


Polymeric Membranes Doped with Halloysite Nanotubes Imaged using Proton Microbeam Microscopy
 ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Sample Preparation
2.1.1. Preparation of PANI-Capped HNTs
2.1.2. Preparation of Undoped and HNT-Doped PES Membranes
2.1.3. Preparation of the Samples (HNTs and PES Membranes) for NM Analysis
2.2. Characterization Analysis and Techniques
2.2.1. Spectroscopies
2.2.2. Atomic Force Microscopy (AFM)
2.2.3. Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM)
2.2.4. Micro Nuclear Microscopy for PIXE and FSTIM Analyses
2.2.5. Water Permeance
2.2.6. Water Contact Angle and Mechanical Properties
2.2.7. Porosity
3. Results and Discussion
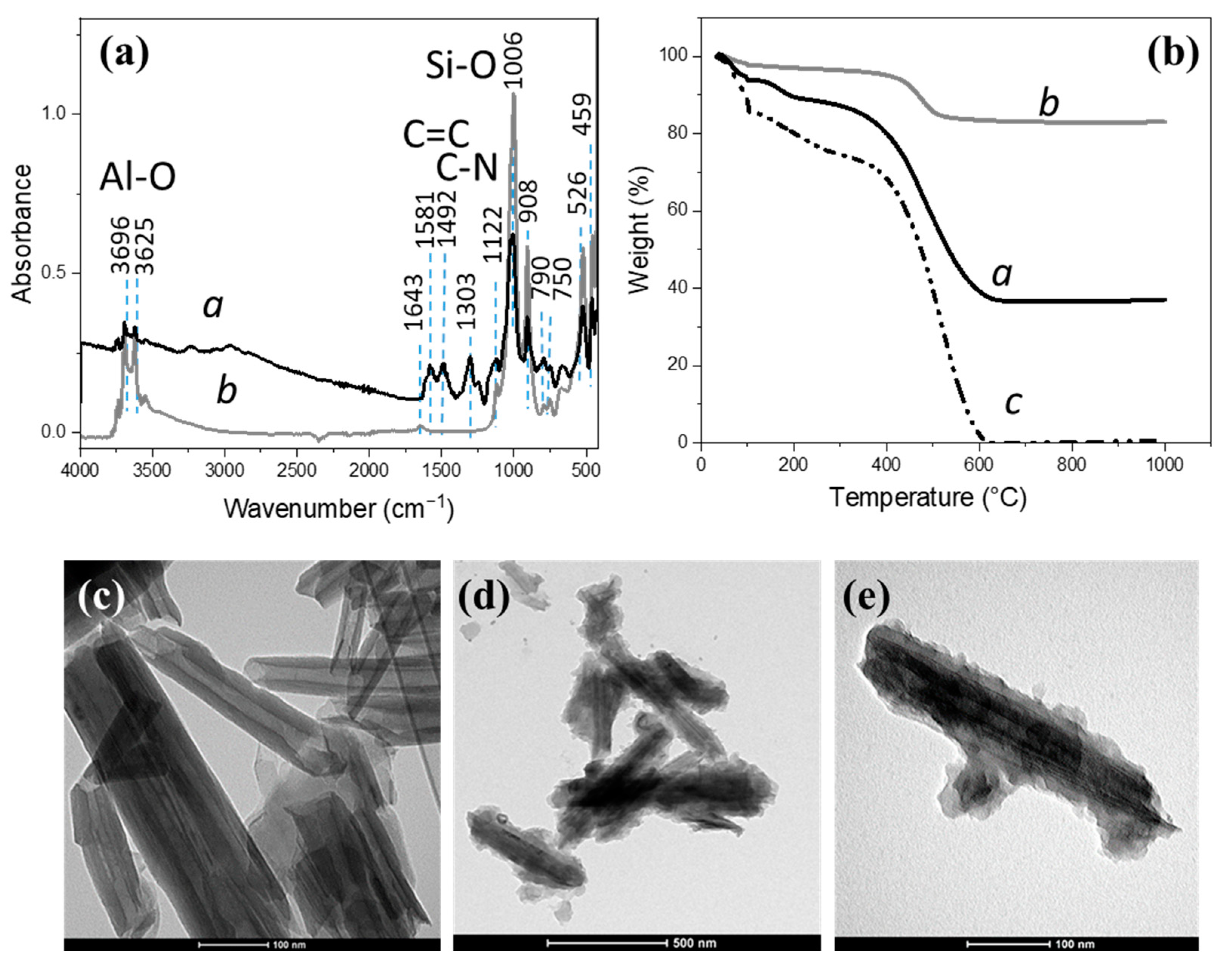
3.1. PANI-Coated HNTs
3.2. Surface and Bulk Properties of Undoped and Doped PES Membranes
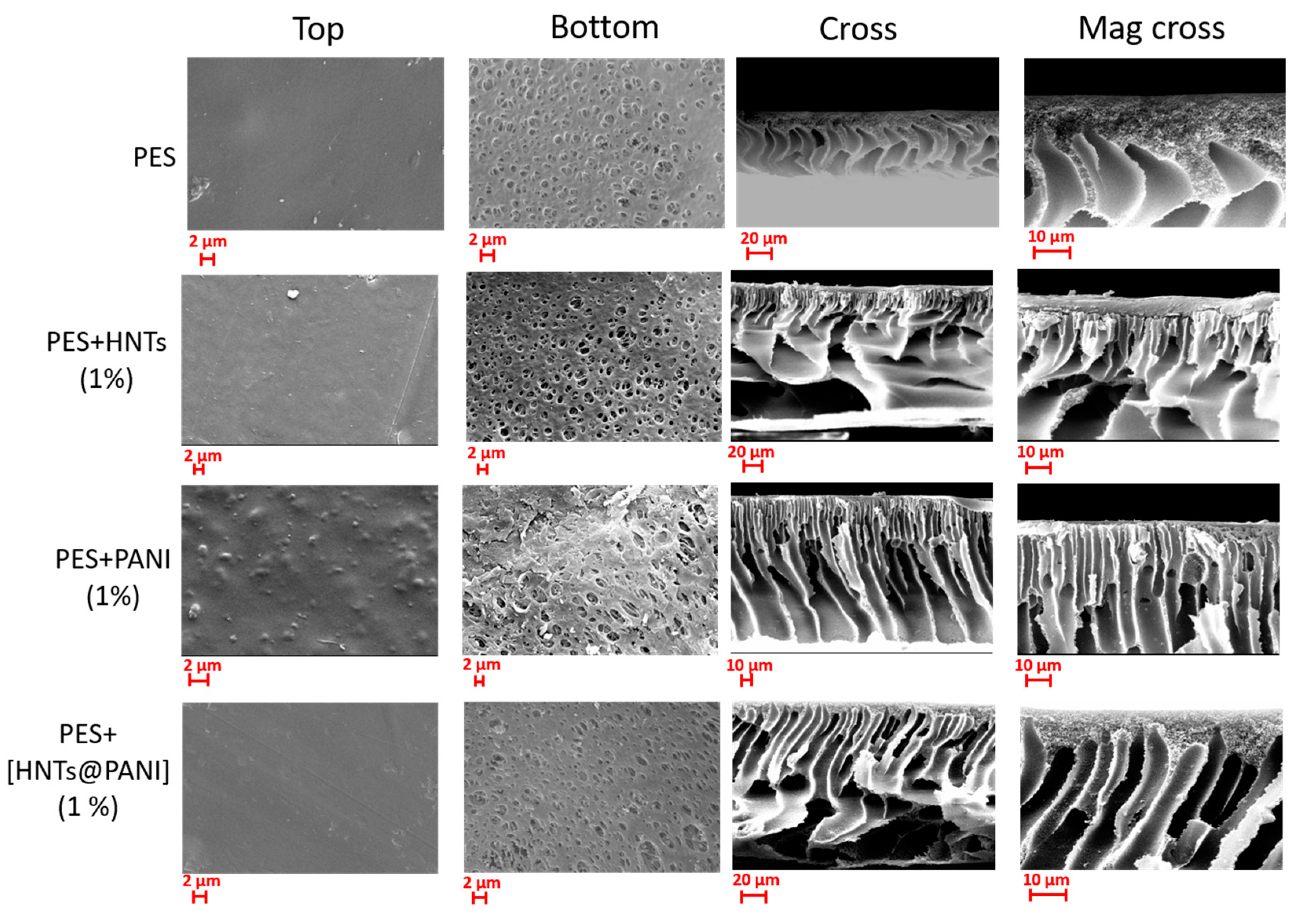
3.3. SEM Investigation of Undoped and Doped PES Membranes
3.4. Proton-Probe-Based Investigation of Doped PES Membranes
3.4.1. Why the Nuclear Microscopy Technique?
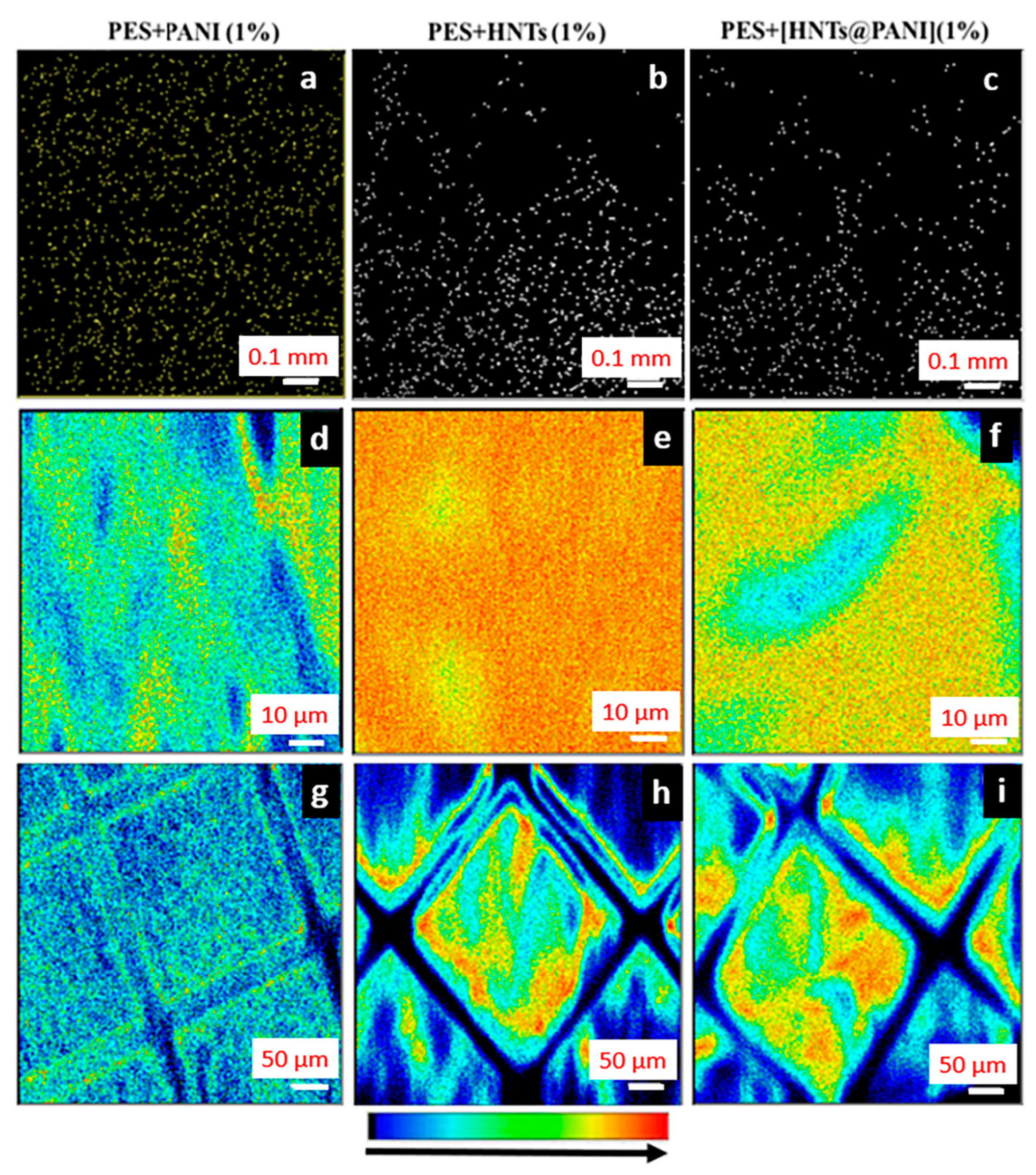
3.4.2. PANI- and HNT-Doped Membranes Investigated using+ the NM Technique
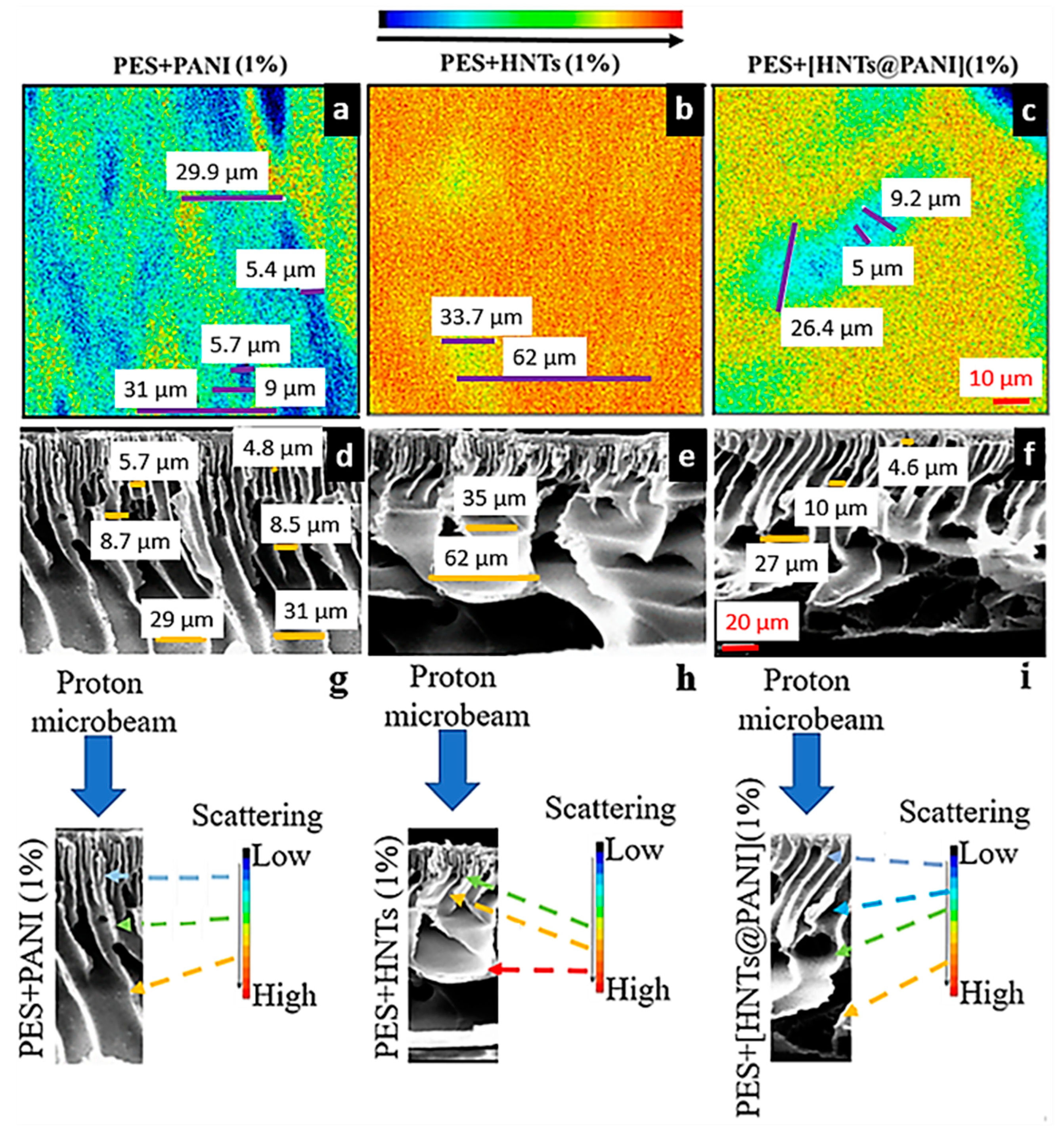
3.4.3. Correlation between FSTIM and SEM Mapping of PANI- and HNT-Doped Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vergaro, V.; Abdullayev, E.; Lvov, Y.M.; Zeitoun, A.; Cingolani, R.; Rinaldi, R.; Leporatti, S. Cytocompatibility and Uptake of Halloysite Clay Nanotubes. Biomacromolecules 2010, 11, 820–826. [Google Scholar] [CrossRef] [PubMed]
- White, R.D.; Bavykin, D.V.; Walsh, F.C. The stability of halloysite nanotubes in acidic and alkaline aqueous suspensions. Nanotechnology 2012, 23, 065705. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Niedenthal, W.; Smarsly, B.M.; Natile, M.M.; Huang, Y.; Carraro, M. Au nanoparticles supported on piranha etched halloysite nanotubes for highly efficient heterogeneous catalysis. Appl. Surf. Sci. 2021, 546, 149100. [Google Scholar] [CrossRef]
- Shchukin, D.G.; Sukhorukov, G.B.; Price, R.R.; Lvov, Y.M. Halloysite Nanotubes as Biomimetic Nanoreactors. Small 2005, 1, 510–513. [Google Scholar] [CrossRef]
- Chao, C.; Liu, J.; Wang, J.; Zhang, Y.; Zhang, B.; Zhang, Y.; Xiang, X.; Chen, R. Surface Modification of Halloysite Nanotubes with Dopamine for Enzyme Immobilization. ACS Appl. Mater. Interfaces 2013, 5, 10559–10564. [Google Scholar] [CrossRef] [PubMed]
- Massaro, M.; Noto, R.; Riela, S. Halloysite Nanotubes: Smart Nanomaterials in Catalysis. Catalysts 2022, 12, 149. [Google Scholar] [CrossRef]
- Wang, R.; Jiang, G.; Ding, Y.; Wang, Y.; Sun, X.; Wang, X.; Chen, W. Photocatalytic Activity of Heterostructures Based on TiO2 and Halloysite Nanotubes. ACS Appl. Mater. Interfaces 2011, 3, 4154–4158. [Google Scholar] [CrossRef] [PubMed]
