
Intranasal Administration of Catechol-Based Pt(IV) Coordination Polymer Nanoparticles for Glioblastoma Therapy


 , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
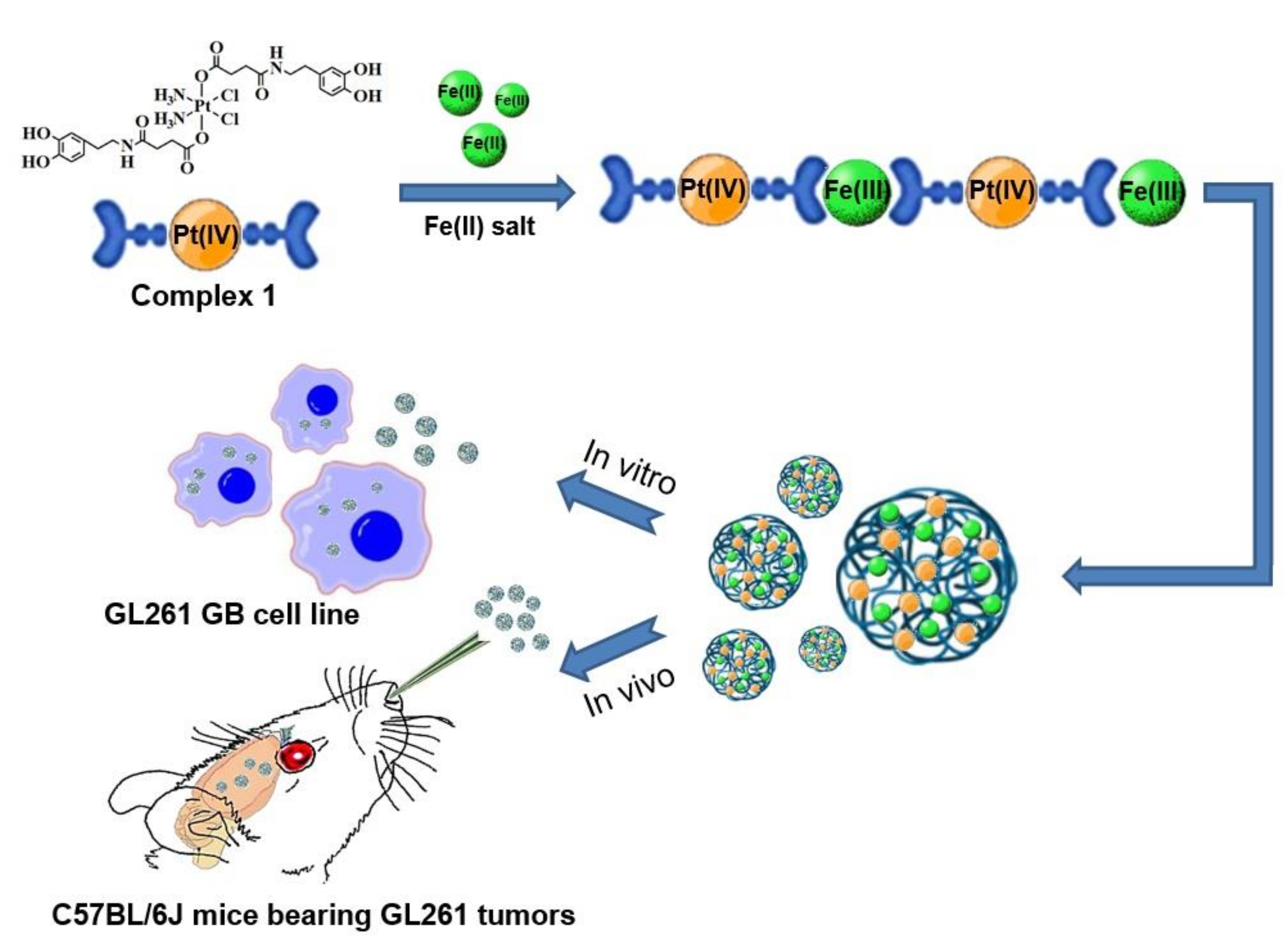
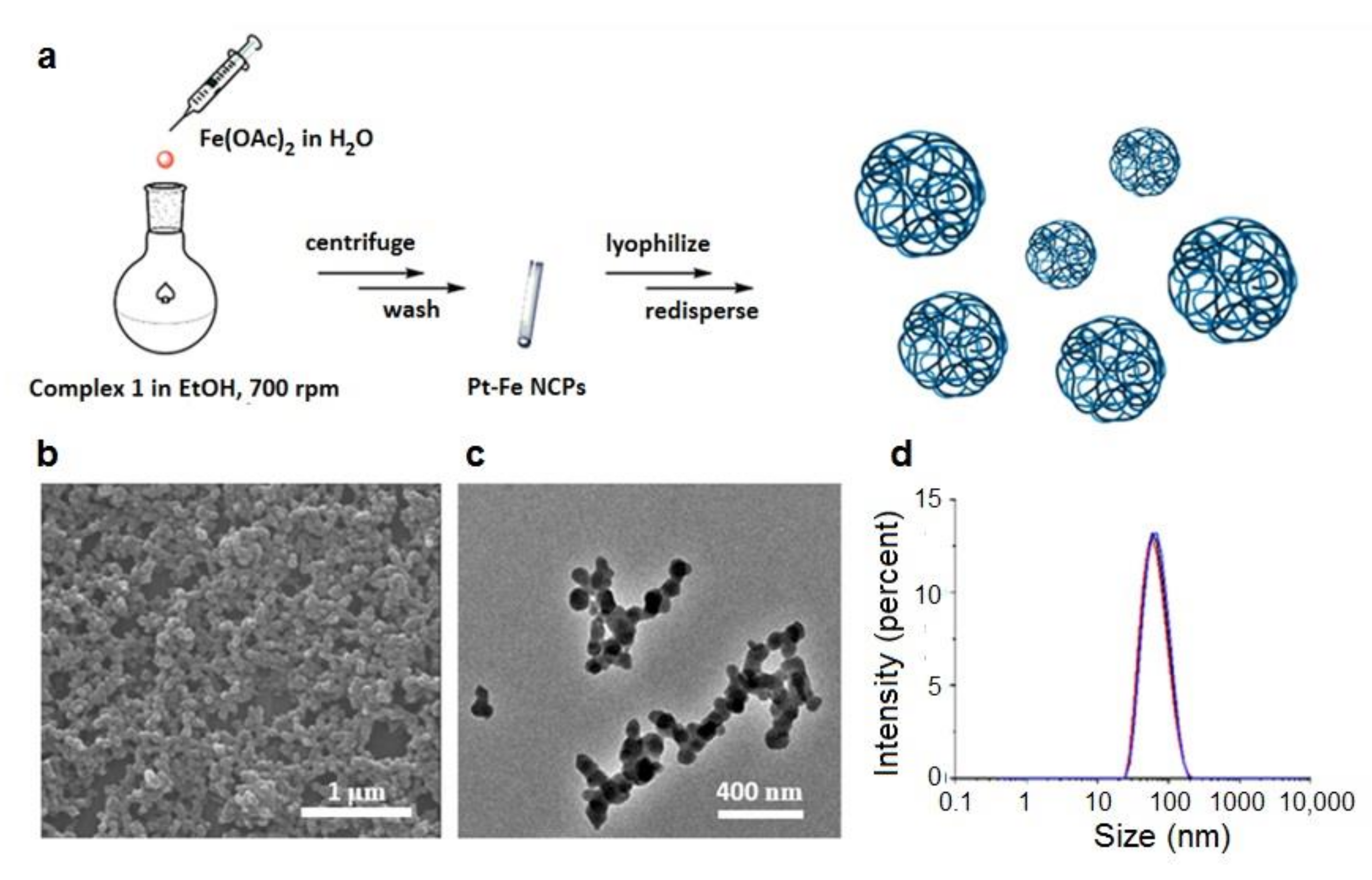
2.2. Synthesis of Pt-Fe NCPs
2.3. Characterization Methods
2.4. In Vitro Magnetic Resonance Imaging Analyses (MRI-Phantoms)
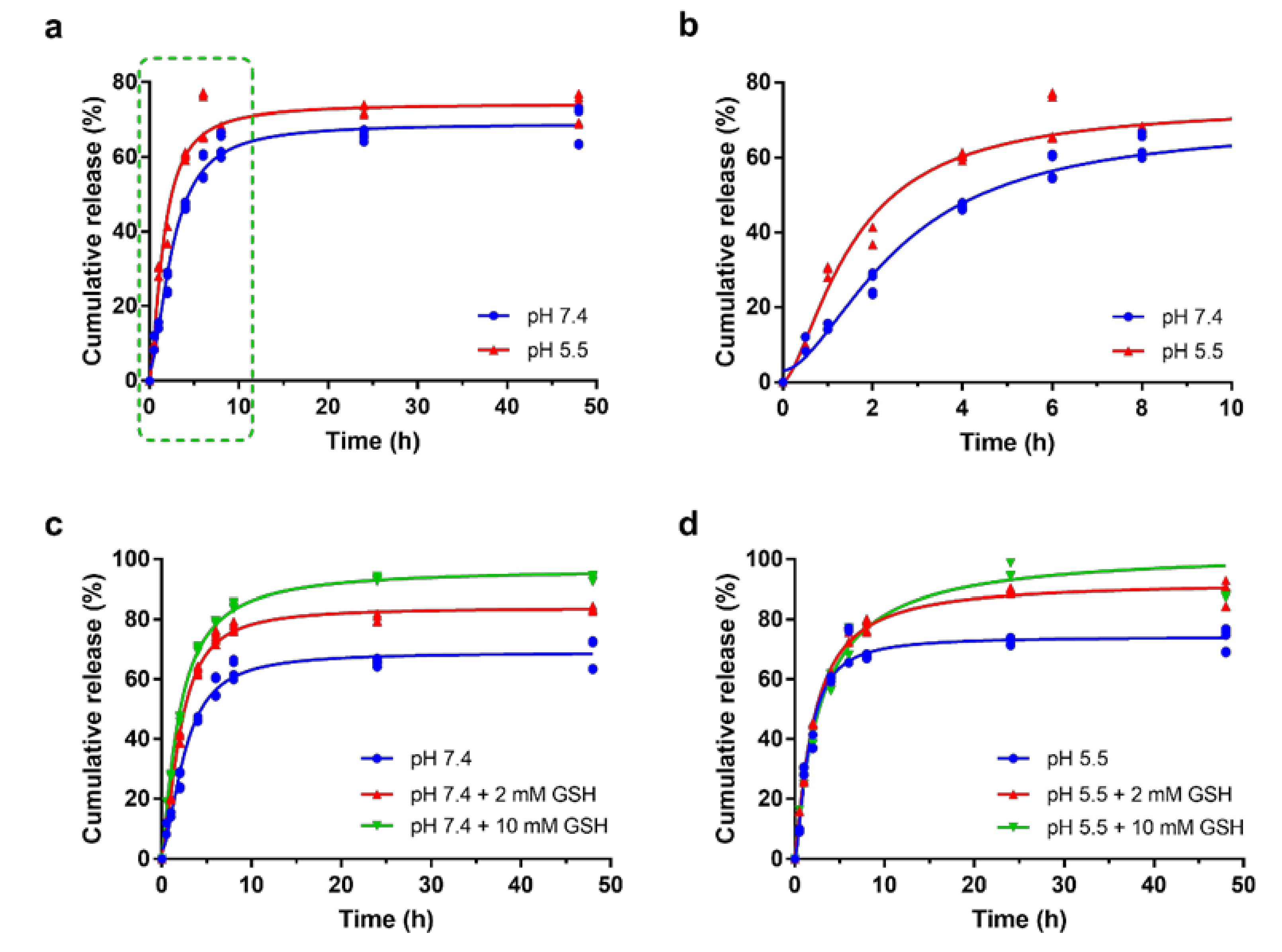
2.5. Drug Release Assay
2.6. Cell Lines Culture
2.7. In Vitro Cytotoxicity Assays
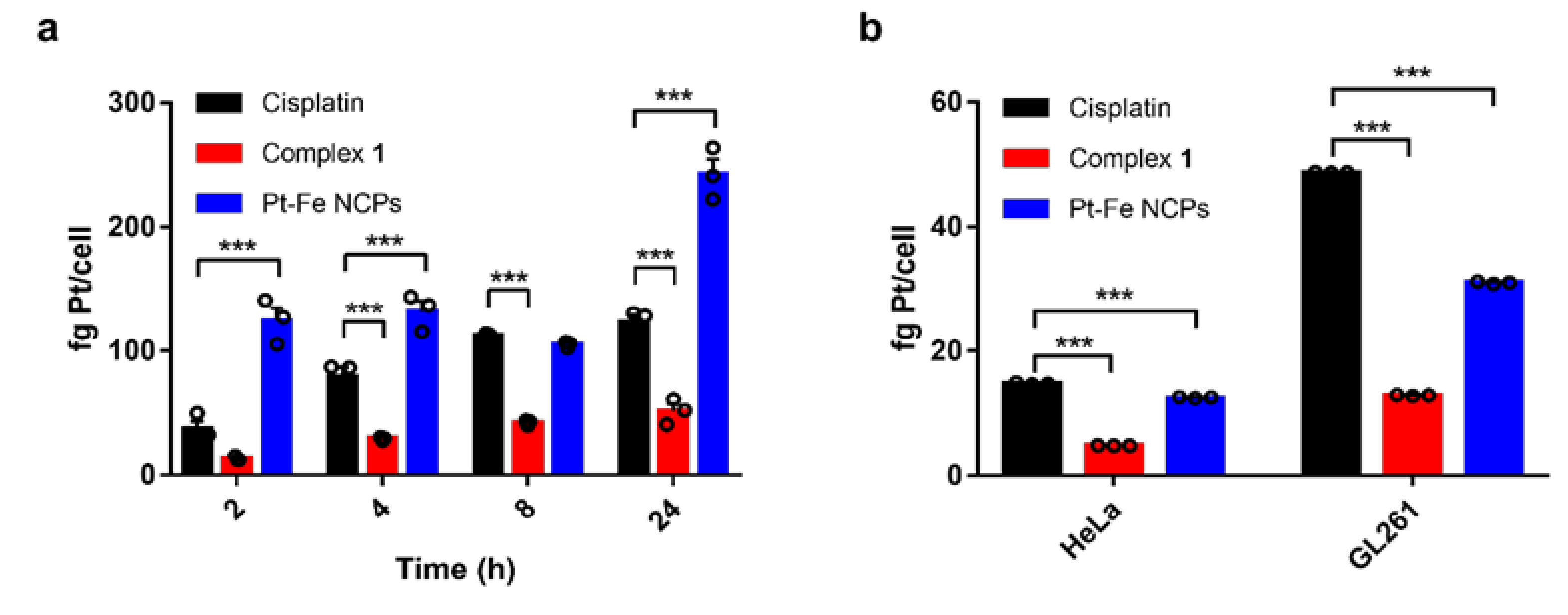
2.8. Cellular Internalization Studies
2.9. DNA-Bound Pt
2.10. Sample Digestion Treatments for ICP-MS Measurement
2.11. Estimation of ROS Formation
2.12. Animal Studies
2.13. GL261 GB Preclinical Model Generation and Animal Treatment
2.13.1. Tumor Generation
2.13.2. Tumor-Bearing Mice Treatment
2.14. Tissue Preservation Procedures
2.15. In Vivo MRI Studies
2.16. Tolerability Assays
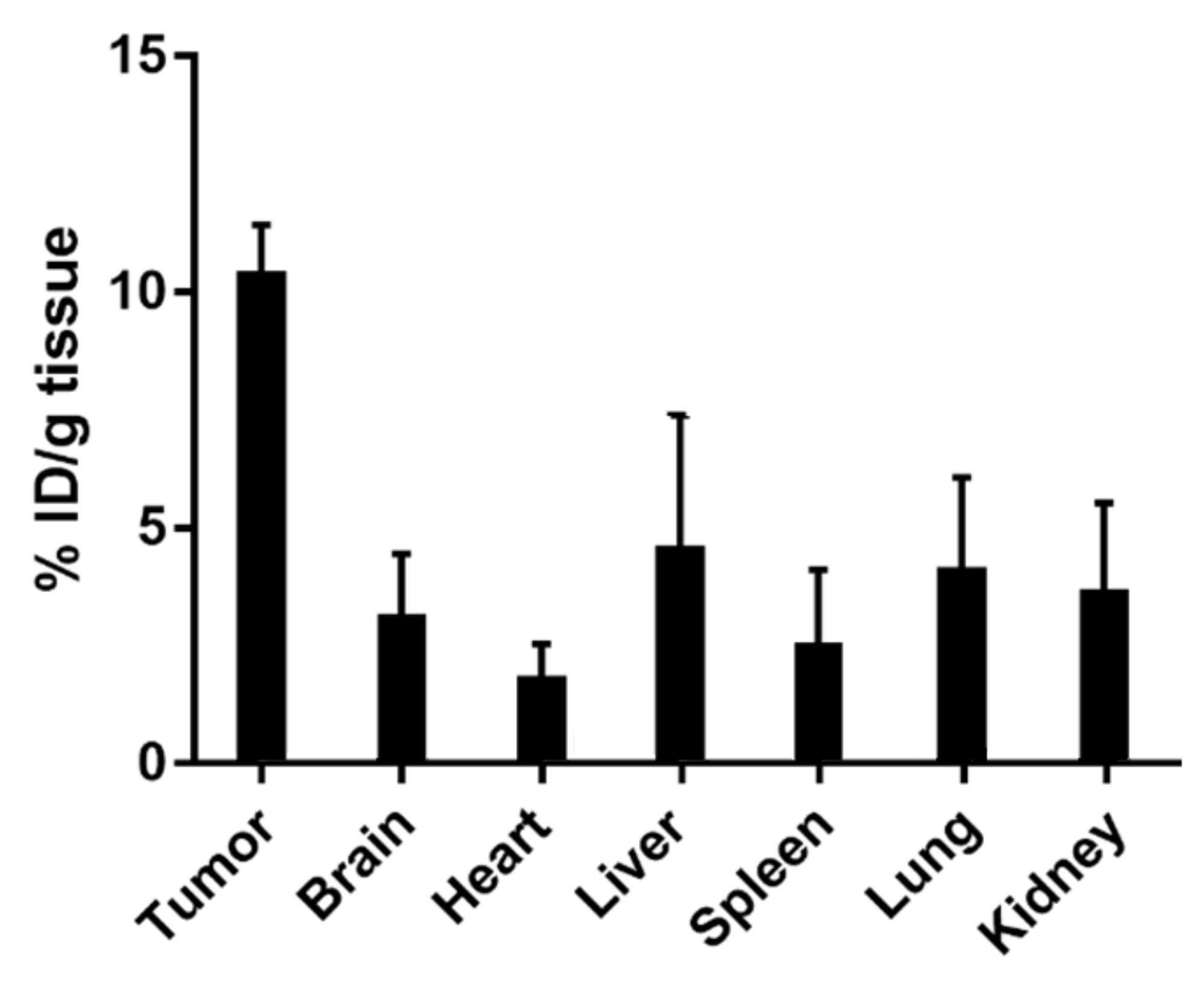
2.17. In Vivo Biodistribution Study
2.18. Assessment of Antitumor Efficacy in Vivo
2.19. Histological Examination
2.20. Statistical Analyses
3. Results
3.1. Synthesis and Characterization of Pt-Fe NCPs
