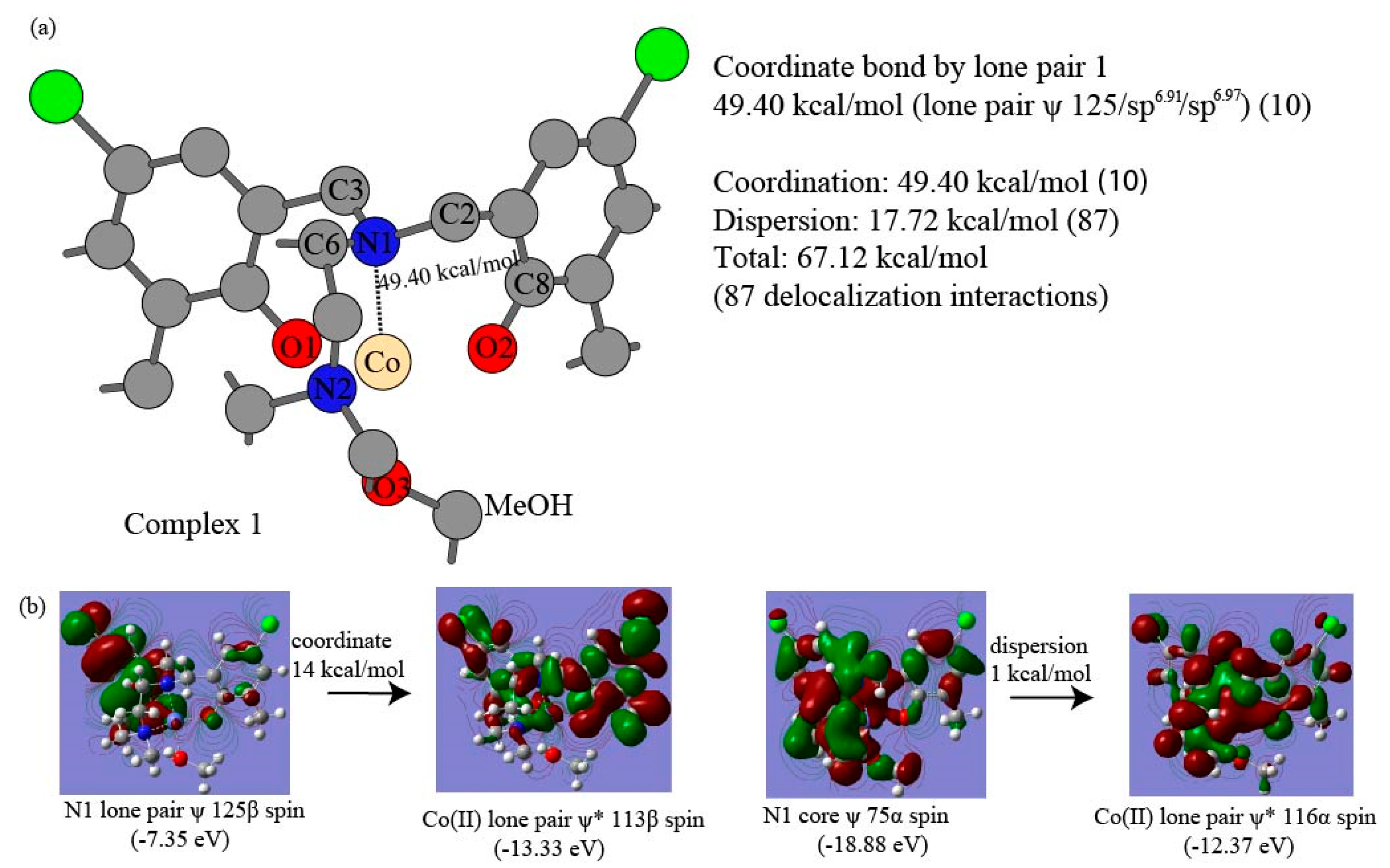
3.2. Intramolecular Correlation Energy of Complexes and NBO-Based Perturbation Theory Energy Analysis
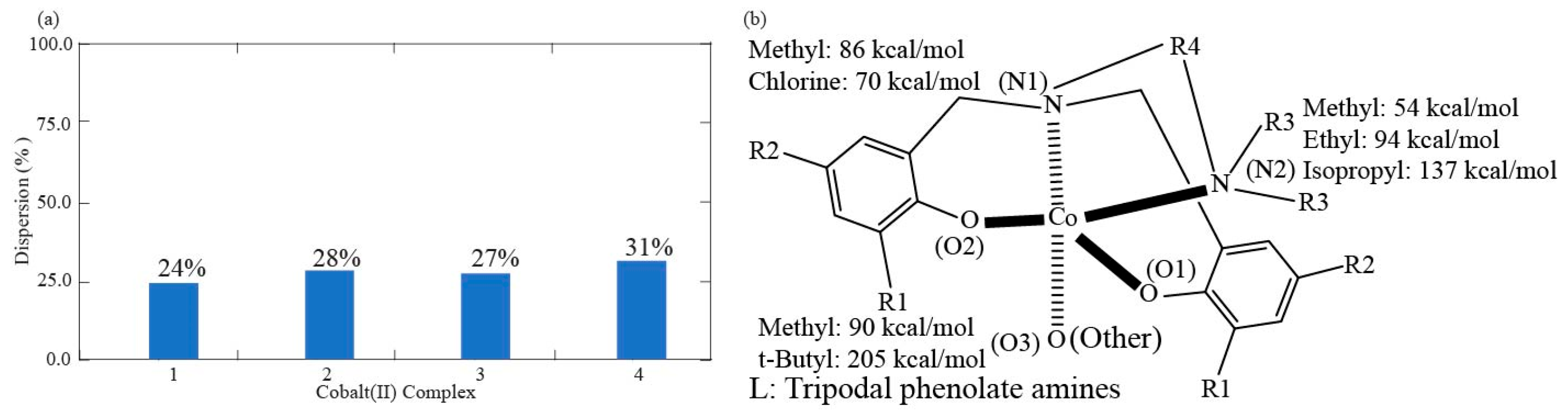
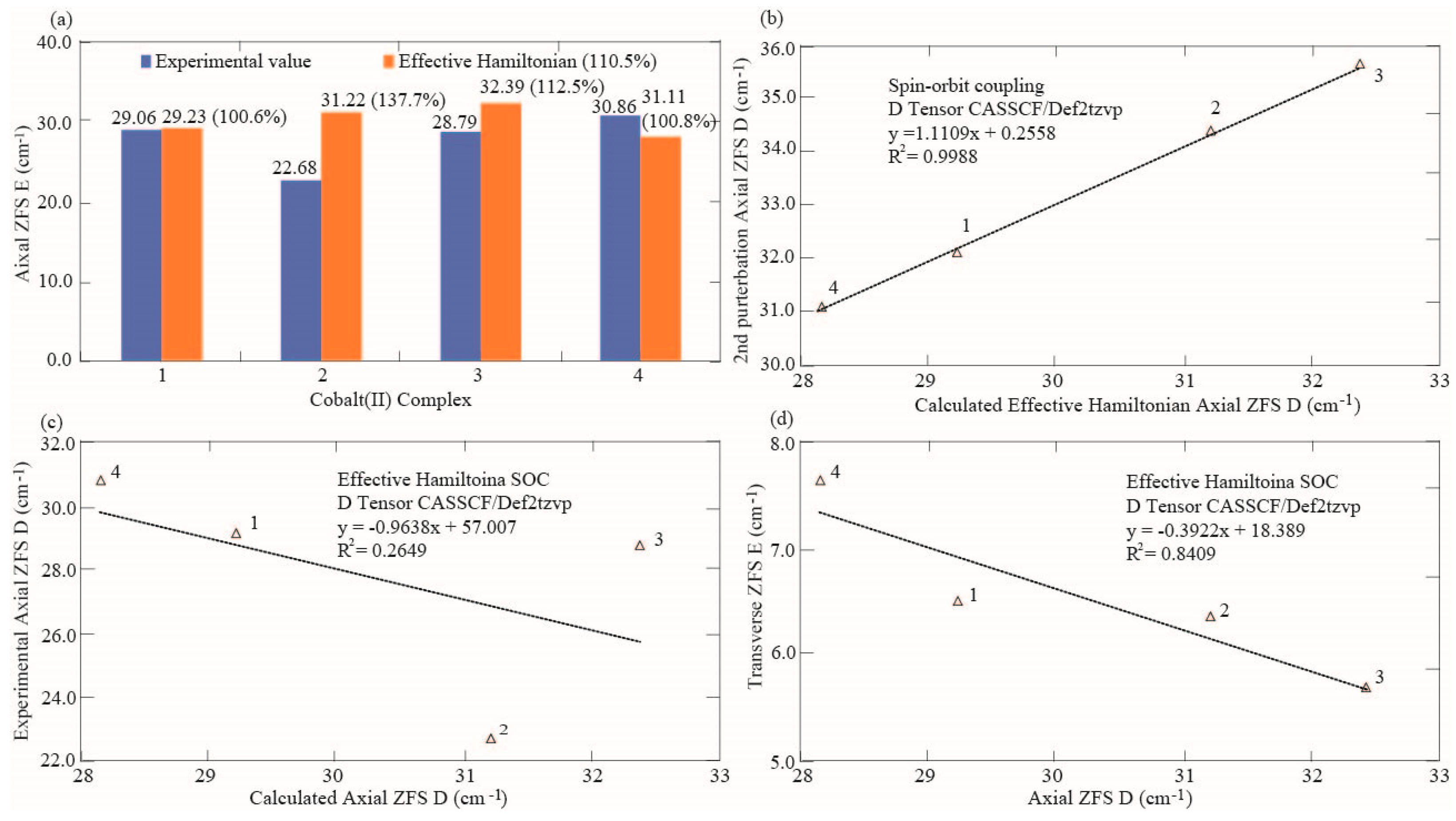
3.3. Effect of Correlation Dispersion Force and Magnetic Axial and Transverse ZFS
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronado, E. Molecular magnetism: From chemical design to spin control in molecules, materials and devices. Nat. Rev. Mater. 2020, 5, 87–104. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, J. Lanthanide single-molecule magnets with high anisotropy barrier: Where to from here? Natl. Sci. Rev. 2022, 9, nwac194. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Guo, M.; Li, X.-L.; Tang, J. Molecular magnetism of lanthanide: Advances and perspectives. Coord. Chem. Rev. 2019, 378, 350–364. [Google Scholar] [CrossRef]
- Mautner, F.A.; Bierbaumer, F.; Fischer, R.C.; Tubau, À.; Speed, S.; Ruiz, E.; Massoud, S.S.; Vicente, R.; Gómez-Coca, S. Insights into the Spin Dynamics of Mononuclear Cerium(III) Single-Molecule Magnets. Inorg. Chem. 2022, 61, 11124–11136. [Google Scholar] [CrossRef]
- Shao, D.; Wang, X.-Y. Development of Single-Molecule Magnets†. Chin. J. Chem. 2020, 38, 1005–1018. [Google Scholar] [CrossRef]
- Feng, M.; Tong, M.-L. Single Ion Magnets from 3d to 5f: Developments and Strategies. Chem. A Eur. J. 2018, 24, 7574–7594. [Google Scholar] [CrossRef]
- Craig, G.A.; Murrie, M. 3d single-ion magnets. Chem. Soc. Rev. 2015, 44, 2135–2147. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron(I) complex. Nat. Chem. 2013, 5, 577–581. [Google Scholar] [CrossRef]
- Kumar Sahu, P.; Kharel, R.; Shome, S.; Goswami, S.; Konar, S. Understanding the unceasing evolution of Co(II) based single-ion magnets. Coord. Chem. Rev. 2023, 475, 214871. [Google Scholar] [CrossRef]
- Guo, F.S.; Day, B.M.; Chen, Y.C.; Tong, M.L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem. Int. Ed. Engl. 2017, 56, 11445–11449. [Google Scholar] [CrossRef]
- Yin, X.; Deng, L.; Ruan, L.; Wu, Y.; Luo, F.; Qin, G.; Han, X.; Zhang, X. Recent Progress for Single-Molecule Magnets Based on Rare Earth Elements. Materials 2023, 16, 3568. [Google Scholar] [CrossRef] [PubMed]
- Sessoli, R.; Gatteschi, D.; Caneschi, A.; Novak, M.A. Magnetic bistability in a metal-ion cluster. Nature 1993, 365, 141–143. [Google Scholar] [CrossRef]
- Wernsdorfer, W. Molecular Nanomagnets. In Advanced Magnetic Nanostructures; Sellmyer, D., Skomski, R., Eds.; Springer: Boston, MA, USA, 2006; pp. 147–181. [Google Scholar]
- Freedman, D.E.; Harman, W.H.; Harris, T.D.; Long, G.J.; Chang, C.J.; Long, J.R. Slow Magnetic Relaxation in a High-Spin Iron(II) Complex. J. Am. Chem. Soc. 2010, 132, 1224–1225. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Sainctavit, P.; Danieli, C.; Otero, E.; Sciancalepore, C.; Talarico, A.M.; Arrio, M.A.; Cornia, A.; Gatteschi, D.; et al. Magnetic memory of a single-molecule quantum magnet wired to a gold surface. Nat. Mater. 2009, 8, 194–197. [Google Scholar] [CrossRef]
- Moreno-Pineda, E.; Wernsdorfer, W. Measuring molecular magnets for quantum technologies. Nat. Rev. Phys. 2021, 3, 645–659. [Google Scholar] [CrossRef]
- Greenspon, A.S.; Marceaux, B.L.; Hu, E.L. Robust lanthanide emitters in polyelectrolyte thin films for photonic applications. Nanotechnology 2018, 29, 075302. [Google Scholar] [CrossRef]
- Boulon, M.-E.; Cucinotta, G.; Luzon, J.; Degl’Innocenti, C.; Perfetti, M.; Bernot, K.; Calvez, G.; Caneschi, A.; Sessoli, R. Magnetic Anisotropy and Spin-Parity Effect Along the Series of Lanthanide Complexes with DOTA. Angew. Chem. Int. Ed. 2013, 52, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef]
- Yatoo, M.; Habib, F.; Ahmad, Z.; Ahmad, S.; Husain, A. A Concise Review of Single-Molecule Magnets, Carbon Nanotubes and Hybrids Between Them. Preprints 2023. [Google Scholar] [CrossRef]
- Duan, Y.; Rosaleny, L.E.; Coutinho, J.T.; Giménez-Santamarina, S.; Scheie, A.; Baldoví, J.J.; Cardona-Serra, S.; Gaita-Ariño, A. Data-driven design of molecular nanomagnets. Nat. Commun. 2022, 13, 7626. [Google Scholar] [CrossRef]
- Etesse, J.; Holzäpfel, A.; Ortu, A.; Afzelius, M. Optical and spin manipulation of non-Kramers rare-earth ions in a weak magnetic field for quantum memory applications. Phys. Rev. A 2021, 103, 022618. [Google Scholar] [CrossRef]
- Mondal, S.; Lunghi, A. Unraveling the Contributions to Spin-Lattice Relaxation in Kramers Single-Molecule Magnets. J. Am. Chem. Soc. 2022, 144, 22965–22975. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.S.; Sigrist, M.; Sørensen, M.A.; Barra, A.-L.; Weyhermüller, T.; Piligkos, S.; Thuesen, C.A.; Vinum, M.G.; Mutka, H.; Weihe, H.; et al. [ReF6]2−: A Robust Module for the Design of Molecule-Based Magnetic Materials. Angew. Chem. Int. Ed. 2014, 53, 1351–1354. [Google Scholar] [CrossRef]
- Sato, R.; Suzuki, K.; Minato, T.; Shinoe, M.; Yamaguchi, K.; Mizuno, N. Field-induced slow magnetic relaxation of octahedrally coordinated mononuclear Fe(iii)-, Co(ii)-, and Mn(iii)-containing polyoxometalates. Chem. Commun. 2015, 51, 4081–4084. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, J.; Castro, I.; Ruiz-García, R.; Cano, J.; Julve, M.; Lloret, F.; De Munno, G.; Wernsdorfer, W.; Pardo, E. Field-Induced Slow Magnetic Relaxation in a Six-Coordinate Mononuclear Cobalt(II) Complex with a Positive Anisotropy. J. Am. Chem. Soc. 2012, 134, 15704–15707. [Google Scholar] [CrossRef]
- Palii, A.V.; Korchagin, D.V.; Yureva, E.A.; Akimov, A.V.; Misochko, E.Y.; Shilov, G.V.; Talantsev, A.D.; Morgunov, R.B.; Aldoshin, S.M.; Tsukerblat, B.S. Single-Ion Magnet Et4N[CoII(hfac)3] with Nonuniaxial Anisotropy: Synthesis, Experimental Characterization, and Theoretical Modeling. Inorg. Chem. 2016, 55, 9696–9706. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Chen, L.; Liu, C.-M.; Zhang, Y.-Q.; Yin, S.-G.; Liu, Q.-Y. Field-Induced Slow Magnetic Relaxation and Gas Adsorption Properties of a Bifunctional Cobalt(II) Compound. Inorg. Chem. 2015, 54, 11362–11368. [Google Scholar] [CrossRef]
