

Strained Monolayer MoTe2 as a Photon Absorber in the Telecom Range



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Unstrained MoTe2: Structural and Electronic Properties
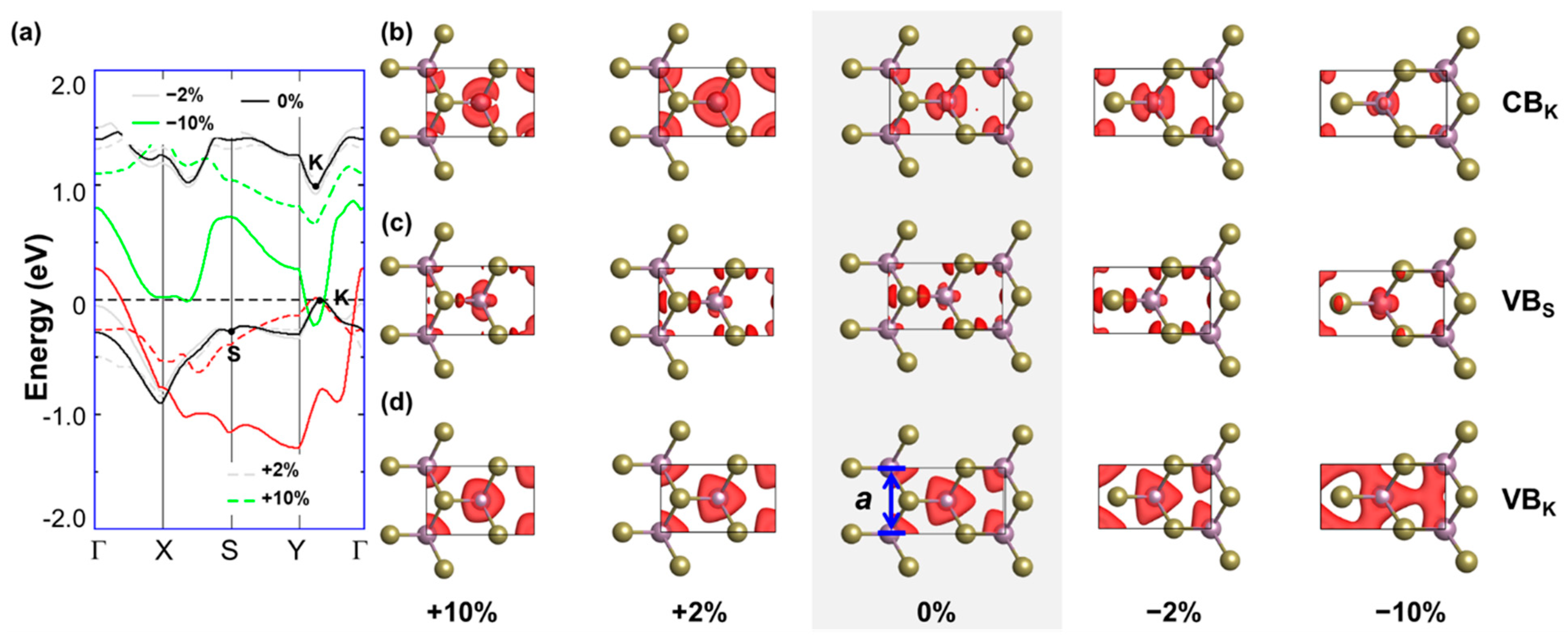
3.2. Electronic Structure of Uniaxially Strained MoTe2
3.3. Charge-Density Distribution and Structure–Property Relationship
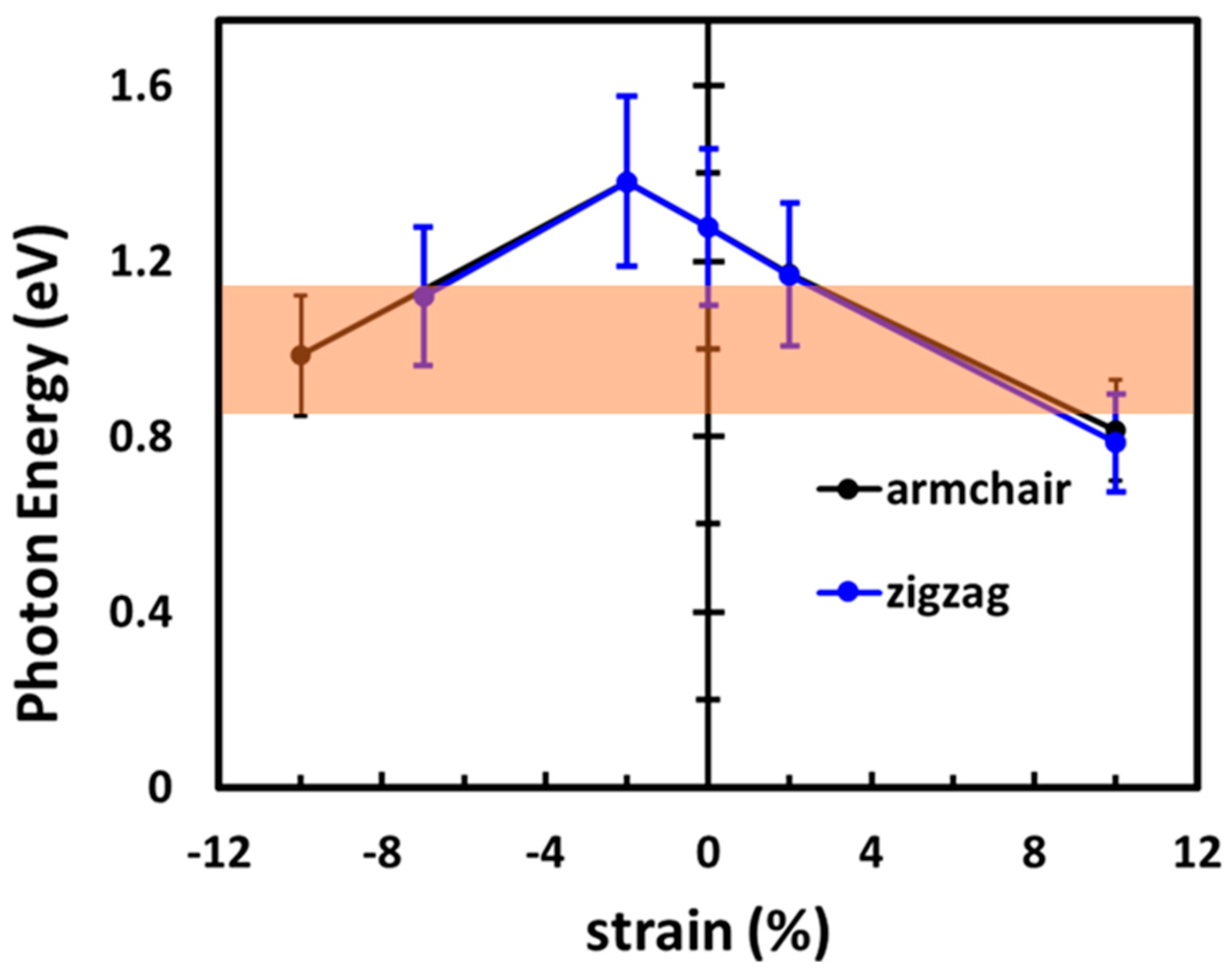
3.4. Photon Absorption in the Telecom Range
4. Discussion
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Nag, A.; Raidongia, K.; Hembram, K.P.S.S.; Datta, R.; Waghmare, U.V.; Rao, C.N.R. Graphene Analogues of BN: Novel Synthesis and Properties. ACS Nano 2010, 4, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kong, X.; Hu, Z.-X.; Yang, F.; Ji, W. High-Mobility Transport Anisotropy and Linear Dichroism in Few-Layer Black Phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef]
- Zhu, Z.; Tománek, D. Semiconducting Layered Blue Phosphorus: A Computational Study. Phys. Rev. Lett. 2014, 112, 176802. [Google Scholar] [CrossRef] [PubMed]
- Bhimanapati, G.R.; Lin, Z.; Meunier, V.; Jung, Y.; Cha, J.; Das, S.; Xiao, D.; Son, Y.; Strano, M.S.; Cooper, V.R.; et al. Recent Advances in Two-Dimensional Materials beyond Graphene. ACS Nano 2015, 9, 11509–11539. [Google Scholar] [CrossRef]
- Heine, T. Transition Metal Chalcogenides: Ultrathin Inorganic Materials with Tunable Electronic Properties. Acc. Chem. Res. 2015, 48, 65–72. [Google Scholar] [CrossRef]
- Lv, R.; Terrones, H.; Elías, A.L.; Perea-López, N.; Gutiérrez, H.R.; Cruz-Silva, E.; Rajukumar, L.P.; Dresselhaus, M.S.; Terrones, M. Two-Dimensional Transition Metal Dichalcogenides: Clusters, Ribbons, Sheets and More. Nano Today 2015, 10, 559–592. [Google Scholar] [CrossRef]
- Choi, W.; Choudhary, N.; Han, G.H.; Park, J.; Akinwande, D.; Lee, Y.H. Recent Development of Two-Dimensional Transition Metal Dichalcogenides and Their Applications. Mater. Today 2017, 20, 116–130. [Google Scholar] [CrossRef]
- VahidMohammadi, A.; Rosen, J.; Gogotsi, Y. The World of Two-Dimensional Carbides and Nitrides (MXenes). Science 2021, 372, eabf1581. [Google Scholar] [CrossRef]