3.2. In Vitro Magnetic Resonance Imaging (MRI) Studies
3.3. Drug Release Profile
3.4. Cytotoxicity Assays
3.5. Cell Internalization and DNA-Bound Pt
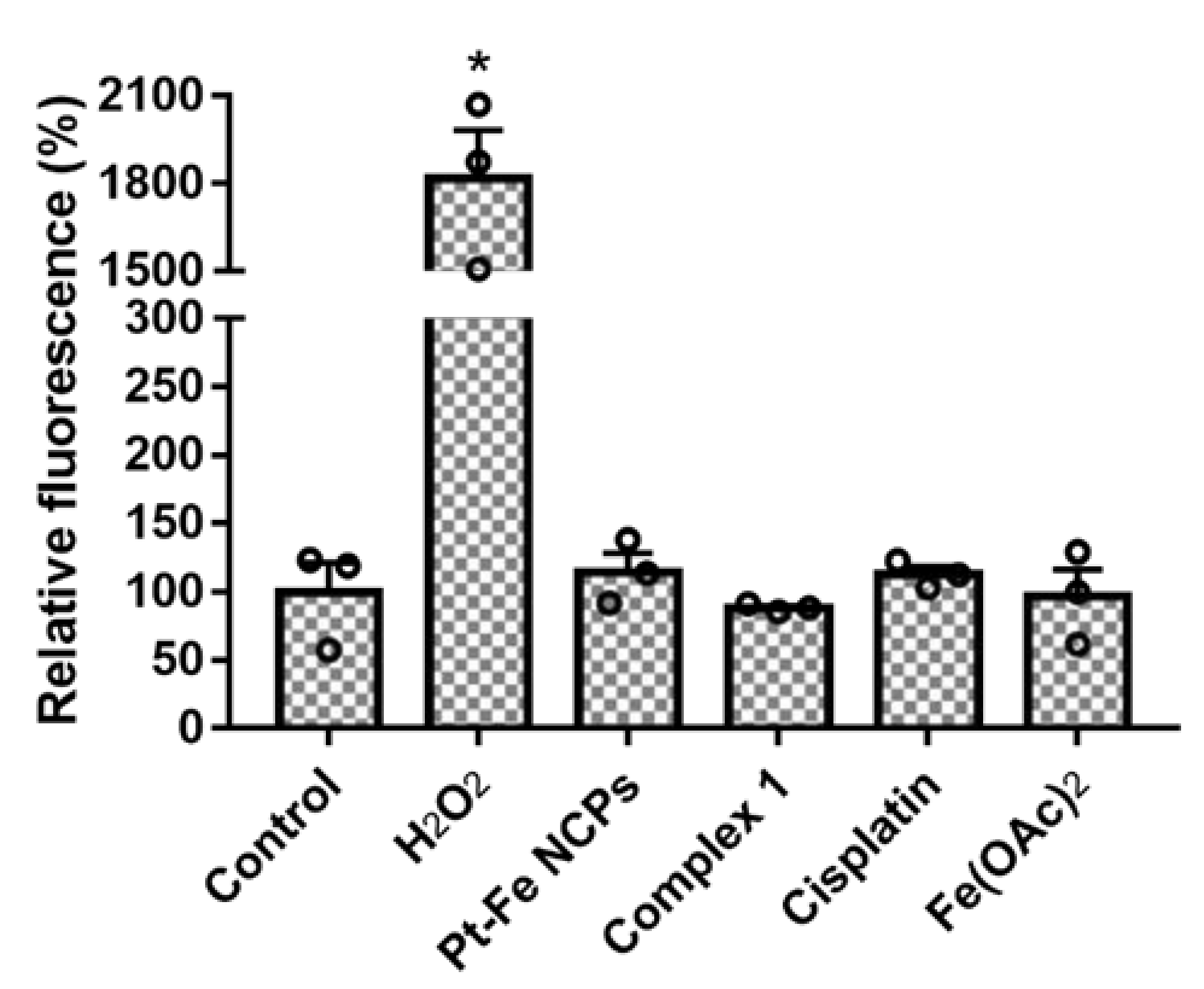
3.6. ROS Production in GL261 cells
3.7. In Vivo Tolerability and Biodistribution via Intranasal Administration
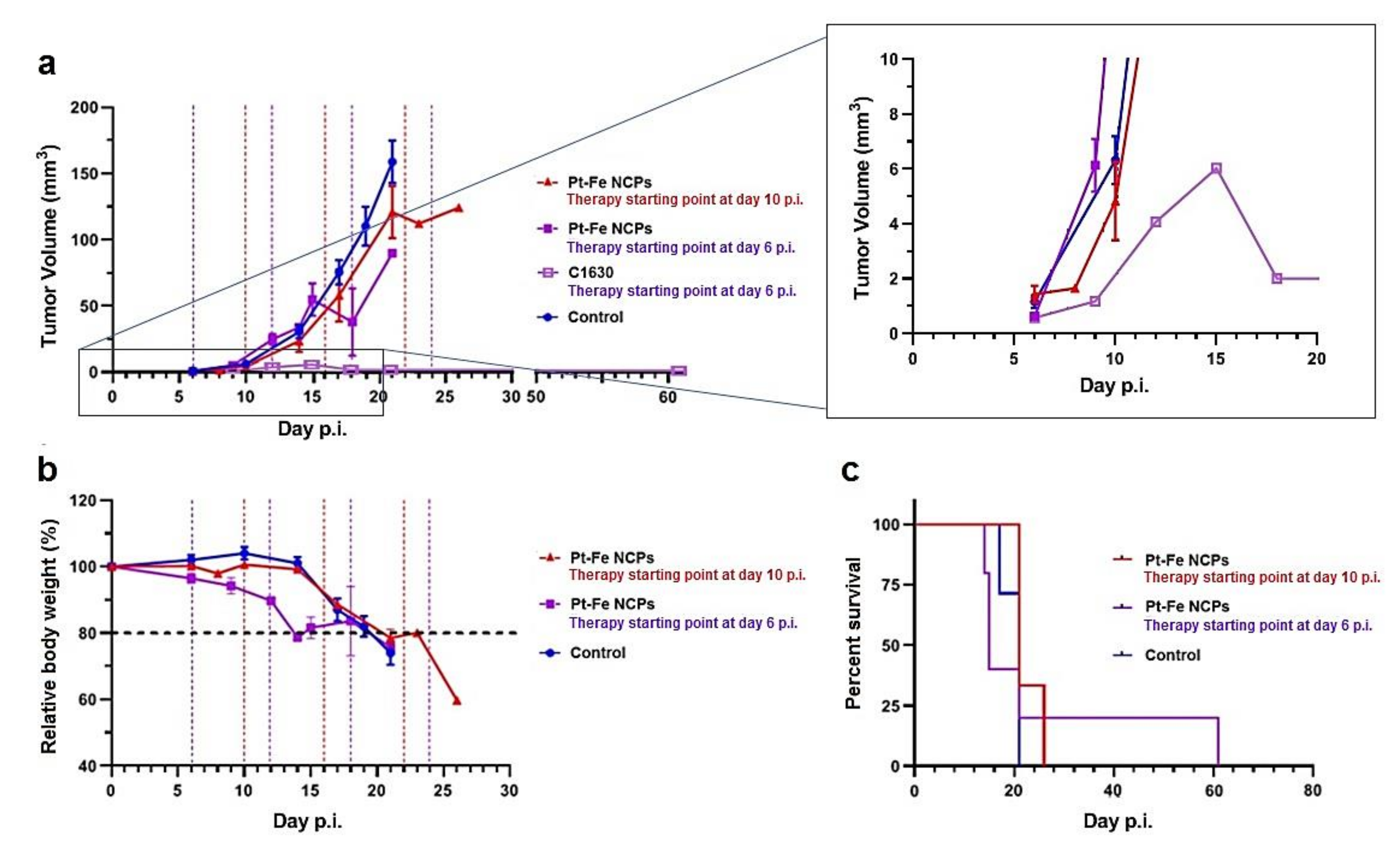
3.8. In Vivo Anticancer Efficacy
4. Discussion
4.1. Advantages of Pt(IV) Prodrug Nanoformulation
4.2. In Vivo Efficacy Is Not Directly Related to the Paradigm “More Is Better”: Words of Caution
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delgado-López, P.D.; Corrales-García, E.M. Survival in Glioblastoma: A Review on the Impact of Treatment Modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Englot, D.J.; Birk, H.; Molinaro, A.M.; Chang, S.M.; Clarke, J.L.; Prados, M.D.; Taylor, J.W.; Berger, M.S.; Butowski, N.A. Impact of Timing of Concurrent Chemoradiation for Newly Diagnosed Glioblastoma: A Critical Review of Current Evidence. Neurosurgery 2015, 62 (Suppl. 1), 160–165. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A comprehensive review in improving delivery of small-molecule chemotherapeutic agents overcoming the blood-brain/brain tumor barriers for glioblastoma treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef]