- Glotov, A.; Vutolkina, A.; Pimerzin, A.; Vinokurov, V.; Lvov, Y. Clay nanotube-metal core/shell catalysts for hydroprocesses. Chem. Soc. Rev. 2021, 50, 9240–9277. [Google Scholar] [CrossRef]
- Naumenko, E.A.; Guryanov, I.D.; Yendluri, R.; Lvov, Y.M.; Fakhrullin, R.F. Clay nanotube–biopolymer composite scaffolds for tissue engineering. Nanoscale 2016, 8, 7257–7271. [Google Scholar] [CrossRef]
- Massaro, M.; Lazzara, G.; Milioto, S.; Noto, R.; Riela, S. Covalently modified halloysite clay nanotubes: Synthesis, properties, biological and medical applications. J. Mater. Chem. B 2017, 5, 2867–2882. [Google Scholar] [CrossRef]
- Lvov, Y.M.; DeVilliers, M.M.; Fakhrullin, R.F. The application of halloysite tubule nanoclay in drug delivery. Expert Opin. Drug Deliv. 2016, 13, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, H.; Zhang, Y.; Zhang, B.; Liu, J. Recent advances in halloysite nanotube derived composites for water treatment. Environ. Sci. Nano 2016, 3, 28–44. [Google Scholar] [CrossRef]
- Attia, N.F.; Ahmed, H.E.; El Ebissy, A.A.; El Ashery, S.E.A. Green and novel approach for enhancing flame retardancy, UV protection and mechanical properties of fabrics utilized in historical textile fabrics conservation. Prog. Org. Coatings 2022, 166, 106822. [Google Scholar] [CrossRef]
- Zahidah, K.A.; Kakooei, S.; Ismail, M.C.; Bothi Raja, P. Halloysite nanotubes as nanocontainer for smart coating application: A review. Prog. Org. Coatings 2017, 111, 175–185. [Google Scholar] [CrossRef]
- Xu, W.; Xu, L.; Pan, H.; Li, K.; Li, J.; Wang, L.; Shen, Y.; Liu, Y.; Li, T. Robust ZnO/HNTs-based superhydrophobic cotton fabrics with UV shielding, self-cleaning, photocatalysis, and oil/water separation. Cellulose 2022, 29, 4021–4037. [Google Scholar] [CrossRef]
- Chao, C.; Zhao, Y.; Guan, H.; Liu, G.; Hu, Z.; Zhang, B. Improved Performance of Immobilized Laccase on Poly(diallyldimethylammonium chloride) Functionalized Halloysite for 2,4-Dichlorophenol Degradation. Environ. Eng. Sci. 2017, 34, 762–770. [Google Scholar] [CrossRef]
- Lvov, Y.; Wang, W.; Zhang, L.; Fakhrullin, R. Halloysite Clay Nanotubes for Loading and Sustained Release of Functional Compounds. Adv. Mater. 2016, 28, 1227–1250. [Google Scholar] [CrossRef]
- Zhou, T.; Li, C.; Jin, H.; Lian, Y.; Han, W. Effective Adsorption/Reduction of Cr(VI) Oxyanion by Halloysite@Polyaniline Hybrid Nanotubes. ACS Appl. Mater. Interfaces 2017, 9, 6030–6043. [Google Scholar] [CrossRef]
- Liu, M.; Jia, Z.; Jia, D.; Zhou, C. Recent advance in research on halloysite nanotubes-polymer nanocomposite. Prog. Polym. Sci. 2014, 39, 1498–1525. [Google Scholar] [CrossRef]
- Gaaz, T.; Sulong, A.; Kadhum, A.; Al-Amiery, A.; Nassir, M.; Jaaz, A. The Impact of Halloysite on the Thermo-Mechanical Properties of Polymer Composites. Molecules 2017, 22, 838. [Google Scholar] [CrossRef]
