
Intrinsic Dynamic and Static Nature of π···π Interactions in Fused Benzene-Type Helicenes and Dimers, Elucidated with QTAIM Dual Functional Analysis

Abstract
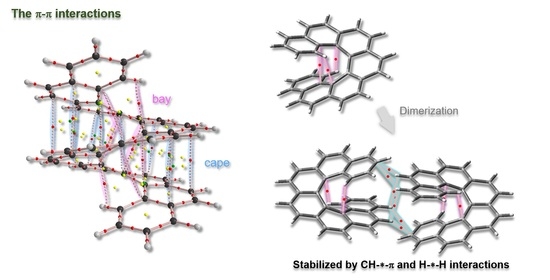
1. Introduction
2. Methodological Details of the Calculations
3. Results and Discussion
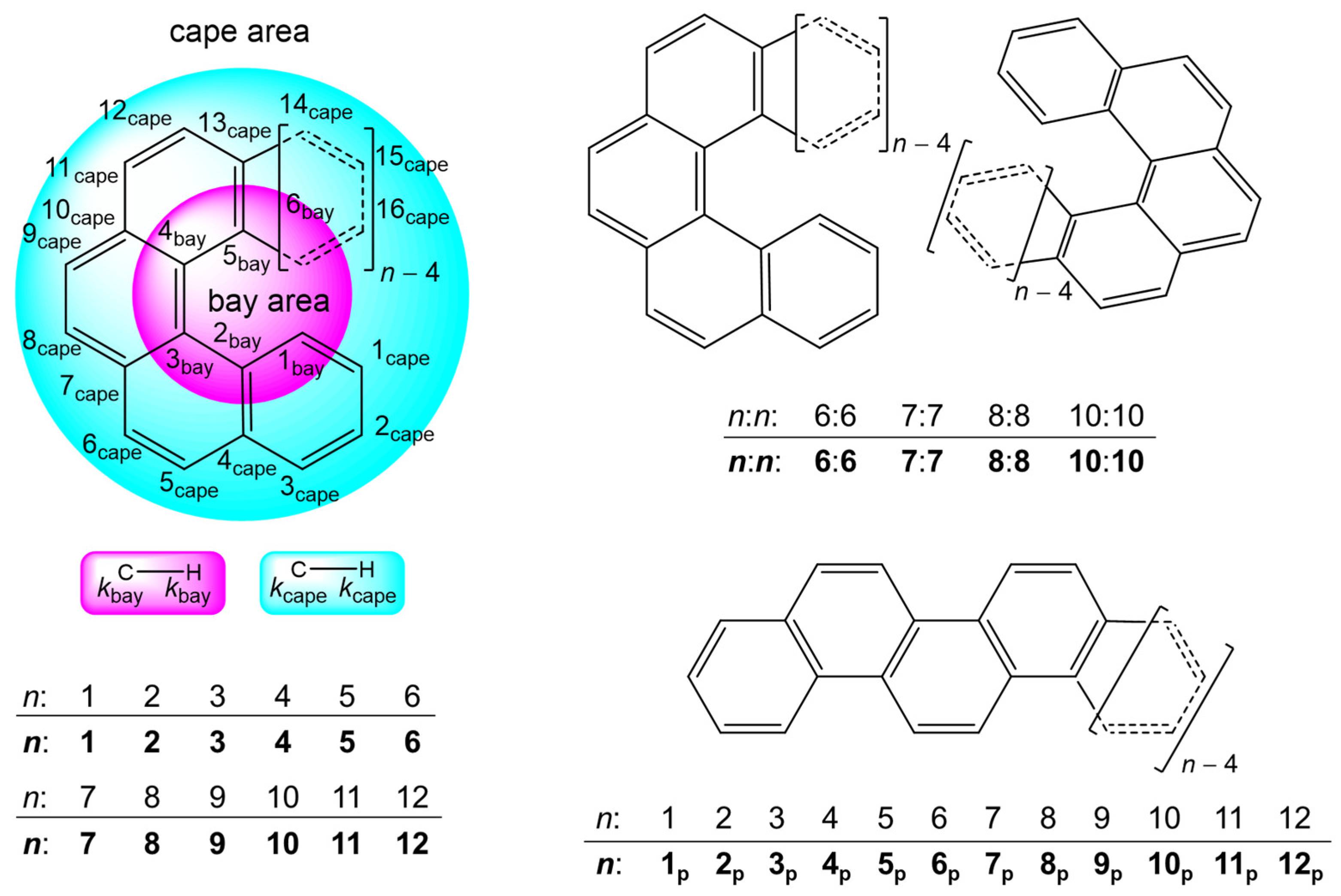
3.1. Structural Features of 1–12 and Their Energy Profile
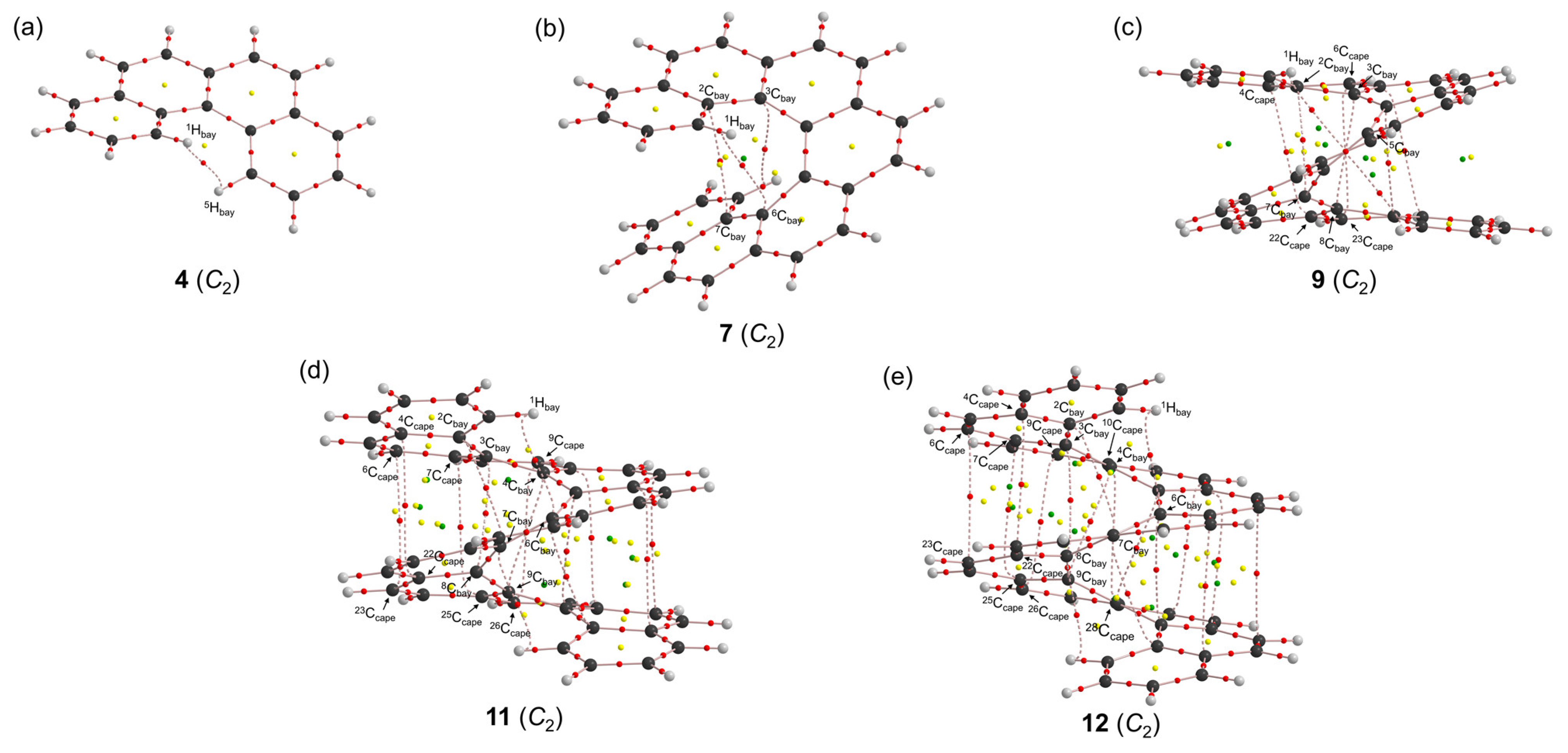
3.2. Survey of X-∗-Y (X, Y = C and H) in 3–12 with the Molecular Graphs
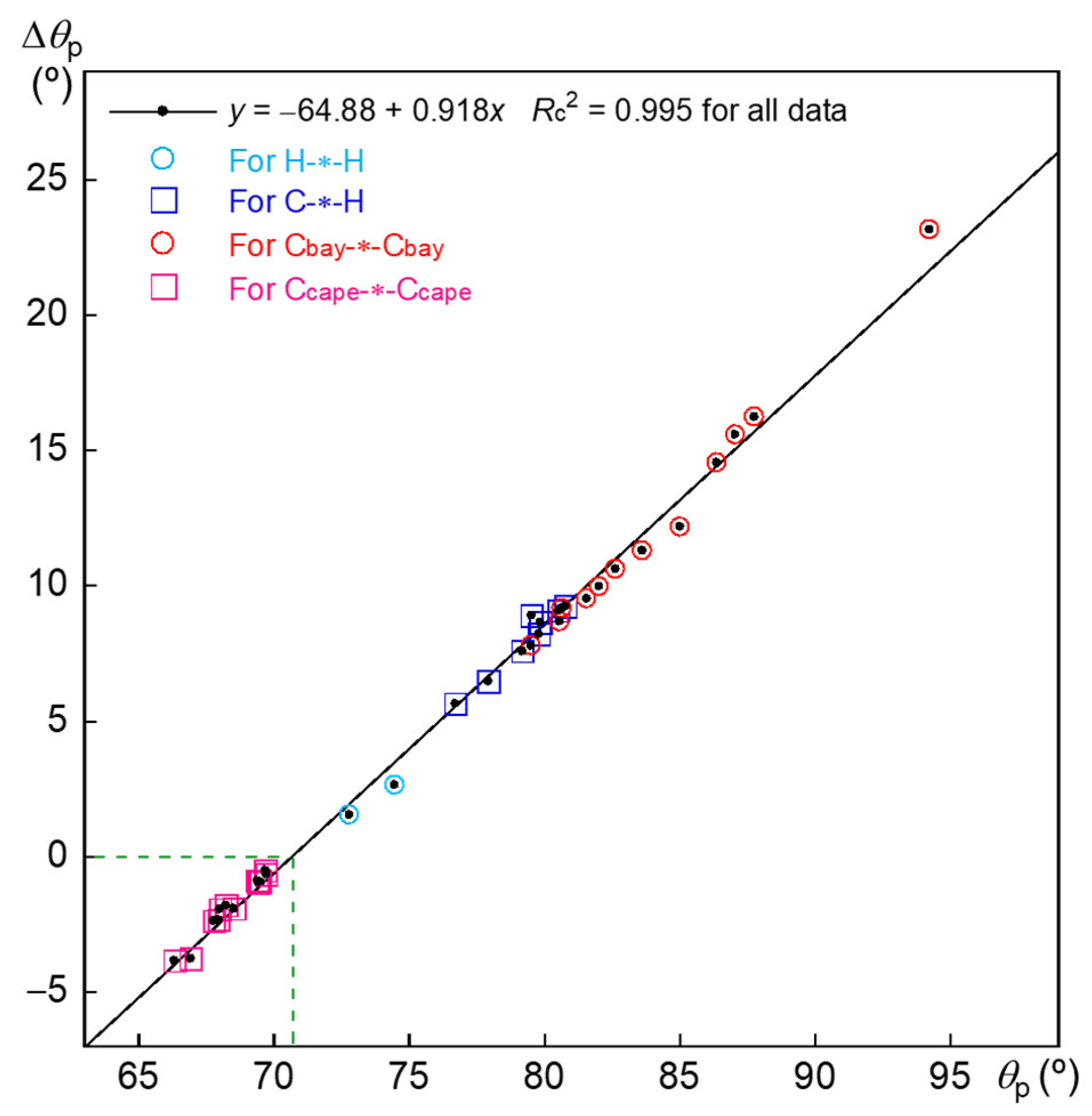
3.3. Nature of Each X-∗-Y in 3–12
3.4. Nature of Each X-∗-Y in 6:6 and 7:7
4. Basis Set and Level Dependence of the Predicted Natures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, Y.; Chen, C.F. Helicenes: Synthesis and Applications. Chem. Rev. 2012, 112, 1463–1535. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.F.; Shen, Y. Helicene Chemistry: From Synthesis to Applications; Springer: Berlin, Germany, 2017. [Google Scholar] [CrossRef]
- Dhbaibi, K.; Favereau, L.; Crassous, J. Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P). Chem. Rev. 2019, 119, 8846–8953. [Google Scholar] [CrossRef] [PubMed]
- Tani, F.; Narita, M.; Murafuji, T. Helicene Radicals: Molecules Bearing a Combination of Helical Chirality and Unpaired Electron Spin. ChemPlusChem 2020, 85, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Jakubec, M.; Storch, J. Recent Advances in Functionalizations of Helicene Backbone. J. Org. Chem. 2020, 85, 13415–13428. [Google Scholar] [CrossRef]
- Ren, M.; Wang, J.; Xie, X.; Zhang, J.; Wang, P. Double-Helicene-Based Hole-Transporter for Perovskite Solar Cells with 22% Efficiency and Operation Durability. ACS Energy Lett. 2019, 4, 2683–2688. [Google Scholar] [CrossRef]
- Xu, N.; Li, Y.; Ricciarelli, D.; Wang, J.; Mosconi, E.; Yuan, Y.; de Angelis, F.; Zakeeruddin, S.M.; Grätzel, M.; Wang, P. An Oxa[5]Helicene-Based Racemic Semiconducting Glassy Film for Photothermally Stable Perovskite Solar Cells. iScience 2019, 15, 234–242. [Google Scholar] [CrossRef]
- Wang, J.; Shi, H.; Xu, N.; Zhang, J.; Yuan, Y.; Lei, M.; Wang, L.; Wang, P. Aza[5]Helicene Rivals N-Annulated Perylene as π-Linker of D-π-D Typed Hole-Transporters for Perovskite Solar Cells. Adv. Funct. Mater. 2020, 30, 2002114. [Google Scholar] [CrossRef]