- Meng, Y.-S.; Mo, Z.; Wang, B.-W.; Zhang, Y.-Q.; Deng, L.; Gao, S. Observation of the single-ion magnet behavior of d8 ions on two-coordinate Co(i)–NHC complexes. Chem. Sci. 2015, 6, 7156–7162. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Zhou, H.; Cen, P.; Jin, X.; Xie, G.; Chen, S.; Hu, Q. Single-Ion-Magnet Behavior in a Two-Dimensional Coordination Polymer Constructed from CoII Nodes and a Pyridylhydrazone Derivative. Inorg. Chem. 2015, 54, 8884–8886. [Google Scholar] [CrossRef]
- Mondal, A.K.; Jover, J.; Ruiz, E.; Konar, S. Investigation of easy-plane magnetic anisotropy in P-ligand square-pyramidal CoII single ion magnets. Chem. Commun. 2017, 53, 5338–5341. [Google Scholar] [CrossRef]
- Mondal, A.K.; Goswami, T.; Misra, A.; Konar, S. Probing the Effects of Ligand Field and Coordination Geometry on Magnetic Anisotropy of Pentacoordinate Cobalt(II) Single-Ion Magnets. Inorg. Chem. 2017, 56, 6870–6878. [Google Scholar] [CrossRef]
- Cahier, B.; Perfetti, M.; Zakhia, G.; Naoufal, D.; El-Khatib, F.; Guillot, R.; Rivière, E.; Sessoli, R.; Barra, A.-L.; Guihéry, N.; et al. Magnetic Anisotropy in Pentacoordinate NiII and CoII Complexes: Unraveling Electronic and Geometrical Contributions. Chem. A Eur. J. 2017, 23, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- El-Khatib, F.; Cahier, B.; Shao, F.; López-Jordà, M.; Guillot, R.; Rivière, E.; Hafez, H.; Saad, Z.; Girerd, J.-J.; Guihéry, N.; et al. Design and Magnetic Properties of a Mononuclear Co(II) Single Molecule Magnet and Its Antiferromagnetically Coupled Binuclear Derivative. Inorg. Chem. 2017, 56, 4601–4608. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Shi, L.; Zhang, S.-L.; Zhao, X.-H.; Wu, D.-Q.; Wei, X.-Q.; Wang, X.-Y. Syntheses, structures, and magnetic properties of three new chain compounds based on a pentagonal bipyramidal Co(ii) building block. CrystEngComm 2016, 18, 4150–4157. [Google Scholar] [CrossRef]
- Świtlicka-Olszewska, A.; Palion-Gazda, J.; Klemens, T.; Machura, B.; Vallejo, J.; Cano, J.; Lloret, F.; Julve, M. Single-ion magnet behaviour in mononuclear and two-dimensional dicyanamide-containing cobalt(ii) complexes. Dalton Trans. 2016, 45, 10181–10193. [Google Scholar] [CrossRef]
- Briganti, M.; Santanni, F.; Tesi, L.; Totti, F.; Sessoli, R.; Lunghi, A. A Complete Ab Initio View of Orbach and Raman Spin–Lattice Relaxation in a Dysprosium Coordination Compound. J. Am. Chem. Soc. 2021, 143, 13633–13645. [Google Scholar] [CrossRef]
- Accorsi, S.; Barra, A.-L.; Caneschi, A.; Chastanet, G.; Cornia, A.; Fabretti, A.C.; Gatteschi, D.; Mortalò, C.; Olivieri, E.; Parenti, F.; et al. Tuning Anisotropy Barriers in a Family of Tetrairon(III) Single-Molecule Magnets with an S = 5 Ground State. J. Am. Chem. Soc. 2006, 128, 4742–4755. [Google Scholar] [CrossRef]
- Lu, F.; Guo, W.-X.; Zhang, Y.-Q. Largely Enhancing the Blocking Energy Barrier and Temperature of a Linear Cobalt(II) Complex through the Structural Distortion: A Theoretical Exploration. Inorg. Chem. 2022, 61, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Murrie, M. Cobalt(ii) single-molecule magnets. Chem. Soc. Rev. 2010, 39, 1986–1995. [Google Scholar] [CrossRef]
- Oshio, H.; Nakano, M. High-spin molecules with magnetic anisotropy toward single-molecule magnets. Chemistry 2005, 11, 5178–5185. [Google Scholar] [CrossRef]
- Fukui, K.; Ohya-Nishiguchi, H.; Hirota, N. The relation between zero-field splittings and distortions along the normal coordinates in transition metal complexes. Mol. Phys. 1990, 71, 1269–1283. [Google Scholar] [CrossRef]
- Murugesan, S.; Stöger, B.; Carvalho, M.D.; Ferreira, L.P.; Pittenauer, E.; Allmaier, G.; Veiros, L.F.; Kirchner, K. Synthesis and Reactivity of Four- and Five-Coordinate Low-Spin Cobalt(II) PCP Pincer Complexes and Some Nickel(II) Analogues. Organometallics 2014, 33, 6132–6140. [Google Scholar] [CrossRef] [PubMed]
- Krzystek, J.; Zvyagin, S.A.; Ozarowski, A.; Fiedler, A.T.; Brunold, T.C.; Telser, J. Definitive Spectroscopic Determination of Zero-Field Splitting in High-Spin Cobalt(II). J. Am. Chem. Soc. 2004, 126, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- Devkota, L.; SantaLucia, D.J.; Wheaton, A.M.; Pienkos, A.J.; Lindeman, S.V.; Krzystek, J.; Ozerov, M.; Berry, J.F.; Telser, J.; Fiedler, A.T. Spectroscopic and Magnetic Studies of Co(II) Scorpionate Complexes: Is There a Halide Effect on Magnetic Anisotropy? Inorg. Chem. 2023, 62, 5984–6002. [Google Scholar] [CrossRef]
- Kremer, S.; Henke, W.; Reinen, D. High-spin-low-spin equilibriums of cobalt(2+) in the terpyridine complexes Co(terpy)2X2.nH2O. Inorg. Chem. 1982, 21, 3013–3022. [Google Scholar] [CrossRef]
- Izumiyama, N.; Fujii, S.; Kato, K.; Tokunaga, R.; Hayami, S.; Nakaya, M. Spin-crossover cobalt(ii) complexes exhibiting temperature- and concentration-dependent optical changes in solution. Dalton Trans. 2024, 53, 9547–9553. [Google Scholar] [CrossRef]
- Craven, M.; Nygaard, M.H.; Zadrozny, J.M.; Long, J.R.; Overgaard, J. Determination of d-Orbital Populations in a Cobalt(II) Single-Molecule Magnet Using Single-Crystal X-ray Diffraction. Inorg. Chem. 2018, 57, 6913–6920. [Google Scholar] [CrossRef]
- Palii, A.V.; Clemente-Juan, J.M.; Coronado, E.; Klokishner, S.I.; Ostrovsky, S.M.; Reu, O.S. Role of Orbital Degeneracy in the Single Molecule Magnet Behavior of a Mononuclear High-Spin Fe(II) Complex. Inorg. Chem. 2010, 49, 8073–8077. [Google Scholar] [CrossRef]
