
Substantial Na-Ion Storage at High Current Rates: Redox-Pseudocapacitance through Sodium Oxide Formation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
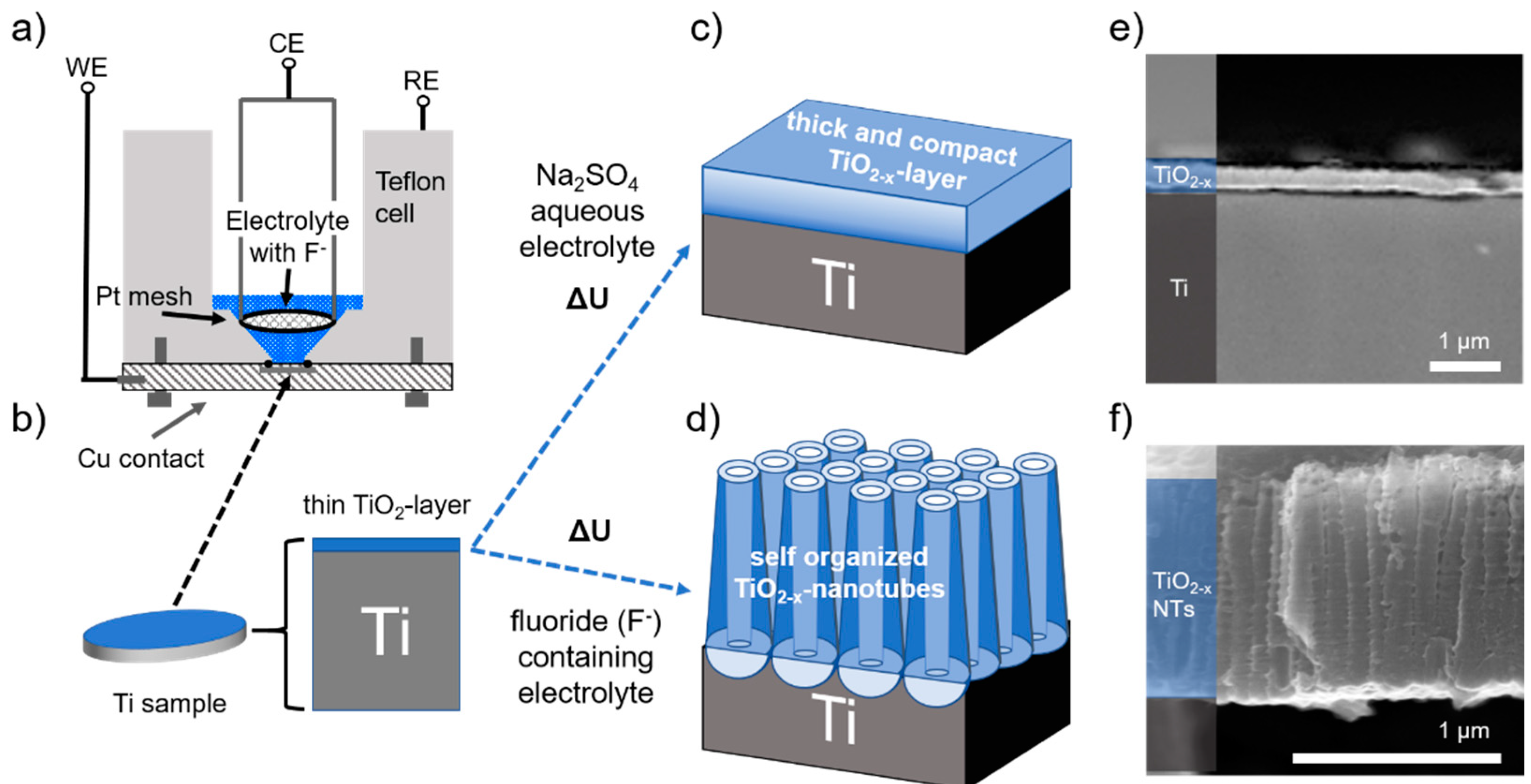
2. Materials and Methods
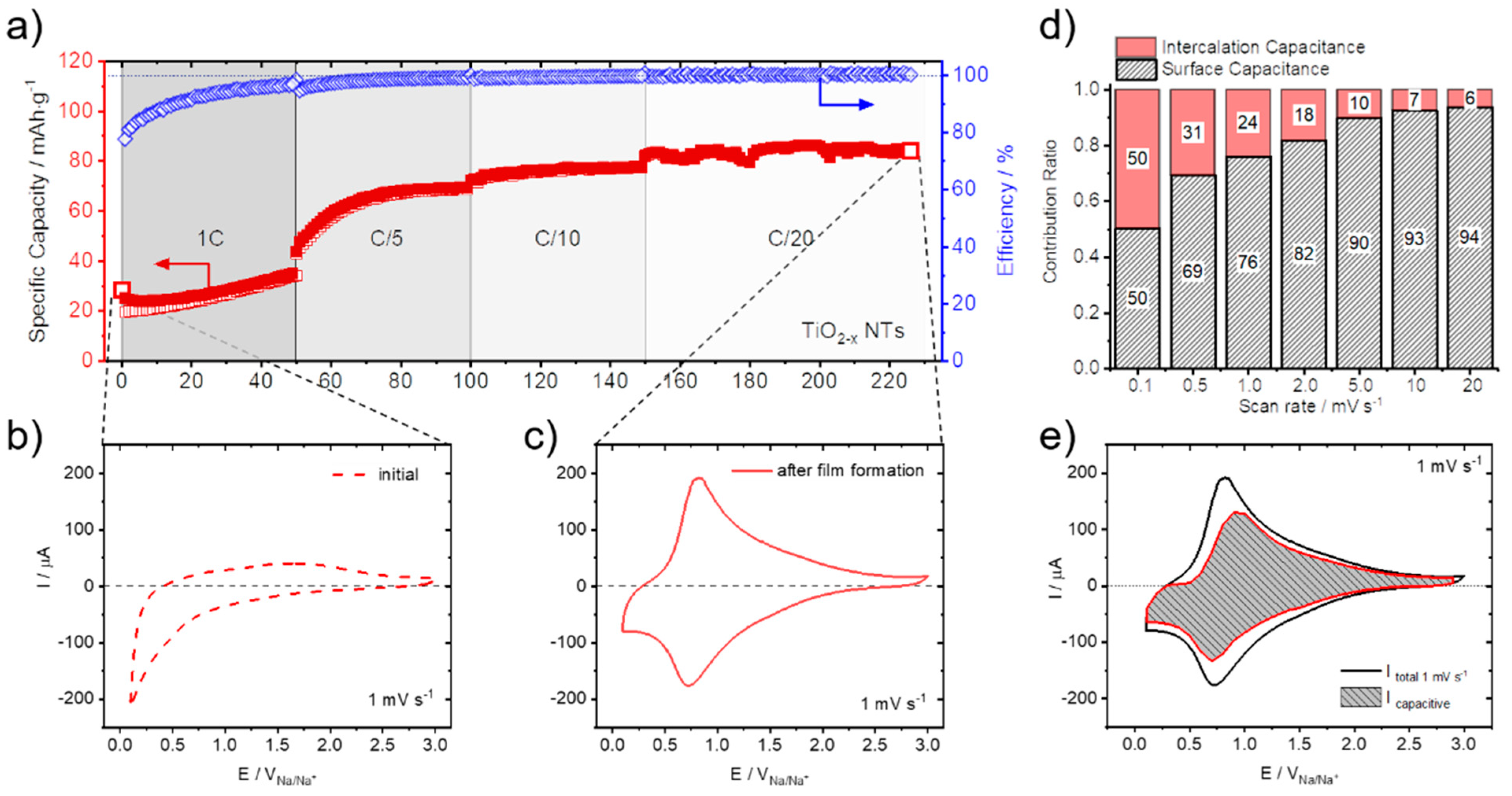
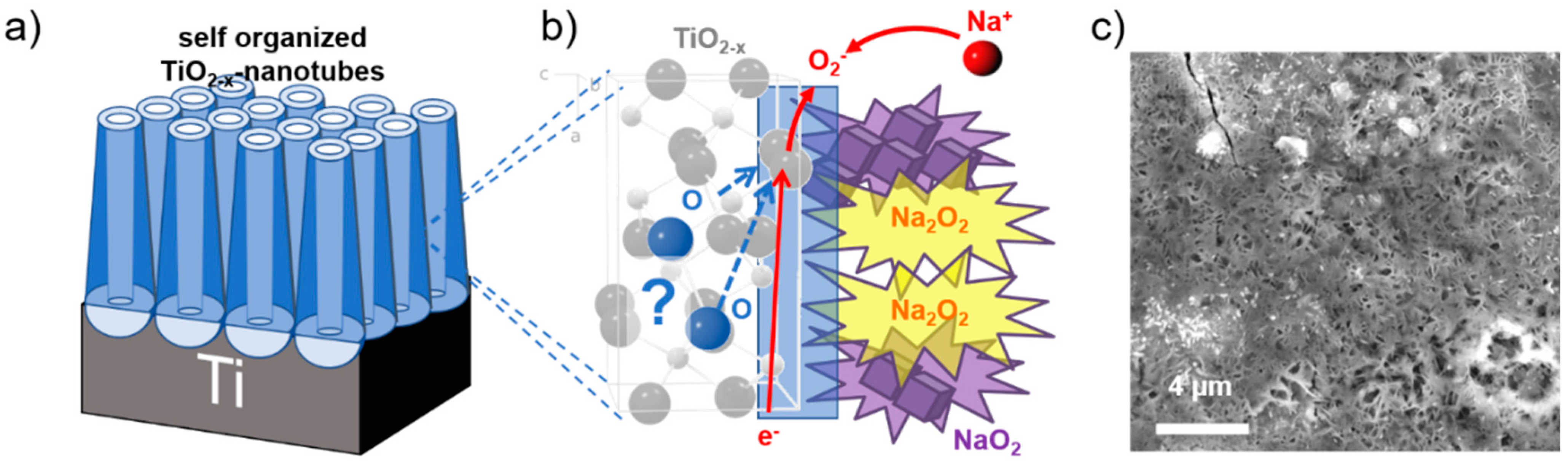
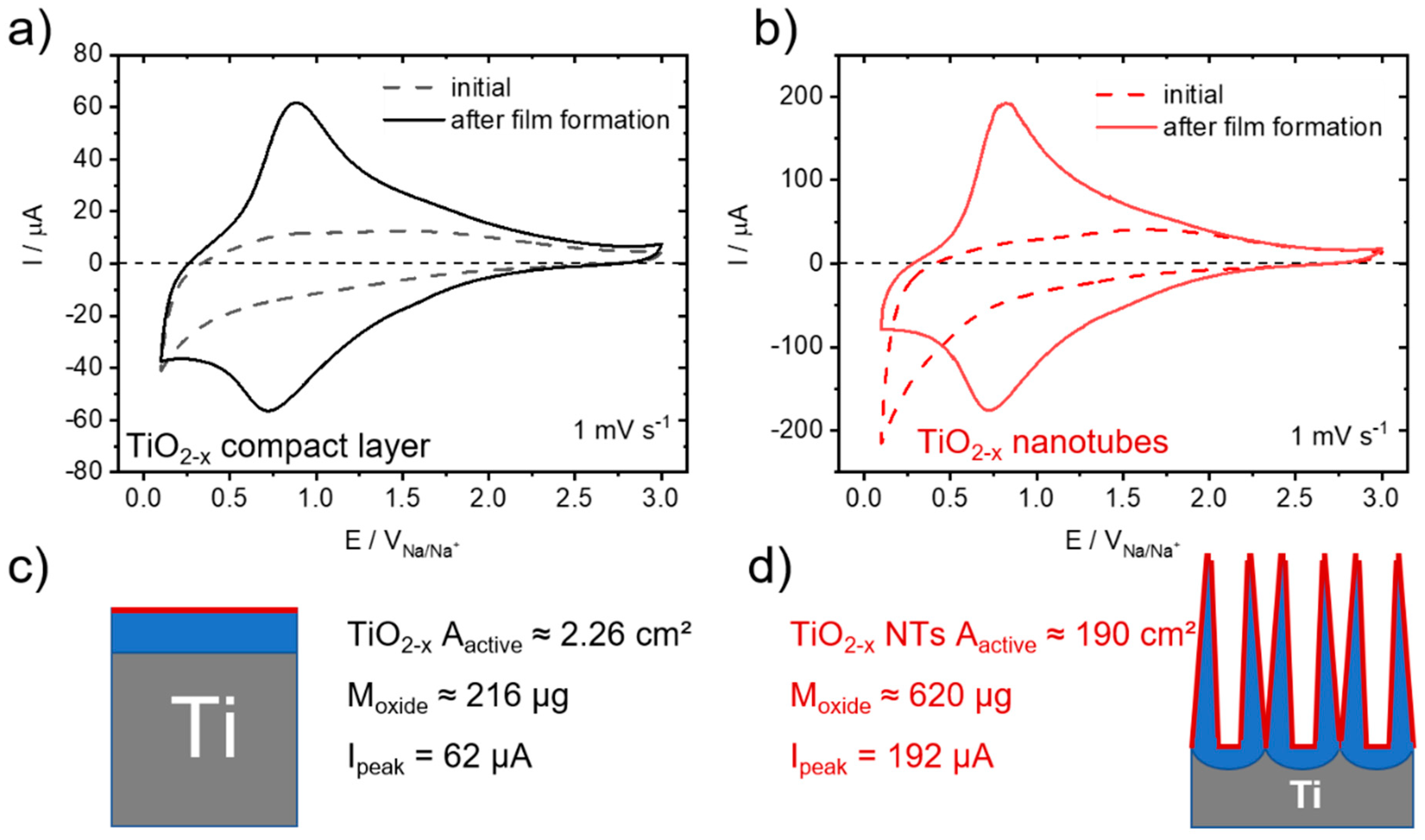
3. Results
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Way, R.; Ives, M.C.; Mealy, P.; Farmer, J.D. Empirically Grounded Technology Forecasts and the Energy Transition. Joule 2022, 6, P2057–P2082. [Google Scholar] [CrossRef]
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M.; Guillén Bolaños, T.; Bindi, M.; Brown, S.; Camilloni, I.A.; Diedhiou, A.; Djalante, R.; Ebi, K.; et al. The Human Imperative of Stabilizing Global Climate Change at 1.5 °C. Science 2019, 365, eaaw6974. [Google Scholar] [CrossRef]