- Galiano, F.; Mancuso, R.; Carraro, M.; Bundschuh, J.; Hoinkis, J.; Bonchio, M.; De Luca, G.; Gabriele, B.; Figoli, A. A polyoxometalate-based self-cleaning smart material with oxygenic activity for water remediation with membrane technology. Appl. Mater. Today 2021, 23, 101002. [Google Scholar] [CrossRef]
- Yu, J.; Boudjelida, S.; Galiano, F.; Figoli, A.; Bonchio, M.; Carraro, M. Porous Polymeric Membranes Doped with Halloysite Nanotubes and Oxygenic Polyoxometalates. Adv. Mater. Interfaces 2022, 9, 2102152. [Google Scholar] [CrossRef]
- Mishra, G.; Mukhopadhyay, M. TiO2 decorated functionalized halloysite nanotubes (TiO2@HNTs) and photocatalytic PVC membranes synthesis, characterization and its application in water treatment. Sci. Rep. 2019, 9, 4345. [Google Scholar] [CrossRef] [PubMed]
- Renz, P.; Kokkinopoulou, M.; Landfester, K.; Lieberwirth, I. Imaging of Polymeric Nanoparticles: Hard Challenge for Soft Objects. Macromol. Chem. Phys. 2016, 217, 1879–1885. [Google Scholar] [CrossRef]
- Garcia-Garcia, D.; Ferri, J.M.; Ripoll, L.; Hidalgo, M.; Lopez-Martinez, J.; Balart, R. Characterization of selectively etched halloysite nanotubes by acid treatment. Appl. Surf. Sci. 2017, 422, 616–625. [Google Scholar] [CrossRef]
- Bertin, N.; Spelman, T.A.; Combriat, T.; Hue, H.; Stéphan, O.; Lauga, E.; Marmottant, P. Bubble-based acoustic micropropulsors: Active surfaces and mixers. Lab Chip 2017, 17, 1515–1528. [Google Scholar] [CrossRef]
- Pacheco, F.; Sougrat, R.; Reinhard, M.; Leckie, J.O.; Pinnau, I. 3D visualization of the internal nanostructure of polyamide thin films in RO membranes. J. Memb. Sci. 2016, 501, 33–44. [Google Scholar] [CrossRef]
- Sundaramoorthi, G.; Hadwiger, M.; Ben-Romdhane, M.; Behzad, A.R.; Madhavan, P.; Nunes, S.P. 3D Membrane Imaging and Porosity Visualization. Ind. Eng. Chem. Res. 2016, 55, 3689–3695. [Google Scholar] [CrossRef]
- Reingruber, H.; Zankel, A.; Mayrhofer, C.; Poelt, P. Quantitative characterization of microfiltration membranes by 3D reconstruction. J. Memb. Sci. 2011, 372, 66–74. [Google Scholar] [CrossRef]
- Calcagnile, L.; Quarta, G.; D’Elia, M.; Muscogiuri, D.; Maruccio, L.; Butalag, K.; Gianfrate, G.; Sanapo, C.; Toma, U. Instrumental developments at the IBA-AMS dating facility at the University of Lecce. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2005, 240, 22–25. [Google Scholar] [CrossRef]
- Calcagnile, L.; Maruccio, L.; Scrimieri, L.; delle Side, D.; Braione, E.; D’Elia, M.; Quarta, G. Development and application of facilities at the Centre for Applied Physics, Dating and Diagnostics (CEDAD) at the University of Salento during the last 15 years. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2019, 456, 252–256. [Google Scholar] [CrossRef]