- Kovacs, Z.I.; Burks, S.R.; Frank, J.A. Focused ultrasound with microbubbles induces sterile inflammatory response proportional to the blood brain barrier opening: Attention to experimental conditions. Theranostics 2018, 8, 2245. [Google Scholar] [CrossRef]
- Patel, M.M.; Patel, B.M. Crossing the blood-brain barrier: Recent advances in drug delivery to the brain. CNS Drugs 2017, 31, 109–133. [Google Scholar] [CrossRef]
- Sharabi, S.; Bresler, Y.; Ravid, O.; Shemesh, C.; Atrakchi, D.; Schnaider-Beeri, M.; Gosselet, F.; Dehouck, L.; Last, D.; Guez, D.; et al. Transient blood–brain barrier disruption is induced by low pulsed electrical fields in vitro: An analysis of permeability and trans-endothelial electric resistivity. Drug Deliv. 2019, 26, 459–469. [Google Scholar] [CrossRef]
- Sheleg, S.; Korotkevich, E.; Zhavrid, E.; Muravskaya, G.; Smeyanovich, A.; Shanko, Y.; Yurkshtovich, T.; Bychkovsky, P.; Belyaev, S. Local chemotherapy with cisplatin-depot for glioblastoma multiforme. J. Neurooncol. 2002, 60, 53–59. [Google Scholar] [CrossRef]