- Zhuang, R.; Cai, S.; Mei, Z.; Liang, H.; Zhao, N.; Mu, H.; Yu, W.; Jiang, Y.; Yuan, J.; Lau, S.; et al. Solution-Grown BiI/BiI3 van Der Waals Heterostructures for Sensitive X-ray Detection. Nat. Commun. 2023, 14, 1621. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Ma, Y.; Li, Y.; Heine, T. GeP3: A Small Indirect Band Gap 2D Crystal with High Carrier Mobility and Strong Interlayer Quantum Confinement. Nano Lett. 2017, 17, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Meng, F.; Wang, H.; Wang, H.; Ni, Y. Novel Two-Dimensional Semiconductor SnP 3: High Stability, Tunable Bandgaps and High Carrier Mobility Explored Using First-Principles Calculations. J. Mater. Chem. A 2018, 6, 11890–11897. [Google Scholar] [CrossRef]
- Ramzan, M.S.; Bacic, V.; Jing, Y.; Kuc, A. Electronic Properties of a New Family of Layered Materials from Groups 14 and 15: First-Principles Simulations. J. Phys. Chem. C 2019, 123, 25470–25476. [Google Scholar] [CrossRef]
- Wang, S.-S.; Sun, W.; Dong, S. Quantum Spin Hall Insulators and Topological Rashba-Splitting Edge States in Two-Dimensional CX3 (X = Sb, Bi). Phys. Chem. Chem. Phys. 2021, 23, 2134–2140. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS_2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-Layer MoS2 Transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Qian, X.; Liu, J.; Fu, L.; Li, J. Quantum Spin Hall Effect in Two-Dimensional Transition Metal Dichalcogenides. Science 2014, 346, 1344–1347. [Google Scholar] [CrossRef]
- Mak, K.F.; Shan, J. Photonics and Optoelectronics of 2D Semiconductor Transition Metal Dichalcogenides. Nat. Photonics 2016, 10, 216–226. [Google Scholar] [CrossRef]
- Xu, R.; Guo, J.; Mi, S.; Wen, H.; Pang, F.; Ji, W.; Cheng, Z. Advanced atomic force microscopies and their applications in two-dimensional materials: A review. Mater. Futures 2022, 1, 032302. [Google Scholar] [CrossRef]
- Li, Y.; An, N.; Lu, Z.; Wang, Y.; Chang, B.; Tan, T.; Guo, X.; Xu, X.; He, J.; Xia, H.; et al. Nonlinear Co-Generation of Graphene Plasmons for Optoelectronic Logic Operations. Nat. Commun. 2022, 13, 3138. [Google Scholar] [CrossRef]
- Guo, Z.; Zeng, Y.; Meng, F.; Qu, H.; Zhang, S.; Hu, S.; Fan, S.; Zeng, H.; Cao, R.; Prasad, P.N.; et al. In-Situ Neutron-Transmutation for Substitutional Doping in 2D Layered Indium Selenide Based Phototransistor. eLight 2022, 2, 9. [Google Scholar] [CrossRef]
- Ahn, G.H.; Amani, M.; Rasool, H.; Lien, D.-H.; Mastandrea, J.P.; Ager III, J.W.; Dubey, M.; Chrzan, D.C.; Minor, A.M.; Javey, A. Strain-Engineered Growth of Two-Dimensional Materials. Nat. Commun. 2017, 8, 608. [Google Scholar] [CrossRef]