- Johansson, S.A.E.; Johansson, T.B. Analytical application of particle induced X-ray emission. Nucl. Instruments Methods 1976, 137, 473–516. [Google Scholar] [CrossRef]
- Lefevre, H.W.; Schofield, R.M.S.; Bench, G.S.; Legge, G.J.F. STIM with energy loss contrast: An imaging modality unique to MeV ions. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 1991, 54, 363–370. [Google Scholar] [CrossRef]
- Chen, X.; Chen, C.-B.; Udalagama, C.N.B.; Ren, M.; Fong, K.E.; Yung, L.Y.L.; Giorgia, P.; Bettiol, A.A.; Watt, F. High-Resolution 3D Imaging and Quantification of Gold Nanoparticles in a Whole Cell Using Scanning Transmission Ion Microscopy. Biophys. J. 2013, 104, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Alsalhy, Q.F.; Salih, H.A.; Simone, S.; Zablouk, M.; Drioli, E.; Figoli, A. Poly(ether sulfone) (PES) hollow-fiber membranes prepared from various spinning parameters. Desalination 2014, 345, 21–35. [Google Scholar] [CrossRef]
- Ursino, C.; Russo, F.; Ferrari, R.M.; De Santo, M.P.; Di Nicolò, E.; He, T.; Galiano, F.; Figoli, A. Polyethersulfone hollow fiber membranes prepared with Polarclean® as a more sustainable solvent. J. Memb. Sci. 2020, 608, 118216. [Google Scholar] [CrossRef]
- Razali, N.F.; Mohammad, A.W.; Hilal, N.; Leo, C.P.; Alam, J. Optimisation of polyethersulfone/polyaniline blended membranes using response surface methodology approach. Desalination 2013, 311, 182–191. [Google Scholar] [CrossRef]
- Yuan, P.; Tan, D.; Aannabi-Bergaya, F.; Yan, W.; Fan, M.; Liu, D.; He, H. Changes in Structure, Morphology, Porosity, and Surface Activity Of Mesoporous Halloysite Nanotubes Under Heating. Clays Clay Miner. 2012, 60, 561–573. [Google Scholar] [CrossRef]
- Pereira, M.R.; Yarwood, J. ATR-FTIR spectroscopic studies of the structure and permeability of sulfonated poly(ether sulfone) membranes. Part 2.—Water diffusion processes. J. Chem. Soc., Faraday Trans. 1996, 92, 2737–2743. [Google Scholar] [CrossRef]
- Belfer, S.; Fainchtain, R.; Purinson, Y.; Kedem, O. Surface characterization by FTIR-ATR spectroscopy of polyethersulfone membranes-unmodified, modified and protein fouled. J. Memb. Sci. 2000, 172, 113–124. [Google Scholar] [CrossRef]
- Galiano, F.; Figoli, A.; Deowan, S.A.; Johnson, D.; Altinkaya, S.A.; Veltri, L.; De Luca, G.; Mancuso, R.; Hilal, N.; Gabriele, B.; et al. A step forward to a more efficient wastewater treatment by membrane surface modification via polymerizable bicontinuous microemulsion. J. Memb. Sci. 2015, 482, 103–114. [Google Scholar] [CrossRef]
- Noel Jacob, K.; Senthil Kumar, S.; Thanigaivelan, A.; Tarun, M.; Mohan, D. Sulfonated polyethersulfone-based membranes for metal ion removal via a hybrid process. J. Mater. Sci. 2014, 49, 114–122. [Google Scholar] [CrossRef]