- Wei, Y.; Zheng, A.; Xie, X.; Zhang, J.; He, L.; Wang, P. A Pyrrole-Bridged Bis(Oxa[5]Helicene)-Based Molecular Semiconductor for Efficient and Durable Perovskite Solar Cells: Microscopic Insights. ACS Mater. Lett. 2021, 3, 947–955. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Xie, X.; Ren, Y.; Zhang, B.; He, L.; Zhang, J.; Wang, L.D.; Wang, P. A Helicene-Based Molecular Semiconductor Enables 85 °C Stable Perovskite Solar Cells. ACS Energy Lett. 2021, 6, 1764–1772. [Google Scholar] [CrossRef]
- Tsujihara, T.; Inada-Nozaki, N.; Takehara, T.; Zhou, D.Y.; Suzuki, T.; Kawano, T. Nickel-Catalyzed Construction of Chiral 1-[6]Helicenols and Application in the Synthesis of [6]Helicene-Based Phosphinite Ligands. Eur. J. Org. Chem. 2016, 2016, 4948–4952. [Google Scholar] [CrossRef]
- Yamamoto, K.; Shimizu, T.; Igawa, K.; Tomooka, K.; Hirai, G.; Suemune, H.; Usui, K. Rational Design and Synthesis of [5]Helicene-Derived Phosphine Ligands and Their Application in Pd-Catalyzed Asymmetric Reactions. Sci. Rep. 2016, 6, 36211. [Google Scholar] [CrossRef]
- Demmer, C.S.; Voituriez, A.; Marinetti, A. Catalytic Uses of Helicenes Displaying Phosphorus Functions. Comptes Rendus Chim. 2017, 20, 860–879. [Google Scholar] [CrossRef]
- Cauteruccio, S.; Licandro, E.; Panigati, M.; D’Alfonso, G.; Maiorana, S. Modifying the Properties of Organic Molecules by Conjugation with Metal Complexes: The Case of Peptide Nucleic Acids and of the Intrinsically Chiral Thiahelicenes. Coord. Chem. Rev. 2019, 386, 119–137. [Google Scholar] [CrossRef]
- Beránek, T.; Žádný, J.; Strašák, T.; Karban, J.; Císařová, I.; Sýkora, J.; Storch, J. Synthesis of a Helical Phosphine and a Catalytic Study of Its Palladium Complex. ACS Omega 2020, 5, 882–892. [Google Scholar] [CrossRef]
- Medena, C.; Aubert, C.; Derat, E.; Fensterbank, L.; Gontard, G.; Khaled, O.; Ollivier, C.; Vanthuyne, N.; Petit, M.; Barbazanges, M. Helical Bisphosphinites in Asymmetric Tsuji-Trost Allylation: A Remarkable P:Pd Ratio Effect. ChemCatChem 2021, 13, 4543–4548. [Google Scholar] [CrossRef]
- Kel, O.; Fürstenberg, A.; Mehanna, N.; Nicolas, C.; Laleu, B.; Hammarson, M.; Albinsson, B.; Lacour, J.; Vauthey, E. Chiral Selectivity in the Binding of [4]Helicene Derivatives to Double-Stranded DNA. Chem. Eur. J. 2013, 19, 7173–7180. [Google Scholar] [CrossRef]
- Kawara, K.; Tsuji, G.; Taniguchi, Y.; Sasaki, S. Synchronized Chiral Induction between [5]Helicene–Spermine Ligand and B-Z DNA Transition. Chem. Eur. J. 2017, 23, 1763–1769. [Google Scholar] [CrossRef]
- Kalachyova, Y.; Guselnikova, O.; Elashnikov, R.; Panov, I.; Žádný, J.; Církva, V.; Storch, J.; Sykora, J.; Zaruba, K.; Švorčík, V.; et al. Helicene-SPP-Based Chiral Plasmonic Hybrid Structure: Toward Direct Enantiomers SERS Discrimination. ACS Appl. Mater. Interfaces 2019, 11, 1555–1562. [Google Scholar] [CrossRef]