- Kou, L.; Frauenheim, T.; Chen, C. Nanoscale Multilayer Transition-Metal Dichalcogenide Heterostructures: Band Gap Modulation by Interfacial Strain and Spontaneous Polarization. J. Phys. Chem. Lett. 2013, 4, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Huang, J.-K.; Chuu, C.-P.; Hsu, W.-T.; Lee, C.-J.; Aljarb, A.; Huang, C.-W.; Chiu, M.-H.; Tang, H.-L.; Lin, C.; et al. Strain-Directed Layer-By-Layer Epitaxy Toward van Der Waals Homo- and Heterostructures. ACS Mater. Lett. 2021, 3, 442–453. [Google Scholar] [CrossRef]
- Ramzan, M.S.; Kunstmann, J.; Kuc, A.B. Tuning Valleys and Wave Functions of van Der Waals Heterostructures by Varying the Number of Layers: A First-Principles Study. Small 2021, 27, 2008153. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Yu, W.; Yuan, J.; Lin, S.; Zhang, G. Interface and Surface Engineering of Black Phosphorus: A Review for Optoelectronic and Photonic Applications. Mater. Futures 2022, 1, 012301. [Google Scholar] [CrossRef]
- Mueller, T.; Malic, E. Exciton Physics and Device Application of Two-Dimensional Transition Metal Dichalcogenide Semiconductors. NPJ 2D Mater. Appl. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Conley, H.J.; Wang, B.; Ziegler, J.I.; Haglund, R.F.; Pantelides, S.T.; Bolotin, K.I. Bandgap Engineering of Strained Monolayer and Bilayer MoS2. Nano Lett. 2013, 13, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.B.; Seol, G.; Kang, J.S.; Fang, H.; Battaglia, C.; Kapadia, R.; Ager, J.W.; Guo, J.; Javey, A. Strain-Induced Indirect to Direct Bandgap Transition in Multilayer WSe2. Nano Lett. 2014, 14, 4592–4597. [Google Scholar] [CrossRef]
- Wang, Y.; Cong, C.; Yang, W.; Shang, J.; Peimyoo, N.; Chen, Y.; Kang, J.; Wang, J.; Huang, W.; Yu, T. Strain-Induced Direct–Indirect Bandgap Transition and Phonon Modulation in Monolayer WS2. Nano Res. 2015, 8, 2562–2572. [Google Scholar] [CrossRef]
- Island, J.O.; Kuc, A.; Diependaal, E.H.; Bratschitsch, R.; Van Der Zant, H.S.J.; Heine, T.; Castellanos-Gomez, A. Precise and Reversible Band Gap Tuning in Single-Layer MoSe2 by Uniaxial Strain. Nanoscale 2016, 8, 2589–2593. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.; Liu, X.; Christopher, J.W.; Cantley, L.; Wadehra, A.; Kim, B.L.; Goldberg, B.B.; Swan, A.K.; Bunch, J.S. Band Gap Engineering with Ultralarge Biaxial Strains in Suspended Monolayer MoS2. Nano Lett. 2016, 16, 5836–5841. [Google Scholar] [CrossRef] [PubMed]
- Aslan, B.; Deng, M.; Heinz, T.F. Strain Tuning of Excitons in Monolayer WSe2. Phys. Rev. B 2018, 98, 115308. [Google Scholar] [CrossRef]