- Gomez-Coca, S.; Cremades, E.; Aliaga-Alcalde, N.; Ruiz, E. Mononuclear single-molecule magnets: Tailoring the magnetic anisotropy of first-row transition-metal complexes. J. Am. Chem. Soc. 2013, 135, 7010–7018. [Google Scholar] [CrossRef]
- Bunting, P.C.; Atanasov, M.; Damgaard-Møller, E.; Perfetti, M.; Crassee, I.; Orlita, M.; Overgaard, J.; van Slageren, J.; Neese, F.; Long, J.R. A linear cobalt(II) complex with maximal orbital angular momentum from a non-Aufbau ground state. Science 2018, 362, eaat7319. [Google Scholar] [CrossRef]
- Yao, X.N.; Du, J.Z.; Zhang, Y.Q.; Leng, X.B.; Yang, M.W.; Jiang, S.D.; Wang, Z.X.; Ouyang, Z.W.; Deng, L.; Wang, B.W.; et al. Two-Coordinate Co(II) Imido Complexes as Outstanding Single-Molecule Magnets. J. Am. Chem. Soc. 2017, 139, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Eichhöfer, A.; Lan, Y.; Mereacre, V.; Bodenstein, T.; Weigend, F. Slow magnetic relaxation in trigonal-planar mononuclear Fe(II) and Co(II) bis(trimethylsilyl)amido complexes--a comparative study. Inorg. Chem. 2014, 53, 1962–1974. [Google Scholar] [CrossRef]
- Rechkemmer, Y.; Breitgoff, F.D.; van der Meer, M.; Atanasov, M.; Hakl, M.; Orlita, M.; Neugebauer, P.; Neese, F.; Sarkar, B.; van Slageren, J. A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier. Nat. Commun. 2016, 7, 10467. [Google Scholar] [CrossRef] [PubMed]
- Fataftah, M.S.; Zadrozny, J.M.; Rogers, D.M.; Freedman, D.E. A Mononuclear Transition Metal Single-Molecule Magnet in a Nuclear Spin-Free Ligand Environment. Inorg. Chem. 2014, 53, 10716–10721. [Google Scholar] [CrossRef]
- Wu, C.-M.; Tsai, J.-E.; Lee, G.-H.; Yang, E.-C. Slow magnetization relaxation in a tetrahedrally coordinated mononuclear Co(ii) complex exclusively ligated with phenanthroline ligands. Dalton Trans. 2020, 49, 16813–16820. [Google Scholar] [CrossRef]
- Cui, H.H.; Lu, F.; Chen, X.T.; Zhang, Y.Q.; Tong, W.; Xue, Z.L. Zero-Field Slow Magnetic Relaxation and Hysteresis Loop in Four-Coordinate Co(II) Single-Ion Magnets with Strong Easy-Axis Anisotropy. Inorg. Chem. 2019, 58, 12555–12564. [Google Scholar] [CrossRef] [PubMed]
- Ziegenbalg, S.; Hornig, D.; Görls, H.; Plass, W. Cobalt(II)-Based Single-Ion Magnets with Distorted Pseudotetrahedral [N2O2] Coordination: Experimental and Theoretical Investigations. Inorg. Chem. 2016, 55, 4047–4058. [Google Scholar] [CrossRef]
- Ferentinos, E.; Tzeli, D.; Sottini, S.; Groenen, E.; Ozerov, M.; Poneti, G.; Kaniewska-Laskowska, K.; Krzystek, J.; Kyritsis, P. Magnetic anisotropy and structural flexibility in the field-induced single ion magnets [Co{(OPPh 2 )(EPPh 2 )N} 2 ], E = S, Se, explored by experimental and computational methods. Dalton Trans. 2023, 52, 2036–2050. [Google Scholar] [CrossRef]
- Massoud, S.S.; Perez, Z.E.; Courson, J.R.; Fischer, R.C.; Mautner, F.A.; Vančo, J.; Čajan, M.; Trávníček, Z. Slow magnetic relaxation in penta-coordinate cobalt(ii) field-induced single-ion magnets (SIMs) with easy-axis magnetic anisotropy. Dalton Trans. 2020, 49, 11715–11726. [Google Scholar] [CrossRef]
- Mora-Fonz, M.J.; Catlow, C.R.; Lewis, D.W. Oligomerization and cyclization processes in the nucleation of microporous silicas. Angew. Chem. Int. Ed. Engl. 2005, 44, 3082–3086. [Google Scholar] [CrossRef]
- Jurca, T.; Farghal, A.; Lin, P.H.; Korobkov, I.; Murugesu, M.; Richeson, D.S. Single-molecule magnet behavior with a single metal center enhanced through peripheral ligand modifications. J. Am. Chem. Soc. 2011, 133, 15814–15817. [Google Scholar] [CrossRef]
- Bamberger, H.; Albold, U.; Dubnická Midlíková, J.; Su, C.-Y.; Deibel, N.; Hunger, D.; Hallmen, P.P.; Neugebauer, P.; Beerhues, J.; Demeshko, S.; et al. Iron(II), Cobalt(II), and Nickel(II) Complexes of Bis(sulfonamido)benzenes: Redox Properties, Large Zero-Field Splittings, and Single-Ion Magnets. Inorg. Chem. 2021, 60, 2953–2963. [Google Scholar] [CrossRef]
- Mitsuhashi, R.; Hosoya, S.; Suzuki, T.; Sunatsuki, Y.; Sakiyama, H.; Mikuriya, M. Hydrogen-bonding interactions and magnetic relaxation dynamics in tetracoordinated cobalt(ii) single-ion magnets. Dalton Trans. 2019, 48, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Böhme, M.; Ziegenbalg, S.; Aliabadi, A.; Schnegg, A.; Görls, H.; Plass, W. Magnetic relaxation in cobalt(ii)-based single-ion magnets influenced by distortion of the pseudotetrahedral [N2O2] coordination environment. Dalton Trans. 2018, 47, 10861–10873. [Google Scholar] [CrossRef] [PubMed]
- Plyuta, N.; Petrusenko, S.; Kokozay, V.N.; Cauchy, T.; Lloret, F.; Julve, M.; Cano, J.; Avarvari, N. Field-induced mononuclear cobalt(II) single-molecule magnet (SMM) based on a benzothiadiazole-ortho-vanillin ligand †. Dalton Trans. 2022, 51, 4760–4771. [Google Scholar] [CrossRef]
- Landart-Gereka, A.; Quesada-Moreno, M.M.; Díaz-Ortega, I.F.; Nojiri, H.; Ozerov, M.; Krzystek, J.; Palacios, M.A.; Colacio, E. Large easy-axis magnetic anisotropy in a series of trigonal prismatic mononuclear cobalt(ii) complexes with zero-field hidden single-molecule magnet behaviour: The important role of the distortion of the coordination sphere and intermolecular interactions in the slow relaxation. Inorg. Chem. Front. 2022, 9, 2810–2831. [Google Scholar] [CrossRef]
- Tupolova, Y.P.; Shcherbakov, I.N.; Korchagin, D.V.; Tkachev, V.V.; Lebedev, V.E.; Popov, L.D.; Zakharov, K.V.; Vasiliev, A.N.; Palii, A.V.; Aldoshin, S.M. Fine-Tuning of Uniaxial Anisotropy and Slow Relaxation of Magnetization in the Hexacoordinate Co(II) Complexes with Acidoligands. J. Phys. Chem. C 2020, 124, 25957–25966. [Google Scholar] [CrossRef]
- Mičová, R.; Rajnák, C.; Titiš, J.; Bieńko, A.; Moncoľ, J.; Samoľová, E.; Boča, R. Easy-axis magnetic anisotropy in tetragonally elongated cobalt(ii) complexes beyond the spin-Hamiltonian formalism. Dalton Trans. 2023, 52, 7885–7892. [Google Scholar] [CrossRef]