- Schulte, T.R.; Holstein, J.J.; Clever, G.H. Chiral Self-Discrimination and Guest Recognition in Helicene-Based Coordination Cages. Angew. Chem. 2019, 131, 5618–5622. [Google Scholar] [CrossRef]
- Petdum, A.; Faichu, N.; Sirirak, J.; Khammultri, P.; Promarak, V.; Panchan, W.; Sooksimuang, T.; Charoenpanich, A.; Wanichacheva, N. [5]Helicene-Rhodamine 6 G Hybrid-Based Sensor for Ultrasensitive Hg2+ Detection and Its Biological Applications. J. Photochem. Photobiol. A 2020, 394, 112473. [Google Scholar] [CrossRef]
- Gu, X.H.; Lei, Y.; Wang, S.; Cao, F.; Zhang, Q.; Chen, S.; Wang, K.P.; Hu, Z.Q. Tetrahydro[5]Helicene Fused Nitrobenzoxadiazole as a Fluorescence Probe for Hydrogen Sulfide, Cysteine/Homocysteine and Glutathione. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 229, 118003. [Google Scholar] [CrossRef]
- Li, J.; Martin, K.; Avarvari, N.; Wäckerlin, C.; Ernst, K.H. Spontaneous Separation of On-Surface Synthesized Tris-Helicenes into Two-Dimensional Homochiral Domains. Chem. Commun. 2018, 54, 7948–7951. [Google Scholar] [CrossRef]
- Shiotari, A.; Tanaka, K.; Nakae, T.; Mori, S.; Okujima, T.; Uno, H.; Sakaguchi, H.; Sugimoto, Y. Chiral Discrimination and Manipulation of Individual Heptahelicene Molecules on Cu(001) by Noncontact Atomic Force Microscopy. J. Phys. Chem. C 2018, 122, 4997–5003. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Shen, C.; Gan, F.; Su, X.; Qiu, H.; Yang, B.; Yu, P. Chiral Recognition of Hexahelicene on a Surface via the Forming of Asymmetric Heterochiral Trimers. Int. J. Mol. Sci. 2019, 20, 2018. [Google Scholar] [CrossRef]
- Irziqat, B.; Berger, J.; Mendieta-Moreno, J.I.; Sundar, M.S.; Bedekar, A.V.; Ernst, K.H. Transition from Homochiral Clusters to Racemate Monolayers during 2D Crystallization of Trioxa[11]Helicene on Ag(100). ChemPhysChem 2021, 22, 293–297. [Google Scholar] [CrossRef]
- Matsuiwa, K.; Hayashi, S.; Nakanishi, W. Dynamic and Static Behavior of Intramolecular π-π Interactions in [2.2]- and [3.3]Cyclophanes, Elucidated by QTAIM Dual Functional Analysis with QC Calculations. ChemistrySelect 2017, 2, 1774–1782. [Google Scholar] [CrossRef]
- Matsuiwa, K.; Hayashi, S.; Nakanishi, W. Intramolecular π-π Interactions in Diethanodihydronaphthalene and Derivatives: Dynamic and Static Behavior of the Interactions Elucidated by QTAIM Dual Functional Analysis. ChemistrySelect 2016, 1, 2344–2353. [Google Scholar] [CrossRef]