- Wenzel, R.N. Resistance of solid surfaces to wetting by water. Ind. Eng. Chem. 1936, 28, 988–994. [Google Scholar] [CrossRef]
- Chu, Z.; Chen, K.; Xiao, C.; Ling, H.; Hu, Z. Performance improvement of polyethersulfone ultrafiltration membrane containing variform inorganic nano-additives. Polymer 2020, 188, 122160. [Google Scholar] [CrossRef]
- Gzara, L.; Rehan, Z.A.; Simone, S.; Galiano, F.; Hassankiadeh, N.T.; Al-Sharif, S.F.; Figoli, A.; Drioli, E. Tailoring PES membrane morphology and properties via selected preparation parameters. J. Polym. Eng. 2017, 37, 69–81. [Google Scholar] [CrossRef]
- Russo, F.; Galiano, F.; Pedace, F.; Aricò, F.; Figoli, A. Dimethyl Isosorbide as a Green Solvent for Sustainable Ultrafiltration and Microfiltration Membrane Preparation. ACS Sustain. Chem. Eng. 2020, 8, 659–668. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Liu, J.; Zhang, H.; Wang, K. Preparation and antibacterial property of polyethersulfone ultrafiltration hybrid membrane containing halloysite nanotubes loaded with copper ions. Chem. Eng. J. 2012, 210, 298–308. [Google Scholar] [CrossRef]
- Kamal, N.; Ahzi, S.; Kochkodan, V. Polysulfone/halloysite composite membranes with low fouling properties and enhanced compaction resistance. Appl. Clay Sci. 2020, 199, 105873. [Google Scholar] [CrossRef]
- Roberge, H.; Moreau, P.; Couallier, E.; Abellan, P. Determination of the key structural factors affecting permeability and selectivity of PAN and PES polymeric filtration membranes using 3D FIB/SEM. J. Memb. Sci. 2022, 653, 120530. [Google Scholar] [CrossRef]
- Drioli, E.; Ali, A.; Simone, S.; MacEdonio, F.; Al-Jlil, S.A.; Al Shabonah, F.S.; Al-Romaih, H.S.; Al-Harbi, O.; Figoli, A.; Criscuoli, A. Novel PVDF hollow fiber membranes for vacuum and direct contact membrane distillation applications. Sep. Purif. Technol. 2013, 115, 27–38. [Google Scholar] [CrossRef]
- Chae Park, H.; Po Kim, Y.; Yong Kim, H.; Soo Kang, Y. Membrane formation by water vapor induced phase inversion. J. Memb. Sci. 1999, 156, 169–178. [Google Scholar] [CrossRef]
- Hołda, A.K.; Vankelecom, I.F.J. Understanding and guiding the phase inversion process for synthesis of solvent resistant nanofiltration membranes. J. Appl. Polym. Sci. 2015, 132, 42130. [Google Scholar] [CrossRef]
- Russo, F.; Tiecco, M.; Galiano, F.; Mancuso, R.; Gabriele, B.; Figoli, A. Launching deep eutectic solvents (DESs) and natural deep eutectic solvents (NADESs), in combination with different harmless co-solvents, for the preparation of more sustainable membranes. J. Memb. Sci. 2022, 649, 120387. [Google Scholar] [CrossRef]
- Kamal, N.; Kochkodan, V.; Zekri, A.; Ahzi, S. Polysulfone Membranes Embedded with Halloysites Nanotubes: Preparation and Properties. Membranes 2019, 10, 2. [Google Scholar] [CrossRef]