- Pilichos, E.; Font-Bardia, M.; Cano, J.; Escuer, A.; Mayans, J. Slow magnetic relaxation for cobalt(ii) complexes in axial bipyramidal environment: An S = 1/2 spin case. Dalton Trans. 2022, 51, 8986–8993. [Google Scholar] [CrossRef]
- Mondal, A.K.; Mondal, A.; Konar, S. Slow Magnetic Relaxation in a One-Dimensional Coordination Polymer Constructed from Hepta-Coordinate Cobalt(II) Nodes. Magnetochemistry 2020, 6, 45. [Google Scholar] [CrossRef]
- Shao, D.; Zhang, S.-L.; Shi, L.; Zhang, Y.-Q.; Wang, X.-Y. Probing the Effect of Axial Ligands on Easy-Plane Anisotropy of Pentagonal-Bipyramidal Cobalt(II) Single-Ion Magnets. Inorg. Chem. 2016, 55, 10859–10869. [Google Scholar] [CrossRef]
- Chen, L.; Wang, J.; Wei, J.-M.; Wernsdorfer, W.; Chen, X.-T.; Zhang, Y.-Q.; Song, Y.; Xue, Z.-L. Slow magnetic relaxation in a mononuclear eight-coordinate cobalt(II) complex. J. Am. Chem. Soc. 2014, 136, 12213–12216. [Google Scholar] [CrossRef] [PubMed]
- Ruamps, R.; Batchelor, L.J.; Maurice, R.; Gogoi, N.; Jiménez-Lozano, P.; Guihéry, N.; de Graaf, C.; Barra, A.-L.; Sutter, J.-P.; Mallah, T. Origin of the Magnetic Anisotropy in Heptacoordinate NiII and CoII Complexes. Chem. A Eur. J. 2013, 19, 950–956. [Google Scholar] [CrossRef]
- Kopotkov, V.A.; Korchagin, D.V.; Sasnovskaya, V.D.; Gilmutdinov, I.F.; Yagubskii, E.B. A Series of Field-Induced Single-Ion Magnets Based on the Seven-Coordinate Co(II) Complexes with the Pentadentate (N3O2) H2dapsc Ligand. Magnetochemistry 2019, 5, 58. [Google Scholar] [CrossRef]
- Habib, F.; Luca, O.R.; Vieru, V.; Shiddiq, M.; Korobkov, I.; Gorelsky, S.I.; Takase, M.K.; Chibotaru, L.F.; Hill, S.; Crabtree, R.H.; et al. Influence of the Ligand Field on Slow Magnetization Relaxation versus Spin Crossover in Mononuclear Cobalt Complexes. Angew. Chem. Int. Ed. 2013, 52, 11290–11293. [Google Scholar] [CrossRef]
- Massoud, S.S.; Mautner, F.A.; Sakiyama, H.; Louka, F.R.; Salem, N.H.M.; Fischer, R.C.; Torvisco, A.; Guizouarn, T.; Velmurugan, G.; Comba, P.; et al. SMM Behavior in Distorted Trigonal Bipyramidal and Tetrahedral Cobalt(II) Complexes Based on Tripodal Tetradentate Phenolic Amines. Eur. J. Inorg. Chem. 2025, 28, e202400777. [Google Scholar] [CrossRef]
- Dong, X.; Xin, Z.; He, D.; Zhang, J.-L.; Lan, Y.-Q.; Zhang, Q.-F.; Chen, Y. Boosting CO2 electroreduction performance over fullerene-modified MOF-545-Co promoted by π–π interaction. Chin. Chem. Lett. 2023, 34, 107459. [Google Scholar] [CrossRef]
- Subedi, D.; Jang, Y.; Ganesan, A.; Schoellhorn, S.; Reid, R.; Verbeck, G.; D’Souza, F. Donor-acceptor conjugates derived from cobalt porphyrin and fullerene via metal-ligand axial coordination: Formation and excited state charge separation. J. Porphyr. Phthalocyanines 2021, 25, 533–546. [Google Scholar] [CrossRef]
- Liu, Y.; Gallo, A.A.; Liu, Y.; Hall, M.B.; Johnson, B.R. QM evaluation of the intramolecular aromatic π-π interactions of Ir(I) complex transition states. J. Mol. Struct. 2023, 1291, 135907. [Google Scholar] [CrossRef]
- Lunghi, A.; Sanvito, S. Computational design of magnetic molecules and their environment using quantum chemistry, machine learning and multiscale simulations. Nat. Rev. Chem. 2022, 6, 761–781. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional calculations of molecular bond energies. J. Chem. Phys. 1986, 84, 4524–4529. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Jung, J.; Atanasov, M.; Neese, F. Ab Initio Ligand-Field Theory Analysis and Covalency Trends in Actinide and Lanthanide Free Ions and Octahedral Complexes. Inorg. Chem. 2017, 56, 8802–8816. [Google Scholar] [CrossRef] [PubMed]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes. J. Comput. Chem. 2013, 34, 1164–1175. [Google Scholar] [CrossRef]
- Glaesemann, K.R.; Schmidt, M.W. On the Ordering of Orbital Energies in High-Spin ROHF. J. Phys. Chem. A 2010, 114, 8772–8777. [Google Scholar] [CrossRef] [PubMed]
- Zanders, D.; Boysen, N.; Land, M.A.; Obenlüneschloß, J.; Masuda, J.D.; Mallick, B.; Barry, S.T.; Devi, A. Co(II) Amide, Pyrrolate, and Aminopyridinate Complexes: Assessment of their Manifold Structural Chemistry and Thermal Properties. Eur. J. Inorg. Chem. 2021, 2021, 5119–5136. [Google Scholar] [CrossRef]
- Kim, H.-C.; Mitra, S.; Veerana, M.; Lim, J.-S.; Jeong, H.-R.; Park, G.; Huh, S.; Kim, S.-J.; Kim, Y. Cobalt(II)-coordination polymers containing glutarates and bipyridyl ligands and their antifungal potential. Sci. Rep. 2019, 9, 14983. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.-Y.; Lin, H.-J.; Hu, Z.; Kuo, C.-Y.; Mikhailova, D.; Lee, J.-M.; Haw, S.-C.; Chen, S.-A.; Schnelle, W.; Ishii, H.; et al. Relation between the Co-O bond lengths and the spin state of Co in layered Cobaltates: A high-pressure study. Sci. Rep. 2017, 7, 3656. [Google Scholar] [CrossRef]
- Buron-Le Cointe, M.; Hébert, J.; Baldé, C.; Moisan, N.; Toupet, L.; Guionneau, P.; Létard, J.F.; Freysz, E.; Cailleau, H.; Collet, E. Intermolecular control of thermoswitching and photoswitching phenomena in two spin-crossover polymorphs. Phys. Rev. B 2012, 85, 064114. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(i) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Alonso, J.A.; Martínez-Lope, M.J.; Casais, M.T.; Fernández-Díaz, M.T. Evolution of the Jahn−Teller Distortion of MnO6 Octahedra in RMnO3 Perovskites (R = Pr, Nd, Dy, Tb, Ho, Er, Y): A Neutron Diffraction Study. Inorg. Chem. 2000, 39, 917–923. [Google Scholar] [CrossRef]
- Marchivie, M.; Guionneau, P.; Letard, J.-F.; Chasseau, D. Photo-induced spin-transition: The role of the iron(II) environment distortion. Acta Crystallogr. Sect. B 2005, 61, 25–28. [Google Scholar] [CrossRef]