- Matsuiwa, K.; Sugibayashi, Y.; Tsubomoto, Y.; Hayashi, S.; Nakanishi, W. Behavior of Intramolecular π-π Interactions with Doubly Degenerated Bond Paths between Carbon Atoms in Opposite Benzene Rings of Diethenodihydronaphthalenes by QTAIM Approach. ChemistrySelect 2017, 2, 90–100. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S.; Narahara, K. Atoms-in-Molecules Dual Parameter Analysis of Weak to Strong Interactions: Behaviors of Electronic Energy Densities versus Laplacian of Electron Densities at Bond Critical Points. J. Phys. Chem. A 2008, 112, 13593–13599. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S.; Narahara, K. Polar Coordinate Representation of Hb(rc) versus (ℏ2/8m)∇2ρb(rc) at BCP in AIM Analysis: Classification and Evaluation of Weak to Strong Interactions. J. Phys. Chem. A 2009, 113, 10050–10057. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S. Atoms-in-Molecules Dual Functional Analysis of Weak to Strong Interactions. Curr. Org. Chem. 2010, 14, 181–197. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S. Dynamic Behaviors of Interactions: Application of Normal Coordinates of Internal Vibrations to AIM Dual Functional Analysis. J. Phys. Chem. A 2010, 114, 7423–7430. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, W.; Hayashi, S.; Matsuiwa, K.; Kitamoto, M. Applications of Normal Coordinates of Internal Vibrations to Generate Perturbed Structures: Dynamic Behavior of Weak to Strong Interactions Elucidated by Atoms-in-Molecules Dual Functional Analysis. Bull. Chem. Soc. Jpn. 2012, 85, 1293–1305. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S. Role of dG/dw and dV/dw in AIM Analysis: An Approach to the Nature of Weak to Strong Interactions. J. Phys. Chem. A 2013, 117, 1795–1803. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Matta, C.F.; Boyd, R.J. An introduction to the quantum theory of atoms in molecules. In The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley: Weinheim, Germany, 2007. [Google Scholar] [CrossRef]
- Nakanishi, W.; Hayashi, S. Perturbed Structures Generated Using Coordinates Derived from Compliance Constants in Internal Vibrations for QTAIM Dual Functional Analysis: Intrinsic Dynamic Nature of Interactions. Int. J. Quantum Chem. 2018, 118, e25590. [Google Scholar] [CrossRef]
- Grunenberg, J.; Brandhorst, K. Compliance 3.0.2 program. Available online: http://www.oc.tu–bs.de/Grunenberg/compliance.html (accessed on 14 December 2021).