- Aslan, B.; Datye, I.M.; Mleczko, M.J.; Sze Cheung, K.; Krylyuk, S.; Bruma, A.; Kalish, I.; Davydov, A.V.; Pop, E.; Heinz, T.F. Probing the Optical Properties and Strain-Tuning of Ultrathin Mo1–xWx Te2. Nano Lett. 2018, 18, 2485–2491. [Google Scholar] [CrossRef]
- Blundo, E.; Felici, M.; Yildirim, T.; Pettinari, G.; Tedeschi, D.; Miriametro, A.; Liu, B.; Ma, W.; Lu, Y.; Polimeni, A. Evidence of the Direct-to-Indirect Band Gap Transition in Strained Two-Dimensional WS2, MoS2, and WSe2. Phys. Rev. Res. 2020, 2, 012024. [Google Scholar] [CrossRef]
- Yu, Y.; Dong, C.-D.; Binder, R.; Schumacher, S.; Ning, C.-Z. Strain-Induced Indirect-to-Direct Bandgap Transition, Photoluminescence Enhancement, and Linewidth Reduction in Bilayer MoTe2. ACS Nano 2023, 17, 4230–4238. [Google Scholar] [CrossRef]
- Maiti, R.; Saadi, M.A.S.R.; Amin, R.; Ozcelik, V.O.; Uluutku, B.; Patil, C.; Suer, C.; Solares, S.; Sorger, V.J. Strain-Induced Spatially Resolved Charge Transport in 2H-MoTe2. ACS Appl. Electron. Mater. 2021, 3, 3781–3788. [Google Scholar] [CrossRef]
- Shafique, A.; Shin, Y.-H. Strain Engineering of Phonon Thermal Transport Properties in Monolayer 2H-MoTe2. Phys. Chem. Chem. Phys. 2017, 19, 32072–32078. [Google Scholar] [CrossRef]
- Maniadaki, A.E.; Kopidakis, G.; Remediakis, I.N. Strain Engineering of Electronic Properties of Transition Metal Dichalcogenide Monolayers. Solid State Commun. 2016, 227, 33–39. [Google Scholar] [CrossRef]
- Kansara, S.; Gupta, S.K.; Sonvane, Y. Effect of Strain Engineering on 2D Dichalcogenides Transition Metal: A DFT Study. Comput. Mater. Sci. 2018, 141, 235–242. [Google Scholar] [CrossRef]
- Johari, P.; Shenoy, V.B. Tuning the Electronic Properties of Semiconducting Transition Metal Dichalcogenides by Applying Mechanical Strains. ACS Nano 2012, 6, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Fan, X.; Singh, D.J.; Chen, H.; Jiang, Q.; Zheng, W.T. Controlling Phase Transition for Single-Layer MTe2 (M = Mo and W): Modulation of the Potential Barrier under Strain. Phys. Chem. Chem. Phys. 2016, 18, 4086–4094. [Google Scholar] [CrossRef] [PubMed]
- Khatibi, Z.; Feierabend, M.; Selig, M.; Brem, S.; Linderälv, C.; Erhart, P.; Malic, E. Impact of Strain on the Excitonic Linewidth in Transition Metal Dichalcogenides. 2D Mater. 2018, 6, 015015. [Google Scholar] [CrossRef]
- Tran, T.N.; Dang, M.T.; Tran, Q.H.; Luong, T.T.; Dinh, V.A. Band Valley Modification under Strain in Monolayer WSe2. AIP Adv. 2022, 12, 115023. [Google Scholar] [CrossRef]