- McCusker, J.K.; Rheingold, A.L.; Hendrickson, D.N. Variable-Temperature Studies of Laser-Initiated 5T2 → 1A1 Intersystem Crossing in Spin-Crossover Complexes: Empirical Correlations between Activation Parameters and Ligand Structure in a Series of Polypyridyl Ferrous Complexes. Inorg. Chem. 1996, 35, 2100–2112. [Google Scholar] [CrossRef]
- Köppel, H.; Barentzen, H. The Jahn-Teller Effect: Fundamentals and Implications for Physics and Chemistry; Springer: Berlin/Heidelberg, Germany, 2009; Volume 97. [Google Scholar]
- Okuniewski, A.; Rosiak, D.; Chojnacki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Šebová, M.; Boca, R.; Dlhan, L.; Nemec, I.; Papankova, B.; Pavlik, J.; Fuess, H. Direct determination of zero-field splitting in Co(II) complexes by FAR infrared spectroscopy. Inorganica Chim. Acta 2012, 383, 143–151. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Mizukami, S.; Houjou, H.; Sugaya, K.; Koyama, E.; Tokuhisa, H.; Sasaki, T.; Kanesato, M. Fluorescence Color Modulation by Intramolecular and Intermolecular π−π Interactions in a Helical Zinc(II) Complex. Chem. Mater. 2005, 17, 50–56. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Murru, S.; Tzeng, N.; Srivastava, R.S. Quantum mechanics study of repulsive π–π interaction and flexibility of phenyl moiety in the iron azodioxide complex. J. Mol. Struct. 2015, 1097, 226–230. [Google Scholar] [CrossRef]
- Ciliberto, E.; Doris, K.A.; Pietro, W.J.; Reisner, G.M.; Ellis, D.E.; Fragala, I.; Herbstein, F.H.; Ratner, M.A.; Marks, T.J. The.pi.-.pi. interactions and bandwidths in molecular metals. A chemical, structural, photoelectron spectroscopic, and Hartree-Fock-Slater study of monomeric and cofacially joined dimeric silicon phthalocyanines. J. Am. Chem. Soc. 1984, 106, 7748–7761. [Google Scholar] [CrossRef]
- Carter-Fenk, K.; Liu, M.; Pujal, L.; Loipersberger, M.; Tsanai, M.; Vernon, R.M.; Forman-Kay, J.D.; Head-Gordon, M.; Heidar-Zadeh, F.; Head-Gordon, T. The Energetic Origins of Pi-Pi Contacts in Proteins. J. Am. Chem. Soc. 2023, 145, 24836–24851. [Google Scholar] [CrossRef]
- Kowalski, P.H.; Krzemińska, A.; Pernal, K.; Pastorczak, E. Dispersion Interactions between Molecules in and out of Equilibrium Geometry: Visualization and Analysis. J. Phys. Chem. A 2022, 126, 1312–1319. [Google Scholar] [CrossRef]
- Khare, E.; Holten-Andersen, N.; Buehler, M.J. Transition-metal coordinate bonds for bioinspired macromolecules with tunable mechanical properties. Nat. Rev. Mater. 2021, 6, 421–436. [Google Scholar] [CrossRef]
- Laporte, O.; Meggers, W.F. Some Rules of Spectral Structure*. J. Opt. Soc. Am. 1925, 11, 459–463. [Google Scholar] [CrossRef]
- Schnaubelt, L.; Petzold, H.; Speck, J.M.; Rüffer, T.; Hörner, G.; Lang, H. Spin Transition and Charge Transfer in Co2+/Co3+ Complexes of Meridional Ligands Holding Nearby Redox-active Triarylamine. Z. Anorg. Allg. Chem. 2018, 644, 1257–1267. [Google Scholar] [CrossRef]
- May, A.M.; Dempsey, J.L. A new era of LMCT: Leveraging ligand-to-metal charge transfer excited states for photochemical reactions. Chem. Sci. 2024, 15, 6661–6678. [Google Scholar] [CrossRef] [PubMed]
- Tuszyński, J.A. First- and second-order contributions to the effective Hamiltonian parameters for Gd3+(8S) and Mn2+(6S). J. Magn. Magn. Mater. 1986, 54–57, 1481–1482. [Google Scholar] [CrossRef]
- Kumar, S.; Arumugam, S.; Schwarz, B.; Ehrenberg, H.; Mondal, K. Static and Dynamic Magnetic Properties of a Co(II)-Complex with N2O2 Donor Set—A Theoretical and Experimental Study. Eur. J. Inorg. Chem. 2023, 26, e202200774. [Google Scholar] [CrossRef]
- Rao, S.V.; Maganas, D.; Sivalingam, K.; Atanasov, M.; Neese, F. Extended Active Space Ab Initio Ligand Field Theory: Applications to Transition-Metal Ions. Inorg. Chem. 2024, 63, 24672–24684. [Google Scholar] [CrossRef]
- Lloret, F.; Julve, M.; Cano, J.; Ruiz-García, R.; Pardo, E. Magnetic properties of six-coordinated high-spin cobalt(II) complexes: Theoretical background and its application. Inorganica Chim. Acta 2008, 361, 3432–3445. [Google Scholar] [CrossRef]
- Khosla, A.L.; Jacko, A.C.; Merino, J.; Powell, B.J. Spin-orbit coupling and strong electronic correlations in cyclic molecules. Phys. Rev. B 2017, 95, 115109. [Google Scholar] [CrossRef]
- Šulka, M.; Šulková, K.; Dubecký, M. Unveiling hidden dynamic correlations in CASSCF correlation energies by Hartree–Fock nodes. J. Chem. Phys. 2024, 161, 114112. [Google Scholar] [CrossRef]
- Landé, A. Über den anomalen Zeemaneffekt (Teil I). Z. Phys. 1921, 5, 231–241. [Google Scholar] [CrossRef]
- Sakiyama, H.; Kimura, R.; Oomiya, H.; Mitsuhashi, R.; Fujii, S.; Kanaizuka, K.; Muddassir, M.; Tamaki, Y.; Asato, E.; Handa, M. Relationship between Structure and Zero-Field Splitting of Octahedral Nickel(II) Complexes with a Low-Symmetric Tetradentate Ligand. Magnetochemistry 2024, 10, 32. [Google Scholar] [CrossRef]
- Pankratova, Y.A.; Nelyubina, Y.V.; Novikov, V.V.; Pavlov, A.A. High-Spin Cobalt(II) Complex with Record-Breaking Anisotropy of the Magnetic Susceptibility According to Paramagnetic NMR Spectroscopy Data. Russ. J. Coord. Chem. 2021, 47, 10–16. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Overall atoms | 57 | 69 | 78 | 84 |
| Total Mass | 486.30 | 542.41 | 515.60 | 539.67 |
| Before annihilation | 3.7575 | 3.7575 | 3.7575 | 3.7575 |
| After annihilation | 3.75 | 3.75 | 3.75 | 3.75 |
| Theoretical value | 3.75 | 3.75 | 3.75 | 3.75 |
| Complex | Mean | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| Co(II)-N1 | 2.15 | 2.19 | 2.17 | 2.16 | 2.06 |
| Co(II)-N2 | 2.15 | 2.12 | 2.20 | 2.11 | 2.06 |
| Co(II)-O1 | 1.94 | 1.95 | 1.95 | 1.97 | 1.89 |
| Co(II)-O2 | 1.93 | 1.94 | 1.94 | 1.93 | 1.90 |
| Co(II)-O3 | 2.12 * | 2.11 | 2.13 | 2.13 |
| Complex | Mean | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| N1-Co(II)-O2 | 91 | 92 | 91 | 91 | 91 |
| N1-Co(II)-O1 | 94 | 91 | 92 | 93 | 99 |