- Brandhorst, K.; Grunenberg, J. Efficient Computation of Compliance Matrices in Redundant Internal Coordinates from Cartesian Hessians for Nonstationary Points. J. Chem. Phys. 2010, 132, 184101. [Google Scholar] [CrossRef]
- Brandhorst, K.; Grunenberg, J. How Strong Is It? The Interpretation of Force and Compliance Constants as Bond Strength Descriptors. Chem. Soc. Rev. 2008, 37, 1558–1567. [Google Scholar] [CrossRef]
- Grunenberg, J. Ill-Defined Concepts in Chemistry: Rigid Force Constants vs. Compliance Constants as Bond Strength Descriptors for the Triple Bond in Diboryne. Chem. Sci. 2015, 6, 4086–4088. [Google Scholar] [CrossRef]
- Grunenberg, J. Direct Assessment of Interresidue Forces in Watson-Crick Base Pairs Using Theoretical Compliance Constants. J. Am. Chem. Soc. 2004, 126, 16310–16311. [Google Scholar] [CrossRef]
- Grunenberg, J.; Barone, G. Are Compliance Constants Ill-Defined Descriptors for Weak Interactions? RSC Adv. 2013, 3, 4757–4762. [Google Scholar] [CrossRef]
- Taylor, W.J.; Pitzer, K.S. Vibrational Frequencies of Semirigid Molecules: A General Method and Values for Ethylbenzene. J. Res. Natl. Bur. Stand. 1947, 38, 1–17. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. I. Derivation of Adiabatic Internal Modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Grunenberg, J.; Goldberg, N. How Strong Is the Gallium≡Gallium Triple Bond? Theoretical Compliance Matrices as a Probe for Intrinsic Bond Strengths. J. Am. Chem. Soc. 2000, 122, 6045–6047. [Google Scholar] [CrossRef]
- Grunenberg, J.; Streubel, R.; Frantzius, G.V.; Marten, W. The Strongest Bond in the Universe? Accurate Calculation of Compliance Matrices for the Ions N2H+, HCO+, and HOC+. J. Chem. Phys. 2003, 119, 165–169. [Google Scholar] [CrossRef]
- Xie, Y.; Schaefer, H.F. The Characterization of Metal–Metal Bonds in Unsaturated Binuclear Homoleptic Transition Metal Carbonyls. The Compliance Matrix. Z. Phys. Chem. 2003, 217, 189–203. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (Revision D.01); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a Long–Range Corrected Hybrid Functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef]
- Hayashi, S.; Nishide, T.; Ueda, K.; Hayami, K.; Nakanishi, W. Effects from Basis Sets and Levels of Calculations on the Nature of Interactions Predicted by QTAIM-Dual Functional Analysis with QTAIM Functions. ChemistrySelect 2019, 4, 6198–6208. [Google Scholar] [CrossRef]
- Ivanov, M.D.; Kirina, Y.V.; Novikov, A.S.; Starova, G.L.; Kukushkin, V.Y. Efficient π–stacking with benzene provides 2D assembly of trans–[PtCl2(p–CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Mukhacheva, A.A.; Komarov, V.Y.; Kokovkin, V.V.; Novikov, A.S.; Abramov, P.A.; Sokolov, M.N. Unusual p-p interactions directed by the [{(C6H6)Ru}2W8O30(OH)2]6− hybrid anion. CrystEngComm 2021, 23, 4125–4135. [Google Scholar] [CrossRef]
- Biegler–König, F.; Schönbohm, J. The AIM2000 Program (Version 2.0). Available online: http://www.aim2000.de (accessed on 14 December 2021).
- Biegler–König, F. Calculation of atomic integration data. J. Comput. Chem. 2000, 21, 1040–1048. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 17.11.14), TK Gristmill Software, Overland Park, KS, USA. 2017. Available online: http://aim.tkgristmill.com (accessed on 14 December 2021).
- Kay, M.I.; Okaya, Y.; Cox, D.E. A Refinement of the Structure of the Room–Temperature Phase of Phenanthrene, C14H10, from X-ray and Neutron Diffraction Data. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1971, 27, 26–33. [Google Scholar] [CrossRef]
- Lakshman, M.K.; Kole, P.L.; Chaturvedi, S.; Saugier, J.H.; Yeh, H.J.C.; Glusker, J.P.; Carrell, H.L.; Katz, A.K.; Afshar, C.E.; Dashwood, W.M.; et al. Methyl Group-Induced Helicity in 1,4-Dimethylbenzo[c]Phenanthrene and Its Metabolites: Synthesis, Physical, and Biological Properties. J. Am. Chem. Soc. 2000, 122, 12629–12636. [Google Scholar] [CrossRef]