- Tabet, A.; Jensen, M.P.; Parkins, C.C.; Patil, P.G.; Watts, C.; Scherman, O.A. Designing Next-Generation Local Drug Delivery Vehicles for Glioblastoma Adjuvant Chemotherapy: Lessons from the Clinic. Adv. Healthc. Mater. 2019, 8, e1801391. [Google Scholar] [CrossRef]
- Zhao, M.; Bozzato, E.; Joudiou, N.; Ghiassinejad, S.; Danhier, F.; Gallez, B.; Préat, V. Codelivery of Paclitaxel and Temozolomide through a Photopolymerizable Hydrogel Prevents Glioblastoma Recurrence after Surgical Resection. J. Control. Release 2019, 309, 72–81. [Google Scholar] [CrossRef]
- Shi, M.; Sanche, L. Convection-Enhanced Delivery in Malignant Gliomas: A Review of Toxicity and Efficacy. J. Oncol. 2019, 2019, 9342796. [Google Scholar] [CrossRef] [PubMed]
- Vogelbaum, M.A.; Aghi, M.K. Convection-Enhanced Delivery for the Treatment of Glioblastoma. Neuro Oncol. 2015, 17 (Suppl. 2), ii3–ii8. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Kaloshi, G.; Sierra del Rio, M.; Ducray, F.; Psimaras, D.; Idbaih, A.; Laigle-Donadey, F.; Taillibert, S.; Houillier, C.; Dehais, C.; Omuro, A.; et al. Nitrosourea-based chemotherapy for low grade gliomas failing initial treatment with temozolomide. J. Neurooncol. 2010, 100, 439–441. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin®) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Jue, T.R.; Sena, E.S.; Macleod, M.R.; McDonald, K.L.; Hirst, T.H. A systematic review and meta-analysis of topoisomerase inhibition in pre-clinical glioma models. Oncotarget 2018, 9, 11387–11401. [Google Scholar] [CrossRef]
- Charests, G.; Sanche, L.; Fortin, D.; Mathieu, D.; Paquette, B. Optimization of the route of platinum drugs administration to optimize the concomitant treatment with radiotherapy for glioblastoma implanted in the Fischer rat brain. J. Neurooncol. 2013, 115, 365–373. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Alphandéry, E. Nano-Therapies for Glioblastoma Treatment. Cancers 2020, 12, 242. [Google Scholar] [CrossRef]
- Ardelean, I.L.; Ficai, D.; Sonmez, M.; Oprea, O.; Nechifor, G.; Andronescu, E.; Ficai, A.; Titu, M.A. Hybrid Magnetic Nanostructures for Cancer Diagnosis and Therapy. Anticancer Agents Med. Chem. 2019, 19, 6–16. [Google Scholar] [CrossRef]
- Evaluate the Safety and Effectiveness of Intranasal Administration of Temozolomide in Patients With Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04091503 (accessed on 24 March 2022).