| N1-Co(II)-N2 | 88 | 82 | 83 | 84 | 101 |
| N2-Co(II)-O1 | 112 | 107 | 108 | 113 | 118 |
| N2-Co(II)-O2 | 123 | 130 | 133 | 120 | 107 |
| O1-Co(II)-O2 | 124 | 123 | 119 | 127 | 126 |
| N1-Co(II)-O3 | 175 | 175 | 179 | 171 | |
| N2-Co(II)-O3 | 94 * | 93 | 96 | 93 | |
| O1-Co(II)-O3 | 91 * | 90 | 88 | 96 | |
| O2-Co(II)-O3 | 88 * | 91 | 89 | 83 |
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| τ4 | 0.75623 | 0.76332 | 0.79877 | 0.82004 |
| τ5 | 0.75015 | 0.76682 | 0.73348 |
| Atom | Mean | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| Co(II) | 2.18 | 2.17 | 2.18 | 2.17 | 2.19 |
| N1 | −0.34 | −0.35 | −0.33 | −0.34 | −0.35 |
| N2 | −0.34 | −0.32 | −0.35 | −0.33 | −0.34 |
| O1 | −0.46 | −0.40 | −0.48 | −0.48 | −0.48 |
| O2 | −0.46 | −0.41 | −0.48 | −0.48 | −0.48 |
| O3 | −0.43 * | −0.42 | −0.41 | −0.47 |
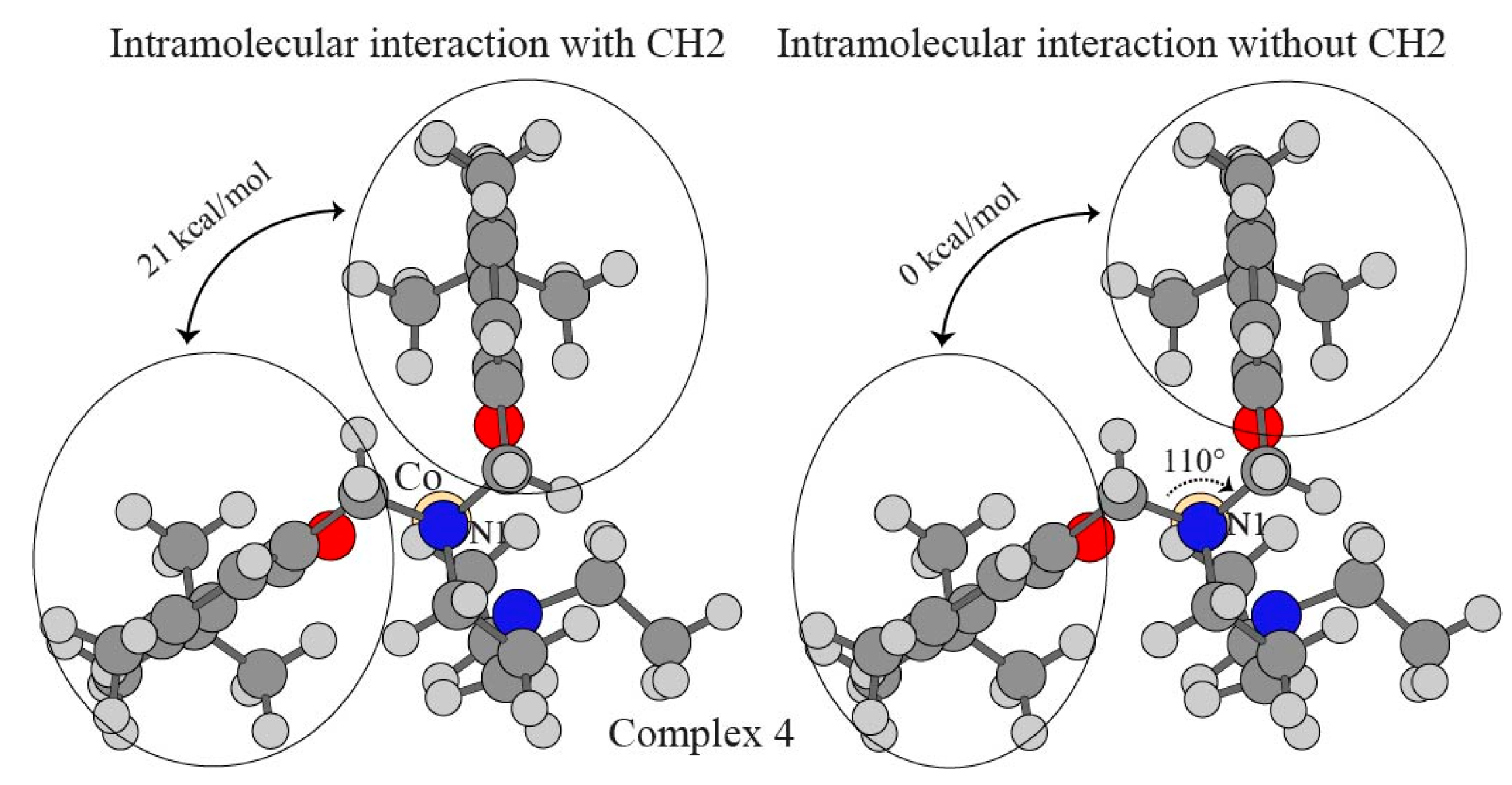
| pi–pi (no CH2) a | ΔEgas b | ΔΔEgasBSSE c | ΔΔEsolv d | ΔE |
|---|---|---|---|---|
| pi–pi (with CH2) | 70.33 | 5.16 | −54.52 | 20.97 |
| pi–pi (no CH2) | −4.23 | 3.35 | 0.98 | 0.10 |
| O2-Co(II) | N1-Co(II) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Donor | Orbital | Acceptor | Orbital | Energy | Donor | Orbital | Acceptor | Orbital | Energy |
| O2 | LP 117α | Co(II) | LP* 113α | 3.70 | N1 | LP 125α | Co(II) | LP* 113α | 3.42 |
| O2 | LP 117α | Co(II) | LP* 114α | 0.78 | N1 | LP 125α | Co(II) | LP* 114α | 0.15 |
| O2 | LP 117α | Co(II) | LP* 115α | 0.84 | N1 | LP 125α | Co(II) | LP* 115α | 3.82 |
| O2 | LP 117α | Co(II) | LP* 116α | 1.13 | N1 | LP 125α | Co(II) | LP* 116α | 12.55 |
| O2 | LP 118α | Co(II) | LP* 113α | 25.26 | |||||
| O2 | LP 118α | Co(II) | LP* 114α | 16.95 | |||||
| O2 | LP 119α | Co(II) | LP* 113α | 18.59 | |||||
| O2 | LP 119α | Co(II) | LP* 114α | 4.29 | |||||
| O2 | LP 119α | Co(II) | LP* 115α | 3.40 | |||||
| O2 | LP 119α | Co(II) | LP* 116α | 0.15 | |||||
| Coordinate | 10 | 75.09 | 4 | 19.94 | |||||
| O2 | LP 117α | Co(II) | RY* 133α | 0.03 | N1 | LP 125α | Co(II) | RY* 134α | 0.06 |
| O2 | LP 117α | Co(II) | RY* 137α | 0.05 | N1-C2 | BD 5α | Co(II) | LP* 113α | 0.07 |
| O2 | LP 118α | Co(II) | RY* 133α | 0.05 | N1-C2 | BD 5α | Co(II) | LP* 114α | 0.38 |
| O2 | LP 118α | Co(II) | RY* 134α | 0.07 | N1-C2 | BD 5α | Co(II) | LP* 115α | 0.61 |
| O2 | LP 119α | Co(II) | RY* 133α | 0.20 | N1-C2 | BD 5α | Co(II) | LP* 116α | 0.79 |
| O2 | LP 119α | Co(II) | RY* 135α | 0.05 | N1-C3 | BD 6α | Co(II) | LP* 113α | 0.04 |
| O2-C8 | BD 1α | Co(II) | LP* 113α | 2.97 | N1-C3 | BD 6α | Co(II) | LP* 114α | 0.37 |
| O2-C8 | BD 1α | Co(II) | LP* 114α | 1.76 | N1-C3 | BD 6α | Co(II) | LP* 115α | 0.53 |
| O2-C8 | BD 1α | Co(II) | LP* 115α | 0.12 | N1-C3 | BD 6α | Co(II) | LP* 116α | 0.71 |
| O2 | CR 72α | Co(II) | LP* 113α | 2.34 | N1-C6 | BD 7α | Co(II) | LP* 113α | 0.03 |
| O2 | CR 72α | Co(II) | LP* 114α | 1.35 | N1-C6 | BD 7α | Co(II) | LP* 114α | 0.34 |
| O2 | CR 72α | Co(II) | LP* 115α | 0.09 | N1-C6 | BD 7α | Co(II) | LP* 115α | 0.59 |
| O2-C8 | BD 1α | Co(II) | RY* 133α | 0.04 | N1-C6 | BD 7α | Co(II) | LP* 116α | 0.77 |
| N1 | CR 75α | Co(II) | LP* 113α | 0.41 | |||||
| N1 | CR 75α | Co(II) | LP* 114α | 0.03 | |||||
| N1 | CR 75α | Co(II) | LP* 115α | 0.34 | |||||
| N1 | CR 75α | Co(II) | LP* 116α | 0.98 | |||||
| N1-C3 | BD 6α | Co(II) | RY* 133α | 0.04 | |||||
| N1-C6 | BD 7α | Co(II) | RY* 133α | 0.03 | |||||
| Dispersion | 13 | 9.12 | 19 | 7.12 | |||||
| O2 | LP 117β | Co(II) | LP* 110β | 5.87 | N1 | LP 125β | Co(II) | LP* 110β | 1.44 |
| O2 | LP 117β | Co(II) | LP* 111β | 0.39 | N1 | LP 125β | Co(II) | LP* 111β | 0.26 |
| O2 | LP 117β | Co(II) | LP* 112β | 0.28 | N1 | LP 125β | Co(II) | LP* 112β | 4.82 |
| O2 | LP 117β | Co(II) | LP* 113β | 1.53 | N1 | LP 125β | Co(II) | LP* 113β | 13.76 |
| O2 | LP 117β | Co(II) | LP* 114β | 0.29 | N1 | LP 125β | Co(II) | LP* 114β | 9.05 |
| O2 | LP 117β | Co(II) | LP* 115β | 0.72 | N1 | LP 125β | Co(II) | LP* 116β | 0.13 |
| O2 | LP 117β | Co(II) | LP* 116β | 1.08 | |||||
| O2 | LP 118β | Co(II) | LP* 110β | 35.90 | |||||
| O2 | LP 118β | Co(II) | LP* 111β | 7.60 | |||||
| O2 | LP 118β | Co(II) | LP* 112β | 0.11 | |||||
| O2 | LP 118β | Co(II) | LP* 114β | 0.50 | |||||
| O2 | LP 118β | Co(II) | LP* 115β | 1.88 | |||||
| O2 | LP 118β | Co(II) | LP* 116β | 0.71 | |||||
| O2 | LP 119β | Co(II) | LP* 110β | 28.07 | |||||
| O2 | LP 119β | Co(II) | LP* 111β | 2.27 | |||||
| O2 | LP 119β | Co(II) | LP* 112β | 2.13 | |||||
| O2 | LP 119β | Co(II) | LP* 114β | 0.59 | |||||
| O2 | LP 119β | Co(II) | LP* 115β | 1.71 | |||||
| O2 | LP 119β | Co(II) | LP* 116β | 0.64 | |||||
| Coordinate | 19 | 92.27 | 6 | 29.46 | |||||
| O2 | LP 117β | Co(II) | RY* 137β | 0.04 | N1-C3 | BD 6β | Co(II) | LP* 133β | 0.03 |
| O2 | LP 119β | Co(II) | RY* 133β | 0.20 | N1-C6 | BD 5β | Co(II) | LP* 110β | 0.41 |
| O2-C8 | BD 1β | Co(II) | LP* 110β | 3.91 | N1-C6 | BD 5β | Co(II) | LP* 111β | 0.12 |
| O2-C8 | BD 1β | Co(II) | LP* 111β | 0.69 | N1-C6 | BD 5β | Co(II) | LP* 112β | 0.48 |
| O2-C8 | BD 1β | Co(II) | LP* 112β | 0.11 | N1-C6 | BD 5β | Co(II) | LP* 113β | 0.77 |
| O2-C8 | BD 1β | Co(II) | LP* 114β | 0.06 | N1-C6 | BD 5β | Co(II) | LP* 114β | 0.09 |
| O2-C8 | BD 1β | Co(II) | LP* 116β | 0.28 | N1-C3 | BD 6β | Co(II) | LP* 110β | 0.34 |
| O2 | CR 72β | Co(II) | LP* 110β | 2.94 | N1-C3 | BD 6β | Co(II) | LP* 111β | 0.10 |
| O2 | CR 72β | Co(II) | LP* 111β | 0.42 | N1-C3 | BD 6β | Co(II) | LP* 112β | 0.45 |
| O2 | CR 72β | Co(II) | LP* 112β | 0.07 | N1-C3 | BD 6β | Co(II) | LP* 113β | 0.72 |
| O2 | CR 72β | Co(II) | LP* 114β | 0.06 | N1-C3 | BD 6β | Co(II) | LP* 114β | 0.10 |
| O2 | CR 72β | Co(II) | LP* 115β | 0.03 | N1-C2 | BD 7β | Co(II) | LP* 110β | 0.40 |
| O2 | CR 72β | Co(II) | LP* 116β | 0.29 | N1-C2 | BD 7β | Co(II) | LP* 111β | 0.07 |