- Subtil, E.L.; Gonçalves, J.; Lemos, H.G.; Venancio, E.C.; Mierzwa, J.C.; dos Santos de Souza, J.; Alves, W.; Le-Clech, P. Preparation and characterization of a new composite conductive polyethersulfone membrane using polyaniline (PANI) and reduced graphene oxide (rGO). Chem. Eng. J. 2020, 390, 124612. [Google Scholar] [CrossRef]
- Poletti, G.; Orsini, F.; Lenardi, C.; Barborini, E. A comparative study between AFM and SEM imaging on human scalp hair. J. Microsc. 2003, 211, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Sadamatsu, S.; Tanaka, M.; Higashida, K.; Matsumura, S. Transmission electron microscopy of bulk specimens over 10 µm in thickness. Ultramicroscopy 2016, 162, 10–16. [Google Scholar] [CrossRef]
- Brandenberger, C.; Clift, M.J.; Vanhecke, D.; Mühlfeld, C.; Stone, V.; Gehr, P.; Rothen-Rutishauser, B. Intracellular imaging of nanoparticles: Is it an elemental mistake to believe what you see? Part. Fibre Toxicol. 2010, 7, 15. [Google Scholar] [CrossRef]
- Wagner, O.; Schultz, M.; Edri, E.; Meir, R.; Barnoy, E.; Meiri, A.; Shpaisman, H.; Sloutskin, E.; Zalevsky, Z. Imaging of nanoparticle dynamics in live and apoptotic cells using temporally-modulated polarization. Sci. Rep. 2019, 9, 1650. [Google Scholar] [CrossRef]
- Michler, G.H. Problems Associated with the Electron Microscopy of Polymers. In Electron Microscopy of Polymers; Springer: Berlin/Heidelberg, Germany, 2016; pp. 175–183. [Google Scholar]
- Michler, G.H.; Lebek, W. Electron Microscopy of Polymers. In Polymer Morphology; Wiley: Hoboken, NJ, USA, 2016; pp. 37–53. [Google Scholar]
- Tajer Mohammad Ghazvini, P.; Ghorbanzadeh Mashkani, S. Screening of bacterial cells for biosorption of oxyanions: Application of micro-PIXE for measurement of biosorption. Hydrometallurgy 2009, 96, 246–252. [Google Scholar] [CrossRef]
- Sohbatzadeh, H.; Keshtkar, A.R.; Safdari, J.; Yousefi, T.; Fatemi, F. Insights into the biosorption mechanisms of U(VI) by chitosan bead containing bacterial cells: A supplementary approach using desorption eluents, chemical pretreatment and PIXE–RBS analyses. Chem. Eng. J. 2017, 323, 492–501. [Google Scholar] [CrossRef]
- He, X.; Shen, H.; Chen, Z.; Rong, C.; Ren, M.; Hou, L.; Wu, C.; Mao, L.; Lu, Q.; Su, B. Element-based prognostics of occupational pneumoconiosis using micro-proton-induced X-ray emission analysis. Am. J. Physiol. Cell Mol. Physiol. 2017, 313, L1154–L1163. [Google Scholar] [CrossRef]
- Ynsa, M.D.; Ripoll-Sau, J.; Osipowicz, T.; Breese, M.B.H.; Ren, M. Micro-PIXE with 3.5 MeV protons for the study of low copper concentrations in atherosclerotic artery. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2021, 497, 34–38. [Google Scholar] [CrossRef]