- Ruppert, C.; Aslan, B.; Heinz, T.F. Optical Properties and Band Gap of Single- and Few-Layer MoTe2 Crystals. Nano Lett. 2014, 14, 6231–6236. [Google Scholar] [CrossRef]
- Edalati-Boostan, S.; Cocchi, C.; Draxl, C. MoTe2 as a Natural Hyperbolic Material across the Visible and the Ultraviolet Region. Phys. Rev. Mater. 2020, 4, 085202. [Google Scholar] [CrossRef]
- Wang, K.; Feng, Y.; Chang, C.; Zhan, J.; Wang, C.; Zhao, Q.; Coleman, J.N.; Zhang, L.; Blau, W.J.; Wang, J. Broadband Ultrafast Nonlinear Absorption and Nonlinear Refraction of Layered Molybdenum Dichalcogenide Semiconductors. Nanoscale 2014, 6, 10530–10535. [Google Scholar] [CrossRef]
- Quan, C.; He, M.; He, C.; Huang, Y.; Zhu, L.; Yao, Z.; Xu, X.; Lu, C.; Xu, X. Transition from Saturable Absorption to Reverse Saturable Absorption in MoTe2 Nano-Films with Thickness and Pump Intensity. Appl. Surf. Sci. 2018, 457, 115–120. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Yin, L.; Huang, Y.; Xu, K.; Wang, F.; Zhan, X.; He, J. Electrostatically Tunable Lateral MoTe2 p–n Junction for Use in High-Performance Optoelectronics. Nanoscale 2016, 8, 13245–13250. [Google Scholar] [CrossRef]
- Bie, Y.-Q.; Grosso, G.; Heuck, M.; Furchi, M.M.; Cao, Y.; Zheng, J.; Bunandar, D.; Navarro-Moratalla, E.; Zhou, L.; Efetov, D.K.; et al. A MoTe2-Based Light-Emitting Diode and Photodetector for Silicon Photonic Integrated Circuits. Nat. Nanotechnol. 2017, 12, 1124–1129. [Google Scholar] [CrossRef]
- Helmrich, S.; Schneider, R.; Achtstein, A.W.; Arora, A.; Herzog, B.; de Vasconcellos, S.M.; Kolarczik, M.; Schöps, O.; Bratschitsch, R.; Woggon, U.; et al. Exciton–Phonon Coupling in Mono- and Bilayer MoTe2. 2D Mater. 2018, 5, 045007. [Google Scholar] [CrossRef]
- Zheng, W.; Bonn, M.; Wang, H.I. Photoconductivity Multiplication in Semiconducting Few-Layer MoTe2. Nano Lett. 2020, 20, 5807–5813. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Liu, F.; Cuadra, J.; Huang, Z.; Li, K.; Rasmita, A.; Srivastava, A.; Liu, Z.; Gao, W.-B. Zeeman Splitting via Spin-Valley-Layer Coupling in Bilayer MoTe2. Nat. Commun. 2017, 8, 802. [Google Scholar] [CrossRef] [PubMed]
- Maiti, R.; Patil, C.; Saadi, M.A.S.R.; Xie, T.; Azadani, J.G.; Uluutku, B.; Amin, R.; Briggs, A.F.; Miscuglio, M.; Van Thourhout, D.; et al. Strain-Engineered High-Responsivity MoTe2 Photodetector for Silicon Photonic Integrated Circuits. Nat. Photonics 2020, 14, 578–584. [Google Scholar] [CrossRef]