| N1-C2 | BD 7β | Co(II) | LP* 112β | 0.46 | |||||
| N1-C2 | BD 7β | Co(II) | LP* 113β | 0.75 | |||||
| N1-C2 | BD 7β | Co(II) | LP* 114β | 0.08 | |||||
| N1 | CR 75β | Co(II) | LP* 110β | 0.32 | |||||
| N1 | CR 75β | Co(II) | LP* 111β | 0.08 | |||||
| N1 | CR 75β | Co(II) | LP* 112β | 0.39 | |||||
| N1 | CR 75β | Co(II) | LP* 113β | 0.96 | |||||
| Dispersion | 13 | 9.10 | 20 | 7.12 | |||||
| O2 LP 117-Co(II) | 11 | 16.61 | N1 LP 125-Co(II) | 10 | 49.40 | ||||
| O2 LP 118-Co(II) | 8 | 88.91 | |||||||
| O2 LP 119-Co(II) | 10 | 61.84 | |||||||
| Co(II) β/α)) | 1.2 | 1.5 | |||||||
| Coordinate Tot | 29 | 167.36 | 10 | 49.40 | |||||
| Dispersion Tot | 26 | 18.22 | 39 | 14.24 | |||||
| Co(II)-O2 | Co(II)-N1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Donor | Orbital | Acceptor | Orbital | Energy | Donor | Orbital | Acceptor | Orbital | Energy |
| Co(II) | CR/LP/LP* | O2 | BD*/RY* | Co(II) | CR/LP/LP* | N1 | BD*/RY* | ||
| α | 23 | 5.49 | 26 | 1.97 | |||||
| β | 17 | 3.22 | 22 | 1.51 | |||||
| Dispersion Tot | 40 | 8.71 | 48 | 3.48 | |||||
| Complex Number of Atoms | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| 57 | 69 | 78 | 84 | ||
| Donor | Acceptor | ||||
| N1 LP | Co(II) α spin LP* | 19.94 | 20.39 | 19.99 | 22.37 |
| N2 LP | Co(II) α spin LP* | 23.37 | 19.78 | 21.68 | 24.61 |
| O1 LP | Co(II) α spin LP* | 75.09 | 63.84 | 67.33 | 68.20 |
| O2 LP | Co(II) α spin LP* | 66.10 | 62.49 | 66.65 | 69.21 |
| Ligand | Co(II) α other | 45.21 | 53.90 | 51.92 | 69.60 |
| N1 LP | Co(II) β spin LP* | 29.46 | 28.54 | 29.73 | 35.99 |
| N2 LP | Co(II) β spin LP* | 32.34 | 30.00 | 34.08 | 35.50 |
| O1 LP | Co(II) β spin LP* | 92.27 | 80.36 | 83.66 | 87.47 |
| O2 LP | Co(II) β spin LP* | 82.74 | 79.52 | 82.59 | 87.66 |
| Ligand | Co(II) β other | 44.76 | 54.22 | 52.28 | 71.38 |
| Co(II) α spin other | N1 other | 1.97 | 1.98 | 2.37 | 2.82 |
| Co(II) α spin other | N2 other | 2.95 | 2.17 | 3.16 | 3.27 |
| Co(II) α spin other | O1 other | 5.49 | 5.08 | 5.52 | 6.12 |
| Co(II) α spin other | O2 other | 5.30 | 5.61 | 5.20 | 5.75 |
| Co(II) α spin other | other atoms | 7.47 | 10.41 | 10.87 | 9.43 |
| Co(II) β spin other | N1 other | 1.51 | 1.13 | 1.58 | 1.95 |
| Co(II) β spin other | N2 other | 1.99 | 1.93 | 1.9 | 1.95 |
| Co(II) β spin other | O1 other | 3.22 | 3.04 | 2.92 | 3.19 |
| Co(II) β spin other | O2 other | 3.35 | 3.35 | 2.63 | 3.13 |
| Co(II) β spin other | Other atoms | 8.01 | 10.54 | 10.51 | 12.61 |
| N1-Co(II) β/α | 1.5 | 1.5 | 1.4 | 1.5 | 1.6 |
| N2-Co(II) β/α | 1.5 | 1.4 | 1.5 | 1.6 | 1.4 |
| O1-Co(II) β/α | 1.3 | 1.2 | 1.3 | 1.2 | 1.3 |
| O2-Co(II) β/α | 1.3 | 1.3 | 1.3 | 1.2 | 1.3 |
| LP1 N1-Co(II) | (Mean) 51.60 | 49.40 | 48.93 | 49.72 | 58.36 |
| LP1 O2-Co(II) | (Mean) 14.55 | 16.61 | 15.38 | 13.13 | 13.08 |
| LP2 O2-Co(II) | (Mean) 80.73 | 88.91 | 85.43 | 64.87 | 83.71 |
| LP3 O2-Co(II) | (Mean) 59.28 | 61.84 | 43.39 | 72.99 | 58.88 |
| N1-Co(II) | (Mean) 51.60 | 49.40 | 48.93 | 49.72 | 58.36 |
| N2-Co(II) | (Mean) 55.34 | 55.71 | 49.78 | 55.76 | 60.11 |
| O1-Co(II) | (Mean) 154.56 | 167.36 | 144.20 | 150.99 | 155.67 |
| O2-Co(II) | (Mean) 149.24 | 148.84 | 142.01 | 149.24 | 156.87 |
| L-M coordination | 410.74 | 421.31 | 384.92 | 405.71 | 431.01 |
| L-M dispersion | 110.82 | 89.97 | 108.12 | 104.2 | 140.98 |
| L-M interaction | 521.56 | 511.28 | 493.04 | 509.91 | 571.99 |
| M-L dispersion | 45.85 | 41.26 | 45.24 | 46.66 | 50.22 |
| L-M/M-L ratio | 11.25 | 12 | 11 | 11 | 11 |
| Dispersion | 156.66 | 131.23 | 153.36 | 150.86 | 191.20 |
| Total interaction | 567.40 | 552.54 | 538.28 | 556.57 | 622.21 |
| coordination % | 72.5 | 76% | 72% | 73% | 69% |
| Dispersion % | 27.5 | 24% | 28% | 27% | 31% |
| Complex Number of Atoms | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| 57 | 69 | 78 | 84 | ||
| Donor | Acceptor | ||||
| O3 LP | Co(II) α spin LP* | 30.18 | 28.52 | 30.76 | |
| MeOH | Co(II) α other | 10.91 | 12.04 | 13.27 | |
| O3 LP | Co(II) β spin LP* | 37.70 | 35.37 | 38.09 | |
| MeOH | Co(II) β other | 11.11 | 12.22 | 13.26 | |
| Co(II) α spin other | MeOH | 4.87 | 4.57 | 2.59 | |
| Co(II) β spin other | MeOH | 4.75 | 4.34 | 2.40 | |
| L-M O3-Co(II) β/α | (Mean) 1.2 | 1.2 | 1.2 | 1.2 | |
| LP(ψ 124)-M O3-Co(II) | 62.17 | 63.10 | 59.18 | 64.24 | |
| LP(ψ 123)-M O3-Co(II) | 4.70 | 4.78 | 4.71 | 4.61 | |
| L-M O3-Co(II) | 66.87 | 67.88 | 63.89 | 68.85 | |
| L-M dispersion | 24.27 | 22.02 | 24.26 | 26.53 | |
| L-M interaction | 91.14 | 89.90 | 88.15 | 95.38 | |
| M-L dispersion | 7.84 | 9.62 | 8.91 | 4.99 | |
| L-M/M-L ratio | 12.67 | 9 | 10 | 19 | |
| Dispersion | 32.45 | 31.79 | 34.17 | 31.40 | |
| Total interaction | 98.98 | 99.52 | 97.06 | 100.37 | |
| coordination % | 67.7 | 68% | 66% | 69% | |
| Dispersion % | 32.3 | 32% | 34% | 31% | |
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| R1 | 89.93 | 89.89 | 205.43 | 204.50 |
| R2 | 70.17 | 70.15 | 86.16 | 85.13 |
| R3 | 55.39 | 136.94 | 54.33 | 94.47 |
| Ligand | 2956.99 | 3056.88 | 2238.29 | 3857.57 |
| Spin | Root | Complex 1 | Complex 2 | Complex 3 | Complex 4 |
|---|---|---|---|---|---|
| 3/2 | 0 | 0.000 | 0.000 | 0.000 | 0.000 |
| 3/2 | 1 | 7.539 | 7.372 | 6.275 | 14.511 |
| 3/2 | 2 | 0.504 | 2.185 | 0.690 | 1.884 |
| 3/2 | 3 | 6.591 | 6.869 | 9.226 | 2.736 |
| 3/2 | 4 | 9.505 | 9.923 | 10.878 | 4.597 |
| 3/2 | 5 | 0.091 | 0.074 | 0.058 | 0.071 |
| 3/2 | 6 | 0.046 | 0.034 | 0.026 | 0.007 |
| 3/2 | 7 | 0.090 | 0.063 | 0.103 | 0.005 |
| 3/2 | 8 | 0.001 | 0.038 | 0.000 | 0.008 |
| 3/2 | 9 | 0.075 | 0.021 | 0.089 | 0.001 |
| 3/2 | Tot | 24.442 | 26.579 | 27.345 | 23.82 |
| 1/2 | 0 | −0.226 | −0.191 | 0.681 | 2.610 |
| 1/2 | 1 | 0.464 | 0.334 | −0.044 | 0.166 |
| 1/2 | 2 | 3.412 | 3.500 | 2.410 | 0.216 |
| 1/2 | 3 | −0.011 | 0.002 | 0.011 | 0.381 |
| 1/2 | 4 | −0.248 | 0.013 | −0.828 | −0.008 |