- Bédard, A.C.; Vlassova, A.; Hernandez-Perez, A.C.; Bessette, A.; Hanan, G.S.; Heuft, M.A.; Collins, S.K. Synthesis, Crystal Structure and Photophysical Properties of Pyrene-Helicene Hybrids. Chem. Eur. J. 2013, 19, 16295–16302. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakamura, Y.; Kishida, H.; Hayama, H.; Nakano, Y.; Yamochi, H.; Saito, G. Racemic Charge–Transfer Complexes of a Helical Polycyclic Aromatic Hydrocarbon Molecule. CrystEngComm 2017, 19, 3626–3632. [Google Scholar] [CrossRef]
- Fujino, S.; Yamaji, M.; Okamoto, H.; Mutai, T.; Yoshikawa, I.; Houjou, H.; Tani, F. Systematic Investigations on Fused π-System Compounds of Seven Benzene Rings Prepared by Photocyclization of Diphenanthrylethenes. Photochem. Photobiol. Sci. 2017, 16, 925–934. [Google Scholar] [CrossRef]
- Mori, K.; Murase, T.; Fujita, M. One-Step Synthesis of [16]Helicene. Angew. Chem. 2015, 127, 6951–6955. [Google Scholar] [CrossRef]
- Bas, G.L.; Navaza, A.; Mauguen, Y.; Rango, C.D. 10-Helicene, C42H24. Cryst. Struct. Commun. 1976, 5, 357–361. [Google Scholar]
- Bas, G.L.; Navaza, A.; Knossow, M.; Rango, C.D. 11-Helicene, C46H26. Cryst. Struct. Commun. 1976, 5, 713–718. [Google Scholar]
- Rulíšek, L.; Exner, O.; Cwiklik, L.; Jungwirth, P.; Starý, I.; Pospíšil, L.; Havias, Z. On the Convergence of the Physicochemical Properties of [n]Helicenes. J. Phys. Chem. C 2007, 111, 14948–14955. [Google Scholar] [CrossRef]
- Krygowski, T.M. Crystallographic Studies of Inter– and Intramolecular Interactions Reflected in Aromatic Character of π–Electron Systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Bader, R.F.W. Bond Paths Are Not Chemical Bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef]
- Grimme, S.; Mück-Lichtenfeld, C.; Erker, G.; Kehr, G.; Wang, H.; Beckers, H.; Willner, H. When Do Interacting Atoms Form a Chemical Bond? Spectroscopic Measurements and Theoretical Analyses of Dideuteriophenanthrene. Angew. Chem. Int. 2009, 48, 2592–2595. [Google Scholar] [CrossRef]
- Cukrowski, I. A Unified Molecular–Wide and Electron Density Based Concept of Chemical Bonding. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, e1579. [Google Scholar] [CrossRef]
- Pendás, A.M.; Francisco, E.; Blanco, M.A.; Gatti, C. Bond Paths as Privileged Exchange Channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef]
- Tognetti, V.; Joubert, L. On the Physical Role of Exchange in the Formation of an Intramolecular Bond Path between Two Electronegative Atoms. J. Chem. Phys. 2013, 138, 024102. [Google Scholar] [CrossRef]
- Cukrowski, I.; de Lange, J.H.; Adeyinka, A.S.; Mangondo, P. Evaluating Common QTAIM and NCI Interpretations of the Electron Density Concentration through IQA Interaction Energies and 1D Cross–Sections of the Electron and Deformation Density Distributions. Comput. Theor. Chem. 2015, 1053, 60–76. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | ρb(rc) | c∇2ρb(rc) 4 | Hb(rc) | R 5 | θ 6 | Cii 7 | θp 8 | κp 9 | Predicted Nature |
|---|---|---|---|---|---|---|---|---|---|
| X-∗-Y | (eao–3) | (au) | (au) | (au) | (°) | (Å mdyn−1) | (°) | (au−1) | |
| 3 (1Hbay-∗-4Hbay) | 0.0130 | 0.0060 | 0.0020 | 0.0063 | 71.3 | 3.29 | 72.8 | 12.4 | p-CS/vdW |
| 4 (1Hbay-∗-5Hbay) | 0.0165 | 0.0078 | 0.0026 | 0.0082 | 71.8 | 6.67 | 74.5 | 11.4 | p-CS/vdW |
| 5 (1Hbay-∗-6Cbay) | 0.0131 | 0.0063 | 0.0021 | 0.0066 | 71.6 | 6.12 | 80.8 | 136 | p-CS/vdW |
| 6 (1Hbay-∗-5Cbay) | 0.0131 | 0.0060 | 0.0020 | 0.0063 | 71.3 | 3.82 | 79.9 | 31.8 | p-CS/vdW |
| 7 (1Hbay-∗-6Cbay) | 0.0135 | 0.0063 | 0.0021 | 0.0067 | 71.5 | 5.47 | 77.9 | 188 | p-CS/vdW |