- Zhao, H.; Pettes, M.T.; Zheng, Y.; Htoon, H. Site-Controlled Telecom-Wavelength Single-Photon Emitters in Atomically-Thin MoTe2. Nat. Commun. 2021, 12, 6753. [Google Scholar] [CrossRef]
- Ye, G.; Xiong, W.; Xie, Y.; Gong, L. The High Photoresponse of Stress-Tuned MoTe2 Optoelectronic Devices in the Telecommunication Band. Phys. Status Solidi (RRL)–Rapid Res. Lett. 2022, 16, 2200276. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Onida, G.; Reining, L.; Rubio, A. Electronic Excitations: Density-Functional versus Many-Body Green’s-Function Approaches. Rev. Mod. Phys. 2002, 74, 601–659. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215, Erratum in J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Shishkin, M.; Kresse, G. Implementation and Performance of the Frequency-Dependent $GW$ Method within the PAW Framework. Phys. Rev. B 2006, 74, 035101. [Google Scholar] [CrossRef]
- Salpeter, E.E.; Bethe, H.A. A Relativistic Equation for Bound-State Problems. Phys. Rev. 1951, 84, 1232–1242. [Google Scholar] [CrossRef]
- Medeiros, P.V.C.; Stafström, S.; Björk, J. Effects of Extrinsic and Intrinsic Perturbations on the Electronic Structure of Graphene: Retaining an Effective Primitive Cell Band Structure by Band Unfolding. Phys. Rev. B 2014, 89, 041407. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, F.W.; Ghosh, A.W. First Principles Study and Empirical Parametrization of Twisted Bilayer MoS2 Based on Band-Unfolding. Appl. Phys. Lett. 2016, 109, 101601. [Google Scholar] [CrossRef]
- Krumland, J.; Cocchi, C. Conditions for Electronic Hybridization between Transition-Metal Dichalcogenide Monolayers and Physisorbed Carbon-Conjugated Molecules. Electron. Struct. 2021, 3, 044003. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Bader, R.F.W. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Oxford University Press: Oxford, UK; New York, NY, USA, 1994; ISBN 978-0-19-855865-1. [Google Scholar]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Trinkle, D.R. Accurate and Efficient Algorithm for Bader Charge Integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Singh, A.K. Semiconductor-Metal Transition in Semiconducting Bilayer Sheets of Transition-Metal Dichalcogenides. Phys. Rev. B 2012, 86, 075454. [Google Scholar] [CrossRef]
- Rasmussen, F.A.; Thygesen, K.S. Computational 2D Materials Database: Electronic Structure of Transition-Metal Dichalcogenides and Oxides. J. Phys. Chem. C 2015, 119, 13169–13183. [Google Scholar] [CrossRef]