| 1/2 | 5 | −0.096 | −0.055 | −0.128 | −0.031 |
| 1/2 | 6 | −0.732 | −0.671 | −0.034 | −1.331 |
| 1/2 | 7 | 0.097 | 0.286 | 8.538 | 7.684 |
| 1/2 | 8 | 6.237 | 5.737 | −1.118 | −1.038 |
| 1/2 | 9 | −0.025 | −0.026 | −0.003 | −0.231 |
| 1/2 | 10 | 0.000 | 0.012 | −0.003 | −0.266 |
| 1/2 | 11 | −0.009 | −0.008 | −0.047 | 0.029 |
| 1/2 | 12 | −0.086 | −0.139 | −0.055 | 0.028 |
| 1/2 | 13 | −0.149 | −0.111 | −0.010 | −0.210 |
| 1/2 | 14 | −0.142 | −0.075 | −0.167 | −0.071 |
| 1/2 | 15 | −0.168 | −0.166 | −0.165 | −0.017 |
| 1/2 | 16 | 0.005 | −0.036 | −0.018 | −0.555 |
| 1/2 | 17 | −0.105 | −0.198 | −0.112 | −0.180 |
| 1/2 | 18 | −0.156 | −0.095 | −0.178 | 0.097 |
| 1/2 | 19 | −0.000 | −0.141 | 0.115 | −0.035 |
| 1/2 | 20 | 0.116 | 0.258 | 0.078 | 0.068 |
| 1/2 | 21 | 0.026 | 0.002 | 0.001 | 0.161 |
| 1/2 | 22 | 0.748 | 0.907 | 0.776 | 0.634 |
| 1/2 | 23 | −0.432 | −0.259 | −0.107 | −0.343 |
| 1/2 | 24 | −0.026 | −0.189 | −0.258 | −0.200 |
| 1/2 | 25 | −0.165 | −0.169 | −0.111 | −0.076 |
| 1/2 | 26 | −0.214 | −0.197 | −0.311 | −0.082 |
| 1/2 | 27 | −0.266 | −0.283 | −0.340 | −0.096 |
| 1/2 | 28 | −0.066 | −0.047 | −0.107 | −0.010 |
| 1/2 | 29 | −0.125 | −0.059 | −0.105 | 0.045 |
| 1/2 | 30 | −0.050 | −0.072 | −0.035 | −0.044 |
| 1/2 | 31 | −0.051 | −0.058 | −0.054 | −0.044 |
| 1/2 | 32 | 0.006 | −0.033 | −0.070 | −0.054 |
| 1/2 | 33 | −0.038 | −0.039 | 0.005 | −0.020 |
| 1/2 | 34 | −0.000 | −0.001 | 0.007 | −0.020 |
| 1/2 | 35 | 0.045 | 0.043 | 0.066 | 0.014 |
| 1/2 | 36 | 0.020 | 0.004 | 0.001 | −0.000 |
| 1/2 | 37 | −0.004 | −0.003 | −0.001 | −0.001 |
| 1/2 | 38 | −0.003 | −0.004 | −0.003 | −0.004 |
| 1/2 | 39 | 0.093 | 0.119 | 0.100 | 0.124 |
| 1/2 | Tot | 7.676 | 7.892 | 8.377 | 7.290 |
| Tot | 32.118 | 34.471 | 35.722 | 31.11 | |
| 3/2 | 76% | 77% | 77% | 77% | |
| 1/2 | 24% | 23% | 23% | 23% |
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Lowest CASSCF | −3444.3676637 | −3600.3760521 | −2799.8676682 | −2840.8954558 |
| HF(Hartree) | −3444.2077057 | −3600.2158344 | −2799.6995722 | −2840.7361725 |
| MP2(Hartree) | −3448.4023597 | −3605.0109351 | −2804.6862648 | −2845.9533635 |
| CASSCF-HF (kcal/mol) | −100.38 | −100.54 | −105.48 | −99.96 |
| MP2-HF (kcal/mol) | −2632.19 | −3008.97 | −3129.20 | −3273.84 |
| Number of electrons | 253 | 285 | 277 | 291 |
| MP2-HF (kcal/mol)/e | −10.40 | −10.56 | −11.30 | −11.25 |
| CASSCF-HF (kcal/mol) | −100.38 | −100.54 | −105.48 | −99.96 |
| Complex | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Number of atoms | 57 | 69 | 78 | 84 |
| 2nd order SOC D | 32.118 | 34.468 | 35.724 | 31.106 |
| SSC D | −0.028 | 0.031 | −0.031 | 0.024 |
| 2nd order D | 32.090 | 34.437 | 35.693 | 31.082 |
| 2nd order SOC E | 6.837 | 6.687 | 5.980 | 8.125 |
| SSC E | −0.007 | 0.009 | −0.008 | 0.004 |
| 2nd order E | 6.830 | 6.696 | 5.972 | 8.121 |
| 2nd order D/E | 5 | 5 | 6 | 4 |
| Effective Hamiltonian D | 29.233 | 31.220 | 32.392 | 28.173 |
| Effective Hamiltonian E | 6.472 | 6.300 | 5.689 | 7.637 |
| Effective Hamiltonian D/E | 5 | 5 | 6 | 4 |
| Experimental D based on (χM) | 29.060 | 22.680 | 28.790 | 30.860 |
| 2nd order perturbation D (%) | 110.4 | 151.8 | 124.0 | 100.7 |
| Effective Hamiltonian D (%) | 100.6 | 137.7 | 112.5 | 91.3 |
| Theoretical iso g factor | 2.216 | 2.225 | 2.219 | 2.220 |
| Theoretical gx factor | 2.046 | 2.040 | 2.028 | 2.052 |
| Theoretical gy factor | 2.229 | 2.248 | 2.251 | 2.221 |
| Theoretical gz factor | 2.372 | 2.387 | 2.376 | 2.386 |
| Experimental g factor | 2.275 | 2.249 | 2.304 | 2.262 |
| Theoretical g factor (%) | 97.4 | 98.9 | 96.3 | 98.1 |
| Complex | 1 | 2 | 3 | 4 | Mean |
|---|---|---|---|---|---|
| Number of Atoms | 57 | 69 | 78 | 84 | |
| 2nd Order Perturbation D 40 Roots | 32.118 | 34.468 | 35.724 | 31.106 | |
| 2nd Order Perturbation D 5 Roots | 28.424 | 30.995 | 30.175 | 27.561 | |
| 2nd Order Perturbation D 5 Roots % | 88.5% | 89.9% | 84.5% | 88.6% | 87.88% |
| Effective Hamiltonian D 40 Roots | 29.233 | 31.220 | 32.392 | 28.173 | |
| Effective Hamiltonian D 5 Roots | 25.896 | 28.196 | 27.249 | 25.040 | |
| Effective Hamiltonian D 5 Roots % | 88.6% | 90.3% | 84.1% | 88.9% | 87.89% |
| 2nd Order Perturbation E 40 Roots | 6.837 | 6.687 | 5.980 | 8.125 | |
| 2nd Order Perturbation E 5 Roots | 6.508 | 6.689 | 4.853 | 8.092 | |
| 2nd Order Perturbation E 5 Roots % | 95.2% | 100% | 81.2% | 99.6% | 94.00% |
| Effective Hamiltonian E 40 Roots | 6.472 | 6.300 | 5.689 | 7.637 | |
| Effective Hamiltonian E 5 Roots | 5.989 | 6.203 | 4.212 | 7.524 | |
| Effective Hamiltonian E 5 Roots % | 92.5% | 98.5% | 74.0% | 98.5% | 97.88% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Massoud, S.S.; Starovoytov, O.N.; Altalhi, T.; Gao, Y.; Yakobson, B.I. Quantum Mechanics MP2 and CASSCF Study of Coordinate Quasi-Double Bonds in Cobalt(II) Complexes as Single Molecule Magnets. Nanomaterials 2025, 15, 938. https://doi.org/10.3390/nano15120938
Liu Y, Massoud SS, Starovoytov ON, Altalhi T, Gao Y, Yakobson BI. Quantum Mechanics MP2 and CASSCF Study of Coordinate Quasi-Double Bonds in Cobalt(II) Complexes as Single Molecule Magnets. Nanomaterials. 2025; 15(12):938. https://doi.org/10.3390/nano15120938
Chicago/Turabian StyleLiu, Yuemin, Salah S. Massoud, Oleg N. Starovoytov, Tariq Altalhi, Yunxiang Gao, and Boris I. Yakobson. 2025. "Quantum Mechanics MP2 and CASSCF Study of Coordinate Quasi-Double Bonds in Cobalt(II) Complexes as Single Molecule Magnets" Nanomaterials 15, no. 12: 938. https://doi.org/10.3390/nano15120938
APA StyleLiu, Y., Massoud, S. S., Starovoytov, O. N., Altalhi, T., Gao, Y., & Yakobson, B. I. (2025). Quantum Mechanics MP2 and CASSCF Study of Coordinate Quasi-Double Bonds in Cobalt(II) Complexes as Single Molecule Magnets. Nanomaterials, 15(12), 938. https://doi.org/10.3390/nano15120938