- Mulware, S.J. The Review of Nuclear Microscopy Techniques: An Approach for Nondestructive Trace Elemental Analysis and Mapping of Biological Materials. J. Biophys. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.; Chen, X.; Chen, C.-B.; Udalagama, C.; van Kan, J.A.; Bettiol, A.A. Whole cell structural imaging at 20 nanometre resolutions using MeV ions. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2013, 306, 6–11. [Google Scholar] [CrossRef]
- Carmona, N.; Ortega-Feliu, I.; Gómez-Tubío, B.; Villegas, M.A. Advantages and disadvantages of PIXE/PIGE, XRF and EDX spectrometries applied to archaeometric characterisation of glasses. Mater. Charact. 2010, 61, 257–267. [Google Scholar] [CrossRef]
- Linke, R.; Demoriter, G.; Schreiner, M.; Alram, M.; Martinek, K.P.; Spindler, P. The superiority of PIXE compared to EDXRF, SEM/EDX and ICP-MS determining the provenance of medieval silver coins. In Proceedings of the 3rd International Symposium on “Nuclear and Related Techniques”, Havanna, Cuba, 22–26 October 2001. [Google Scholar]
- Yamamoto, Y.; Yamamoto, K. Precise XPS depth profile of soda-lime-silica glass using C60 ion beam. J. Non. Cryst. Solids 2010, 356, 14–18. [Google Scholar] [CrossRef]
- Sotiropoulou, S.; Papliaka, Z.E.; Vaccari, L. Micro FTIR imaging for the investigation of deteriorated organic binders in wall painting stratigraphies of different techniques and periods. Microchem. J. 2016, 124, 559–567. [Google Scholar] [CrossRef]
- Barceló, D.; Farré, M. Analysis and Risk of Nanomaterials in Environmental and Food Samples, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2012; ISBN 9780444563286978-0-444-56328-6. [Google Scholar]
- Dubes, A.; Parrot-Lopez, H.; Abdelwahed, W.; Degobert, G.; Fessi, H.; Shahgaldian, P.; Coleman, A.W. Scanning electron microscopy and atomic force microscopy imaging of solid lipid nanoparticles derived from amphiphilic cyclodextrins. Eur. J. Pharm. Biopharm. 2003, 55, 279–282. [Google Scholar] [CrossRef]
- Ansari, K.; van Kan, J.A.; Bettiol, A.A.; Watt, F. Fabrication of high aspect ratio 100nm metallic stamps for nanoimprint lithography using proton beam writing. Appl. Phys. Lett. 2004, 85, 476–478. [Google Scholar] [CrossRef]
- Khare, S.P. Introduction to the Theory of Collisions of Electrons with Atoms and Molecules; Springer: Boston, MA, USA, 2001; ISBN 978-1-4613-5157-3. [Google Scholar]
- van Kan, J.A.; Shao, P.G.; Ansari, K.; Bettiol, A.A.; Osipowicz, T.; Watt, F. Proton beam writing: A tool for high-aspect ratio mask production. Microsyst. Technol. 2007, 13, 431–434. [Google Scholar] [CrossRef]
- Furuta, Y.; Nishikawa, H.; Satoh, T.; Ishii, Y.; Kamiya, T.; Nakao, R.; Uchida, S. Applications of microstructures fabricated by proton beam writing to electric-micro filters. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2009, 267, 2285–2288. [Google Scholar] [CrossRef]