| 7 (2Cbay-∗-7Cbay) | 0.0114 | 0.0051 | 0.0017 | 0.0054 | 71.9 | 3.33 | 80.6 | 128 | p-CS/vdW |
| 8 (1Hbay-∗-6Cbay) | 0.0130 | 0.0061 | 0.0021 | 0.0065 | 71.1 | 5.51 | 76.7 | 189 | p-CS/vdW |
| 8 (2Cbay-∗-7Cbay) | 0.0117 | 0.0052 | 0.0017 | 0.0055 | 71.8 | 2.01 | 86.4 | 28.5 | p-CS/vdW |
| 9 (1Hbay-∗-5Cbay) | 0.0134 | 0.0062 | 0.0022 | 0.0066 | 70.7 | 3.40 | 79.5 | 627 | p-CS/vdW |
| 9 (2Cbay-∗-7Cbay) | 0.0113 | 0.0050 | 0.0016 | 0.0053 | 72.1 | 1.83 | 81.6 | 119 | p-CS/vdW |
| 9 (3Cbay-∗-8Cbay) | 0.0122 | 0.0053 | 0.0016 | 0.0056 | 72.9 | 1.87 | 85.0 | 196 | p-CS/vdW |
| 9 (4Ccape-∗-22Ccape) 10 | 0.0055 | 0.0020 | 0.0007 | 0.0022 | 70.7 | 5.25 | 66.9 | 135 | p-CS/vdW |
| 9 (6Ccape-∗-23Ccape) | 0.0061 | 0.0021 | 0.0007 | 0.0022 | 70.4 | 8.51 | 69.4 | 37.7 | p-CS/vdW |
| 10 (1Hbay-∗-6Cbay) | 0.0137 | 0.0064 | 0.0021 | 0.0067 | 71.6 | 5.74 | 79.2 | 123 | p-CS/vdW |
| 10 (2Cbay-∗-7Cbay) 11 | 0.0113 | 0.0050 | 0.0018 | 0.0053 | 70.5 | 1.86 | 94.2 | 2890 | p-CS/t-HBnc |
| 10 (3Cbay-∗-8Cbay) | 0.0114 | 0.0050 | 0.0016 | 0.0053 | 72.1 | 1.78 | 82.0 | 182 | p-CS/vdW |
| 10 (6Ccape-∗-23Ccape) | 0.0059 | 0.0020 | 0.0007 | 0.0021 | 70.4 | 9.02 | 69.7 | 7.0 | p-CS/vdW |
| 10 (7Ccape-∗-25Ccape) | 0.0061 | 0.0022 | 0.0008 | 0.0024 | 70.3 | 3.42 | 68.0 | 10.3 | p-CS/vdW |
| 11 (1Hbay-∗-6Cbay) | 0.0136 | 0.0063 | 0.0021 | 0.0067 | 71.6 | 5.41 | 79.8 | 113 | p-CS/vdW |
| 11 (2Cbay-∗-7Cbay) | 0.0116 | 0.0051 | 0.0017 | 0.0054 | 71.5 | 1.84 | 87.1 | 142 | p-CS/vdW |
| 11 (3Cbay-∗-8Cbay) | 0.0115 | 0.0050 | 0.0016 | 0.0053 | 72.0 | 1.91 | 82.6 | 166 | p-CS/vdW |
| 11 (4Cbay-∗-9Cbay) | 0.0111 | 0.0049 | 0.0016 | 0.0052 | 71.7 | 1.69 | 79.5 | 155 | p-CS/vdW |
| 11 (4Ccape-∗-22Ccape) | 0.0053 | 0.0019 | 0.0007 | 0.0020 | 70.1 | 6.81 | 68.3 | 13.5 | p-CS/vdW |
| 11 (6Ccape-∗-23Ccape) | 0.0059 | 0.0020 | 0.0007 | 0.0021 | 70.2 | 10.21 | 69.7 | 7.5 | p-CS/vdW |
| 11 (7Ccape-∗-25Ccape) | 0.0059 | 0.0022 | 0.0008 | 0.0023 | 70.2 | 3.58 | 67.8 | 7.2 | p-CS/vdW |
| 11 (9Ccape-∗-26Ccape) | 0.0062 | 0.0021 | 0.0007 | 0.0022 | 70.4 | 5.80 | 69.5 | 64.7 | p-CS/vdW |
| 12 (1Hbay-∗-6Cbay) | 0.0136 | 0.0063 | 0.0021 | 0.0067 | 71.5 | 4.84 | 80.5 | 103 | p-CS/vdW |
| 12 (2Cbay-∗-7Cbay) | 0.0115 | 0.0051 | 0.0017 | 0.0053 | 71.5 | 1.77 | 87.8 | 349 | p-CS/vdW |
| 12 (3Cbay-∗-8Cbay) | 0.0117 | 0.0051 | 0.0016 | 0.0053 | 72.3 | 1.74 | 83.6 | 241 | p-CS/vdW |
| 12 (4Cbay-∗-9Cbay) | 0.0110 | 0.0048 | 0.0016 | 0.0051 | 71.5 | 1.72 | 80.7 | 87.8 | p-CS/vdW |
| 12 (4Ccape-∗-22Ccape) | 0.0055 | 0.0020 | 0.0007 | 0.0022 | 70.2 | 4.80 | 66.3 | 697 | p-CS/vdW |
| 12 (6Ccape-∗-23Ccape) | 0.0060 | 0.0021 | 0.0008 | 0.0022 | 70.0 | 6.78 | 68.0 | 8.6 | p-CS/vdW |
| 12 (7Ccape-∗-25Ccape) | 0.0059 | 0.0022 | 0.0008 | 0.0023 | 70.0 | 3.41 | 68.2 | 3.3 | p-CS/vdW |
| 12 (9Ccape-∗-26Ccape) | 0.0059 | 0.0020 | 0.0007 | 0.0021 | 70.3 | 6.95 | 69.4 | 24.7 | p-CS/vdW |
| 12 (10Ccape-∗-28Ccape) | 0.0056 | 0.0020 | 0.0007 | 0.0022 | 70.5 | 4.09 | 68.6 | 65.4 | p-CS/vdW |
| Species | ρb(rc) | c∇2ρb(rc) | Hb(rc) | R | θ | Cii | θp | κp | Predicted Nature |
|---|---|---|---|---|---|---|---|---|---|
| X-∗-Y | (eao–3) | (au) | (au) | (au) | (°) | (Å mdyn−1) | (°) | (au−1) | |
| 6:6 (1Hbay-∗-17’Hcape) 4 | 0.0045 | 0.0018 | 0.0006 | 0.0019 | 72.6 | 25.02 | 88.1 | 5163 | p-CS/vdW |
| 6:6 (1Hcape-∗-17’Hcape) | 0.0061 | 0.0022 | 0.0006 | 0.0023 | 74.3 | 35.63 | 76.2 | 184.2 | p-CS/vdW |
| 6:6 (1Hcape-∗-16’Hcape) | 0.0051 | 0.0019 | 0.0007 | 0.0020 | 70.9 | 42.01 | 74.9 | 69.9 | p-CS/vdW |
| 6:6 (15Ccape-∗-17’Ccape) 5 | 0.0065 | 0.0025 | 0.0009 | 0.0027 | 69.2 | 12.64 | 70.8 | 1066 | p-CS/vdW |
| 6:6 (16Ccape-∗-17’Ccape) | 0.0066 | 0.0026 | 0.0010 | 0.0028 | 68.3 | 8.63 | 68.0 | 53.5 | p-CS/vdW |
| 6:6 (1Hbay-∗-5Cbay) | 0.0128 | 0.0057 | 0.0019 | 0.0060 | 71.9 | 3.817 | 78.3 | 32.3 | p-CS/vdW |