- Filip, M.R.; Haber, J.B.; Neaton, J.B. Phonon Screening of Excitons in Semiconductors: Halide Perovskites and Beyond. Phys. Rev. Lett. 2021, 127, 067401. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Zhu, H.; Li, Y.; Alsaid, Y.; Fong, K.Y.; Zhou, Y.; Wang, S.; Shi, W.; Wang, Y.; et al. Structural Phase Transition in Monolayer MoTe2 Driven by Electrostatic Doping. Nature 2017, 550, 487–491. [Google Scholar] [CrossRef]
- Duerloo, K.-A.N.; Li, Y.; Reed, E.J. Structural Phase Transitions in Two-Dimensional Mo- and W-Dichalcogenide Monolayers. Nat. Commun. 2014, 5, 4214. [Google Scholar] [CrossRef] [PubMed]
- Blundo, E.; Di Giorgio, C.; Pettinari, G.; Yildirim, T.; Felici, M.; Lu, Y.; Bobba, F.; Polimeni, A. Engineered Creation of Periodic Giant, Nonuniform Strains in MoS2 Monolayers. Adv. Mater. Interfaces 2020, 7, 2000621. [Google Scholar] [CrossRef]
- Darlington, T.P.; Carmesin, C.; Florian, M.; Yanev, E.; Ajayi, O.; Ardelean, J.; Rhodes, D.A.; Ghiotto, A.; Krayev, A.; Watanabe, K.; et al. Imaging Strain-Localized Excitons in Nanoscale Bubbles of Monolayer WSe2 at Room Temperature. Nat. Nanotechnol. 2020, 15, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lü, T.; Myint, Y.W.; Pei, J.; Macdonald, D.; Zheng, J.-C.; Lu, Y. Robust Excitons and Trions in Monolayer MoTe2. ACS Nano 2015, 9, 6603–6609. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Picard, R.; Lagarde, D.; Wang, G.; Echeverry, J.P.; Cadiz, F.; Renucci, P.; Högele, A.; Amand, T.; Marie, X.; et al. Excitonic Properties of Semiconducting Monolayer and Bilayer MoT e2. Phys. Rev. B 2016, 94, 155425. [Google Scholar] [CrossRef]
- Biswas, S.; Champagne, A.; Haber, J.B.; Pokawanvit, S.; Wong, J.; Akbari, H.; Krylyuk, S.; Watanabe, K.; Taniguchi, T.; Davydov, A.V.; et al. Rydberg Excitons and Trions in Monolayer MoTe2. ACS Nano 2023, 17, 7685–7694. [Google Scholar] [CrossRef]
- Köster, J.; Ghorbani-Asl, M.; Komsa, H.-P.; Lehnert, T.; Kretschmer, S.; Krasheninnikov, A.V.; Kaiser, U. Defect Agglomeration and Electron-Beam-Induced Local-Phase Transformations in Single-Layer MoTe2. J. Phys. Chem. C 2021, 125, 13601–13609. [Google Scholar] [CrossRef]
- Schulzetenberg, A.; Johns, J. Chemical Defects Control the Exciton Lifetime in CVD Grown, Few-Layer MoTe2. Electron. Struct. 2021, 3, 025001. [Google Scholar] [CrossRef]