- Safety and Efficacy Study in Recurrent Grade IV Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT02704858 (accessed on 24 March 2022).
- Bruinsmann, F.A.; Richter Vaz, G.; de Cristo Soares Alves, A.; Aguirre, T.; Raffin Pohlmann, A.; Stanisçuaski Guterres, S.; Sonvico, F. Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules 2019, 24, 4312. [Google Scholar] [CrossRef] [PubMed]
- Louis, N.; Liu, S.; He, X.; Drummond, D.C.; Noble, C.O.; Goldman, S.; Mueller, S.; Bankiewicz, K.; Gupta, N.; Hashizume, R. New therapeutic approaches for brainstem tumors: A comparison of delivery routes using nanoliposomal irinotecan in an animal model. J. Neuro Oncol. 2018, 136, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Wu, S.; Calero-Pérez, P.; Candiota, A.P.; Alfonso, P.; Bruna, J.; Yuste, V.J.; Lorenzo, J.; Novio, F.; Ruiz-Molina, D. Synthesis and validation of a bioinspired catechol -functionalized Pt(IV) prodrug for preclinical intranasal glio-blastoma treatment. Cancers 2022, 14, 410. [Google Scholar] [CrossRef]
- García-Pardo, J.; Novio, F.; Nador, F.; Cavaliere, I.; Suárez-García, S.; Lope-Piedrafita, S.; Candiota, A.P.; Romero-Gimenez, J.; Rodríguez-Galván, B.; Bové, J.; et al. Bioinspired Theranostic Coordination Polymer Nanoparticles for Intranasal Dopamine Replacement in Parkinson’s Disease. ACS Nano 2021, 15, 8592–8609. [Google Scholar] [CrossRef]
- Adarsh, N.N.; Frias, C.; Ponnoth Lohidakshan, T.M.; Lorenzo, J.; Novio, F.; Garcia-Pardo, J.; Ruiz-Molina, D. Pt(Iv)-Based Nanoscale Coordination Polymers: Antitumor Activity, Cellular Uptake and Interactions with Nuclear DNA. Chem. Eng. J. 2018, 340, 94–102. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-Based Drugs: Past, Present and Future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Kuang, X.; Chi, D.; Li, J.; Guo, C.; Yang, Y.; Zhou, S.; Luo, C.; Liu, H.; He, Z.; Wang, Y. Disulfide bond based cascade reduction-responsive Pt(IV) nanoassemblies for improved anti-tumor efficiency and biosafety. Colloids Surf. B Biointerfaces 2021, 203, 111766. [Google Scholar] [CrossRef]
- Garofalo, S.; Porzia, A.; Mainiero, F.; Di Angelantonio, S.; Cortese, B.; Basilico, B.; Pagani, F.; Cignitti, G.; Chece, G.; Maggio, R.; et al. Environmental stimuli shape microglial plasticity in glioma. eLife 2017, 6, e33415. [Google Scholar] [CrossRef]
- Saldaña-Ruíz, S.; Soler-Martín, C.; Llorens, J. Role of Cyp2e1-Mediated Metabolism in the Acute and Vestibular Toxicities of Nineteen Nitriles in the Mouse. Toxicol. Lett. 2012, 208, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Guidance on Dose Level Selection for Regulatory General Toxicology Studies for Pharmaceuticals. Available online: http://www.lasa.co.uk/PDF/LASA-NC3RsDoseLevelSelection.pdf (accessed on 24 March 2022).
- Jo, D.-H.; Chiou, Y.-M.; Que, L. Models for Extradiol Cleaving Catechol Dioxygenases: Syntheses, Structures, and Reactivities of Iron(II)−Monoanionic Catecholate Complexes. Inorg. Chem. 2001, 40, 3181–3190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sababi, M.; Brinck, T.; Persson, D.; Pan, J.; Claesson, P.M. In Situ Investigations of Fe3+ Induced Complexation of Adsorbed Mefp-1 Protein Film on Iron Substrate. J. Colloid Interface Sci. 2013, 404, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Nador, F.; Novio, F.; Ruiz-Molina, D. Coordination Polymer Particles with Ligand-Centred pH-Responses and Spin Transition. Chem. Commun. 2014, 50, 14570–14572. [Google Scholar] [CrossRef] [PubMed]
- Amorín-Ferré, L.; Busqué, F.; Bourdelande, J.L.; Ruiz-Molina, D.; Hernando, J.; Novio, F. Encapsulation and Release Mechanisms in Coordination Polymer Nanoparticles. Chem. Eur. J. 2013, 19, 17508–17516. [Google Scholar] [CrossRef] [PubMed]
- Suárez-García, S.; Esposito, T.V.F.; Neufeld-Peters, J.; Bergamo, M.; Yang, H.; Saatchi, K.; Schaffer, P.; Häfeli, U.O.; Ruiz-Molina, D.; Rodríguez-Rodríguez, C.; et al. Hybrid Metal−Phenol Nanoparticles with Polydopamine-like Coating for PET/SPECT/CT Imaging. ACS Appl. Mater. Interfaces 2021, 13, 10705–10718. [Google Scholar] [CrossRef] [PubMed]
- Lajous, H.; Riva, R.; Lelievre, B.; Tetaud, C.; Avril, S.; Hindre, F.; Boury, F.; Jerome, C.; Lecomte, P.; Garcion, E. Hybrid Gd3+/Cisplatin Cross-Linked Polymer Nanoparticles Enhance Platinum Accumulation and Formation of DNA Adducts in Glioblastoma Cell Lines. Biomater. Sci. 2018, 6, 2386–2409. [Google Scholar] [CrossRef]
- Ebrahimi Shahmabadi, H.; Movahedi, F.; Koohi Moftakhari Esfahani, M.; Alavi, S.E.; Eslamifar, A.; Mohammadi Anaraki, G.; Akbarzadeh, A. Efficacy of Cisplatin-Loaded Polybutyl Cyanoacrylate Nanoparticles on the Glioblastoma. Tumour Biol. 2014, 35, 4799–4806. [Google Scholar] [CrossRef]
- Suárez-García, S.; Arias-Ramos, N.; Frias, C.; Candiota, A.P.; Arús, C.; Lorenzo, J.; Ruiz-Molina, D.; Novio, F. Dual T1/T2 Nanoscale Coordination Polymers as Novel Contrast Agents for MRI: A Preclinical Study for Brain Tumor. ACS Appl. Mater. Interfaces 2018, 10, 38819–38832. [Google Scholar] [CrossRef]