| 6:6 (3Cbay-∗-7Hbay) | 0.0134 | 0.0061 | 0.0020 | 0.0064 | 71.7 | 3.414 | 79.3 | 29.5 | p-CS/vdW |
| 7:7 (20Hcape-∗-18’Hcape) | 0.0073 | 0.0031 | 0.0012 | 0.0033 | 68.9 | 14.61 | 75.5 | 24.6 | p-CS/vdW |
| 7:7 (20Hcape-∗-20’Hcape) | 0.0054 | 0.0022 | 0.0008 | 0.0024 | 70.0 | 27.90 | 72.7 | 36.5 | p-CS/vdW |
| 7:7 (20Hcape-∗-2’Ccape) | 0.0079 | 0.0030 | 0.0010 | 0.0032 | 71.4 | 10.01 | 73.3 | 125.7 | p-CS/vdW |
| 7:7 (18Hcape-∗-3’Ccape) | 0.0050 | 0.0016 | 0.0005 | 0.0016 | 73.8 | 17.36 | 73.2 | 181.8 | p-CS/vdW |
| 7:7 (1Hbay-∗-6Cbay) | 0.0132 | 0.0062 | 0.0021 | 0.0065 | 70.8 | 5.557 | 80.0 | 93.5 | p-CS/vdW |
| 7:7 (3Cbay-∗-8Hbay) | 0.0138 | 0.0065 | 0.0021 | 0.0068 | 71.9 | 5.024 | 78.8 | 154.1 | p-CS/vdW |
| Species | r(X···Y) | R | θ | Cii | θp | κp | Predicted Nature |
|---|---|---|---|---|---|---|---|
| X-∗-Y | (Å) | (au) | (°) | (Å mdyn−1) | (°) | (au−1) | |
| 7 (M06-2X/6-311+G(3d,p): ν1 = 43.4 cm−1) | |||||||
| 1Hbay-∗-6Cbay | 2.5902 | 0.0067 | 71.5 | 5.47 | 77.9 | 187.5 | p-CS/vdW |
| 2Cbay-∗-7Cbay | 2.9751 | 0.0054 | 71.9 | 3.33 | 80.6 | 128.2 | p-CS/vdW |
| 7 (M06-2X/6-311+G(2d,p): ν1 = 46.6 cm−1) | |||||||
| 1Hbay-∗-6Cbay | 2.5896 | 0.0067 | 70.4 | 5.49 | 81.6 | 39.9 | p-CS/vdW |
| 2Cbay-∗-7Cbay | 3.0025 | 0.0054 | 69.9 | 3.06 | 78.7 | 120.2 | p-CS/vdW |
| 7 (LC-ωPBE/6-311+G(2d,p): ν1 = 40.1 cm−1) 3,4 | |||||||
| 1Hbay-∗-5Cbay | 2.4681 | 0.0066 | 70.5 | 3.72 | 82.2 | 46.4 | p-CS/vdW |
| 7:7 (M06-2X/6-311+G(3d,p): ν1 = 14.0 cm−1; ν4 = 24.2 cm−1; ν5 = 29.3 cm−1; ν11 = 81.2 cm−1) | |||||||
| 20Hcape-∗-18’Hcape | 2.5423 | 0.0033 | 68.9 | 14.61 | 75.5 | 24.6 | p-CS/vdW |
| 20Hcape-∗-20’Hcape | 2.7155 | 0.0024 | 70.0 | 27.90 | 72.7 | 36.5 | p-CS/vdW |
| 20Hcape-∗-2’Ccape | 2.6769 | 0.0032 | 71.4 | 10.01 | 73.3 | 125.7 | p-CS/vdW |
| 18Hcape-∗-3’Ccape | 2.9640 | 0.0016 | 73.8 | 17.36 | 73.2 | 181.8 | p-CS/vdW |
| 7:7 (M06-2X/6-311+G(2d,p): ν1 = 13.0 cm−1; ν4 = 22.1 cm−1; ν5 = 27.3 cm−1; ν11 = 78.6 cm−1) | |||||||
| 20Hcape-∗-18’Hcape | 2.5643 | 0.0032 | 67.9 | 16.63 | 75.2 | 60.1 | p-CS/vdW |
| 20Hcape-∗-20’Hcape | 2.7287 | 0.0024 | 68.6 | 29.20 | 71.9 | 42.2 | p-CS/vdW |
| 20Hcape-∗-2’Ccape | 2.6881 | 0.0031 | 70.2 | 11.44 | 69.4 | 12.4 | p-CS/vdW |
| 18Hcape-∗-3’Ccape | 2.9857 | 0.0016 | 73.0 | 31.43 | 73.2 | 181.9 | p-CS/vdW |
| 7:7 (LC-ωPBE/6-311+G(2d,p): ν1 = 2.1 cm−1; ν5 = 15.8 cm−1; ν6 = 28.8 cm−1; ν8 = 48.2 cm−1) 5,6 | |||||||
| 20Ccape-∗-18’Hcape | 3.2357 | 0.0015 | 63.8 | 65.63 | 67.4 | 40.4 | p-CS/vdW |
| 20Hcape-∗-20’Hcape | 3.0148 | 0.0014 | 63.6 | 96.42 | 66.9 | 90.7 | p-CS/vdW |
| 20Hcape-∗-2’Ccape | 3.0656 | 0.0014 | 67.2 | 52.66 | 70.0 | 10.9 | p-CS/vdW |
| 18Hcape-∗-3’Ccape | 3.6594 | 0.0005 | 62.9 | 73.99 | 73.9 | 394.0 | p-CS/vdW |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishide, T.; Hayashi, S. Intrinsic Dynamic and Static Nature of π···π Interactions in Fused Benzene-Type Helicenes and Dimers, Elucidated with QTAIM Dual Functional Analysis. Nanomaterials 2022, 12, 321. https://doi.org/10.3390/nano12030321
Nishide T, Hayashi S. Intrinsic Dynamic and Static Nature of π···π Interactions in Fused Benzene-Type Helicenes and Dimers, Elucidated with QTAIM Dual Functional Analysis. Nanomaterials. 2022; 12(3):321. https://doi.org/10.3390/nano12030321
Chicago/Turabian StyleNishide, Taro, and Satoko Hayashi. 2022. "Intrinsic Dynamic and Static Nature of π···π Interactions in Fused Benzene-Type Helicenes and Dimers, Elucidated with QTAIM Dual Functional Analysis" Nanomaterials 12, no. 3: 321. https://doi.org/10.3390/nano12030321
APA StyleNishide, T., & Hayashi, S. (2022). Intrinsic Dynamic and Static Nature of π···π Interactions in Fused Benzene-Type Helicenes and Dimers, Elucidated with QTAIM Dual Functional Analysis. Nanomaterials, 12(3), 321. https://doi.org/10.3390/nano12030321