- Zhang, Y.; Ugeda, M.M.; Jin, C.; Shi, S.-F.; Bradley, A.J.; Martín-Recio, A.; Ryu, H.; Kim, J.; Tang, S.; Kim, Y.; et al. Electronic Structure, Surface Doping, and Optical Response in Epitaxial WSe2 Thin Films. Nano Lett. 2016, 16, 2485–2491. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; He, S.; Ma, F. Semiconductor-Semimetal Transition of MoTe2 Monolayer Modulated by Charge-Injection and Strain Engineering. Chem. Phys. Lett. 2021, 770, 138473. [Google Scholar] [CrossRef]
- Karpińska, M.; Jasiński, J.; Kempt, R.; Ziegler, J.D.; Sansom, H.; Taniguchi, T.; Watanabe, K.; Snaith, H.J.; Surrente, A.; Dyksik, M.; et al. Interlayer Excitons in MoSe2/2D Perovskite Hybrid Heterostructures—The Interplay between Charge and Energy Transfer. Nanoscale 2022, 14, 8085–8095. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Cheng, H.-C.; Wang, Y.; Zhou, B.; Li, D.; Wang, G.; Liu, Y.; Guo, J.; Wu, H.; Kang, D.J.; et al. Lead Halide Perovskite Sensitized WSe2 Photodiodes with Ultrahigh Open Circuit Voltages. eLight 2023, 3, 8. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Zhu, Z.Y.; Tahir, M.; Schwingenschlögl, U. Spin-orbit–induced spin splittings in polar transition metal dichalcogenide monolayers. EPL 2013, 102, 57001. [Google Scholar] [CrossRef]
- Lee, J.H.; Delaney, K.T.; Bousquet, E.; Spaldin, N.A.; Rabe, K.M. Strong coupling of Jahn-Teller distortion to oxygen-octahedron rotation and functional properties in epitaxially strained orthorhombic LaMnO3. Phys. Rev. B 2013, 88, 174426. [Google Scholar] [CrossRef]
- Garcia-Castro, A.C.; Romero, A.H.; Bousquet, E. Strain-Engineered Multiferroicity in P n m a NaMnF3 Fluoroperovskite. Phys. Rev. Lett. 2016, 116, 117202. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramzan, M.S.; Cocchi, C. Strained Monolayer MoTe2 as a Photon Absorber in the Telecom Range. Nanomaterials 2023, 13, 2740. https://doi.org/10.3390/nano13202740
Ramzan MS, Cocchi C. Strained Monolayer MoTe2 as a Photon Absorber in the Telecom Range. Nanomaterials. 2023; 13(20):2740. https://doi.org/10.3390/nano13202740
Chicago/Turabian StyleRamzan, Muhammad Sufyan, and Caterina Cocchi. 2023. "Strained Monolayer MoTe2 as a Photon Absorber in the Telecom Range" Nanomaterials 13, no. 20: 2740. https://doi.org/10.3390/nano13202740
APA StyleRamzan, M. S., & Cocchi, C. (2023). Strained Monolayer MoTe2 as a Photon Absorber in the Telecom Range. Nanomaterials, 13(20), 2740. https://doi.org/10.3390/nano13202740