- Borges, M.; Yu, S.; Laromaine, A.; Roig, A.; Suárez-García, S.; Lorenzo, J.; Ruiz-Molina, D.; Novio, F. Dual T1/T2 MRI contrast agent based on hybrid SPION@coordination polymer nanoparticles. RSC Adv. 2015, 5, 86779. [Google Scholar] [CrossRef]
- Portakal, Z.G.; Shermer, S.; Jenkins, C.; Spezi, E.; Perrett, T.; Tuncel, N.; Phillips, J. Design and Characterization of Tissue-Mimicking Gel Phantoms for Diffusion Kurtosis Imaging. Med. Phys. 2018, 45, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, P.; Xiao, Y.; Li, J.; Yang, F.; Liu, Y.; Zhang, J.-R.; Zhu, J.-J. Acid-Degradable Gadolinium-Based Nanoscale Coordination Polymer: A Potential Platform for Targeted Drug Delivery and Potential Magnetic Resonance Imaging. Nano Res. 2018, 11, 929–939. [Google Scholar] [CrossRef]
- Srivastava, S.; Awasthi, R.; Tripathi, D.; Rai, M.K.; Agarwal, V.; Agrawal, V.; Gajbhiye, N.S.; Gupta, R.K. Magnetic-Nanoparticle-Doped Carbogenic Nanocomposite: An Effective Magnetic Resonance/Fluorescence Multimodal Imaging Probe. Small 2012, 8, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Solórzano, R.; Tort, O.; García-Pardo, J.; Escribà, T.; Lorenzo, J.; Arnedo, M.; Ruiz-Molina, D.; Alibés, R.; Busqué, F.; Novio, F. Versatile iron–catechol-based nanoscale coordination polymers with antiretroviral ligand functionalization and their use as efficient carriers in HIV/AIDS therapy. Biomater. Sci. 2019, 7, 178–186. [Google Scholar] [CrossRef]
- Shen, J.; Wang, Q.; Fang, J.; Shen, W.; Wu, D.; Tang, G.; Yang, J. Therapeutic Polymeric Nanomedicine: Gsh-Responsive Release Promotes Drug Release for Cancer Synergistic Chemotherapy. RSC Adv. 2019, 9, 37232–37240. [Google Scholar] [CrossRef]
- Gao, C.; Tang, F.; Zhang, J.; Lee, S.M.Y.; Wang, R. Glutathione-Responsive Nanoparticles Based on a Sodium Alginate Derivative for Selective Release of Doxorubicin in Tumor Cells. J. Mater. Chem. B 2017, 5, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.; Blanco-Fernández, B.; Costoya, A.; Concheiro, A.; Puoci, F.; Alvarez-Lorenzo, C. Glucose Cryoprotectant Affects Glutathione-Responsive Antitumor Drug Release from Polysaccharide Nanoparticles. Eur. J. Pharm. Biopharm. 2015, 93, 281–292. [Google Scholar] [CrossRef]
- Shukla, D.; Mandal, P.K.; Tripathi, M.; Vishwakarma, G.; Mishra, R.; Sandal, K. Quantitation of in Vivo Brain Glutathione Conformers in Cingulate Cortex among Age-Matched Control, Mci, and Ad Patients Using Mega-Press. Hum. Brain Mapp. 2020, 41, 194–217. [Google Scholar] [CrossRef]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and Cancer: Recent Insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Voth, B.; Nagasawa, D.T.; Pelargos, P.E.; Chung, L.K.; Ung, N.; Gopen, Q.; Tenn, S.; Kamei, D.T.; Yang, I. Transferrin Receptors and Glioblastoma Multiforme: Current Findings and Potential for Treatment. J. Clin. Neurosci. 2015, 22, 1071–1076. [Google Scholar] [CrossRef]
- Puskas, J.E.; Chen, Y.; Kulbaba, K.; Kaszas, G. Comparison of the Molecular Weight and Size Measurement of Polyisobutylenes by Size Exclusion Chromatography/Multi-Angle Laser Light Scattering and Viscometry. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 1777–1783. [Google Scholar] [CrossRef]
- Paine, P.L.; Moore, L.C.; Horowitz, S.B. Nuclear Envelope Permeability. Nature 1975, 254, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Keminer, O.; Peters, R. Permeability of Single Nuclear Pores. Biophys. J. 1999, 77, 217–228. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Hetzer, M.W. Structure, Dynamics and Function of Nuclear Pore Complexes. Trends Cell Biol. 2008, 18, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Wu, S.; Calero-Pérez, P.; Villamañan, L.; Arias-Ramos, N.; Pumarola, M.; Ortega-Martorell, S.; Julià-Sapé, M.; Arús, C.; Candiota, A.P. Anti-Tumour Immune Response in Gl261 Glioblastoma Generated by Temozolomide Immune-Enhancing Metronomic Schedule Monitored with MRSI-Based Nosological Images. NMR Biomed. 2020, 33, e4229. [Google Scholar] [CrossRef]
- Wu, S.; Calero-Pérez, P.; Arús, C.; Candiota, A.P. Anti-Pd-1 Immunotherapy in Preclinical Gl261 Glioblastoma: Influence of Therapeutic Parameters and Non-Invasive Response Biomarker Assessment with Mrsi-Based Approaches. Int. J. Mol. Sci. 2020, 21, 8775. [Google Scholar] [CrossRef]
- Arias-Ramos, N.; Ferrer-Font, L.; Lope-Piedrafita, S.; Mocioiu, V.; Julià-Sapé, M.; Pumarola, M.; Arús, C.; Candiota, A.P. Metabolomics of Therapy Response in Preclinical Glioblastoma: A Multi-Slice Mrsi-Based Volumetric Analysis for Noninvasive Assessment of Temozolomide Treatment. Metabolites 2017, 7, 20. [Google Scholar] [CrossRef]
- Kay, K.; Dolcy, K.; Bies, R.; Shah, D.K. Estimation of Solid Tumor Doubling Times from Progression-Free Survival Plots Using a Novel Statistical Approach. AAPS J. 2019, 21, 27. [Google Scholar] [CrossRef]