- Shah, N.; Singh, D.; Shah, S.; Qureshi, A.; Singh, N.L.; Singh, K.P. Study of microhardness and electrical properties of proton irradiated polyethersulfone (PES). Bull. Mater. Sci. 2007, 30, 477–480. [Google Scholar] [CrossRef]
- Seguchi, T.; Sasuga, T.; Kawakami, W.; Hagiwar, M.; Kohno, I.; Kamitsubo, H. Proton irradiation effects on organic polymers. In Proceedings of the Eleventh International Conference on Cyclotrons and Their Applications, Tokyo, Japan, 13–17 October 1986. [Google Scholar]
- Abramoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane (Dopant wt%) | |||||
|---|---|---|---|---|---|
| PES | PES + PANI (1%) | PES + HNTs (1%) | PES + [HNTs@PANI] (1%) | ||
| Thickness (μm) | 50 | 110 | 80 | 129 | |
| WCA (°) | 70 ± 3 | 68 ± 1 | 69 ± 1 | 73 ± 3 | |
| Pore size (μm) | 0.14 ± 0.01 | 0.52 ± 0.07 | 0.74 ± 0.01 | 1.91 ± 0.10 | |
| Porosity (%) | 90 ± 1 | 86 ± 2 | 92 ± 1 | 89 ± 2 | |
| Mechanical properties | Young’s Modulus (MPa) | 45 ± 6 | 54 ± 4 | 44 ± 3 | 43 ± 6 |
| Elongation at break (%) | 4 ± 1 | 9 ± 2 | 7 ± 2 | 5 ± 2 | |
| Water permeance (L/m2 h bar) | 1.8 ± 1.0 | 1.4 ± 0.5 | 1.6 ± 0.7 | 3.4 ± 0.5 | |
| µ-PIXE Area | Volume (%) | Density (%) | ||
|---|---|---|---|---|
| PES + HNTs (1%) | PES + [HNTs@PANI] (1%) | PES + HNTs (1%) | PES + [HNTs@PANI] (1%) | |
| 1 mm × 1 mm | 6% | 5% | 0.75 | 0.45 |
| 500 µm × 500 µm | 6% | 5% | 0.75 | 0.45 |
| 100 µm × 100µm | 5% | 5% | 0.62 | 0.45 |
| 50 µm × 50 µm | 5% | 4% | 0.62 | 0.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasco, G.; Arima, V.; Boudjelida, S.; Carraro, M.; Bianco, M.; Zizzari, A.; Perrone, E.; Galiano, F.; Figoli, A.; Cesaria, M. Polymeric Membranes Doped with Halloysite Nanotubes Imaged using Proton Microbeam Microscopy. Nanomaterials 2023, 13, 2970. https://doi.org/10.3390/nano13222970
Vasco G, Arima V, Boudjelida S, Carraro M, Bianco M, Zizzari A, Perrone E, Galiano F, Figoli A, Cesaria M. Polymeric Membranes Doped with Halloysite Nanotubes Imaged using Proton Microbeam Microscopy. Nanomaterials. 2023; 13(22):2970. https://doi.org/10.3390/nano13222970
Chicago/Turabian StyleVasco, Giovanna, Valentina Arima, Soufiane Boudjelida, Mauro Carraro, Monica Bianco, Alessandra Zizzari, Elisabetta Perrone, Francesco Galiano, Alberto Figoli, and Maura Cesaria. 2023. "Polymeric Membranes Doped with Halloysite Nanotubes Imaged using Proton Microbeam Microscopy" Nanomaterials 13, no. 22: 2970. https://doi.org/10.3390/nano13222970
APA StyleVasco, G., Arima, V., Boudjelida, S., Carraro, M., Bianco, M., Zizzari, A., Perrone, E., Galiano, F., Figoli, A., & Cesaria, M. (2023). Polymeric Membranes Doped with Halloysite Nanotubes Imaged using Proton Microbeam Microscopy. Nanomaterials, 13(22), 2970. https://doi.org/10.3390/nano13222970