- Tritz, Z.P.; Ayasoufi, K.; Johnson, A.J. Anti-PD-1 checkpoint blockade monotherapy in the orthotopic GL261 glioma model: The devil is in the detail. Neurooncol. Adv. 2021, 3, vdab066. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, Z.; Zhu, G. Emerging platinum(IV) prodrugs to combat cisplatin resistance: From isolated cancer cells to tumor microenvironment. Dalton Trans. 2019, 48, 2536–2544. [Google Scholar] [CrossRef] [PubMed]
- Feldhaeusser, B.; Platt, S.R.; Marrache, S.; Kolishetti, N.; Pathak, R.K.; Montgomery, D.J.; Reno, L.R.; Howerth, E.; Dhar, S. Evaluation of nanoparticle delivered cisplatin in beagles. Nanoscale 2015, 7, 13822–13830. [Google Scholar] [CrossRef] [PubMed]
- Arduino, I.; Depalo, N.; Re, F.; Dal Magro, R.; Panniello, A.; Margiotta, N.; Fanizza, E.; Lopalco, A.; Laquintana, V.; Cutrignelli, A.; et al. Pegylated Solid Lipid Nanoparticles for Brain Delivery of Lipophilic Kiteplatin Pt(IV) Prodrugs: An in Vitro Study. Int. J. Pharm. 2020, 583, 119351. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8+ T-cell responses and immune memory. Oncoimmunology 2015, 4, e1005521. [Google Scholar] [CrossRef]
- Ferrer-Font, L.; Villamañan, L.; Arias-Ramos, N.; Vilardell, J.; Plana, M.; Ruzzene, M.; Pinna, L.A.; Itarte, E.; Arús, C.; Candiota, A.P. Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice. Pharmaceuticals 2017, 10, 24. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of Immunotherapy Resistance: Lessons from Glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic Chemotherapy: Dose and Schedule Dependence and Combination with Immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Di Ianni, N.; Maffezzini, M.; Eoli, M.; Pellegatta, S. Revisiting the Immunological Aspects of Temozolomide Considering the Genetic Landscape and the Immune Microenvironment Composition of Glioblastoma. Front. Oncol. 2021, 11, 747690. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35 (Suppl. 1), S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.B.; Wadajkar, A.S.; Winkles, J.A.; Davila, E.; Kim, A.J.; Woodworth, G.F. Repurposing Platinum-Based Chemotherapies for Multi-Modal Treatment of Glioblastoma. Oncoimmunology 2016, 5, e1208876. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hato, S.V.; Khong, A.; de Vries, I.J.; Lesterhuis, W.J. Molecular Pathways: The Immunogenic Effects of Platinum-Based Chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautès-Fridman, C.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014, 3, e27878. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Enríquez Pérez, J.; Fritzell, S.; Kopecky, J.; Visse, E.; Darabi, A.; Siesjö, P. The Effect of Locally Delivered Cisplatin Is Dependent on an Intact Immune Function in an Experimental Glioma Model. Sci. Rep. 2019, 9, 5632. [Google Scholar] [CrossRef]
- Ferrer-Font, L.; Arias-Ramos, N.; Lope-Piedrafita, S.; Julià-Sapé, M.; Pumarola, M.; Arús, C.; Candiota, A.P. Metronomic treatment in immunocompetent preclinical GL261 glioblastoma: Effects of cyclophosphamide and temozolomide. NMR Biomed. 2017, 30, e3748. [Google Scholar] [CrossRef]
- Avan, A.; Postma, T.J.; Ceresa, C.; Avan, A.; Cavaletti, G.; Giovannetti, E.; Peters, G.J. Platinum-induced neurotoxicity and preventive strategies: Past, present, and future. Oncologist 2015, 20, 411–432. [Google Scholar] [CrossRef]
- Aldossary, S.A. Review on Pharmacology of Cisplatin: Clinical Use, Toxicity and Mechanism of Resistance of Cisplatin. Biomed. Pharmacol. J. 2019, 12, 7–15. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) a | ||||
|---|---|---|---|---|
| Cell Line (24 h) | Cell Line (72 h) | |||
| Compound | HeLa | GL261 | HeLa | GL261 |
| Pt-Fe NCPs | 31.45 ± 1.10 | 13.48 ± 0.90 | 2.56 ± 0.63 | 2.00 ± 0.18 |
| Complex 1 | 29.94 ± 1.04 | 17.40 ± 1.08 | 1.85 ± 0.36 | 4.17 ± 0.12 |
| Cisplatin | 15.98 ± 1.04 | 5.61 ± 0.28 | 2.34 ± 0.30 | 2.16 ± 0.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, X.; Calero-Pérez, P.; Montpeyó, D.; Bruna, J.; Yuste, V.J.; Candiota, A.P.; Lorenzo, J.; Novio, F.; Ruiz-Molina, D. Intranasal Administration of Catechol-Based Pt(IV) Coordination Polymer Nanoparticles for Glioblastoma Therapy. Nanomaterials 2022, 12, 1221. https://doi.org/10.3390/nano12071221
Mao X, Calero-Pérez P, Montpeyó D, Bruna J, Yuste VJ, Candiota AP, Lorenzo J, Novio F, Ruiz-Molina D. Intranasal Administration of Catechol-Based Pt(IV) Coordination Polymer Nanoparticles for Glioblastoma Therapy. Nanomaterials. 2022; 12(7):1221. https://doi.org/10.3390/nano12071221
Chicago/Turabian StyleMao, Xiaoman, Pilar Calero-Pérez, David Montpeyó, Jordi Bruna, Victor J. Yuste, Ana Paula Candiota, Julia Lorenzo, Fernando Novio, and Daniel Ruiz-Molina. 2022. "Intranasal Administration of Catechol-Based Pt(IV) Coordination Polymer Nanoparticles for Glioblastoma Therapy" Nanomaterials 12, no. 7: 1221. https://doi.org/10.3390/nano12071221
APA StyleMao, X., Calero-Pérez, P., Montpeyó, D., Bruna, J., Yuste, V. J., Candiota, A. P., Lorenzo, J., Novio, F., & Ruiz-Molina, D. (2022). Intranasal Administration of Catechol-Based Pt(IV) Coordination Polymer Nanoparticles for Glioblastoma Therapy. Nanomaterials, 12(7), 1221. https://doi.org/10.3390/nano12071221