- Byers, E.; Gidden, M.; Leclère, D.; Balkovic, J.; Burek, P.; Ebi, K.; Greve, P.; Grey, D.; Havlik, P.; Hillers, A.; et al. Global Exposure and Vulnerability to Multi-Sector Development and Climate Change Hotspots. Environ. Res. Lett. 2018, 13, 055012. [Google Scholar] [CrossRef]
- Tollefson, J. World’s Oceans Are Losing Power to Stall Climate Change. Nature 2019, 574, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Tarascon, J.-M.; Armand, M. Issues and Challenges Facing Rechargeable Lithium Batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Amine, K.; Kanno, R.; Tzeng, Y. Rechargeable Lithium Batteries and beyond: Progress, Challenges, and Future Directions. MRS Bull. 2014, 39, 395–401. [Google Scholar] [CrossRef]
- Martin, G.; Rentsch, L.; Höck, M.; Bertau, M. Lithium Market Research—Global Supply, Future Demand and Price Development. Energy Storage Mater. 2017, 6, 171–179. [Google Scholar] [CrossRef]
- Vaalma, C.; Buchholz, D.; Weil, M.; Passerini, S. A Cost and Resource Analysis of Sodium-Ion Batteries. Nat. Rev. Mater. 2018, 3, 18013. [Google Scholar] [CrossRef]
- Slater, M.D.; Kim, D.; Lee, E.; Johnson, C.S. Sodium-Ion Batteries. Adv. Funct. Mater. 2013, 23, 947–958. [Google Scholar] [CrossRef]
- Nayak, P.K.; Yang, L.; Brehm, W.; Adelhelm, P. From Lithium-Ion to Sodium-Ion Batteries: Advantages, Challenges, and Surprises. Angew. Chemie Int. Ed. 2018, 57, 102–120. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y.; Dunn, B. Where Do Batteries End and Supercapacitors Begin? Science 2014, 343, 1210–1211. [Google Scholar] [CrossRef]
- Choi, C.; Ashby, D.S.; Butts, D.M.; DeBlock, R.H.; Wei, Q.; Lau, J.; Dunn, B. Achieving High Energy Density and High Power Density with Pseudocapacitive Materials. Nat. Rev. Mater. 2020, 5, 5–19. [Google Scholar] [CrossRef]
- Augustyn, V.; Simon, P.; Dunn, B. Pseudocapacitive Oxide Materials for High-Rate Electrochemical Energy Storage. Energy Environ. Sci. 2014, 7, 1597. [Google Scholar] [CrossRef]
- Hwang, H.; Kim, H.; Cho, J. MoS 2 Nanoplates Consisting of Disordered Graphene-like Layers for High Rate Lithium Battery Anode Materials. Nano Lett. 2011, 11, 4826–4830. [Google Scholar] [CrossRef]
- Gund, G.S.; Dubal, D.P.; Jambure, S.B.; Shinde, S.S.; Lokhande, C.D. Temperature Influence on Morphological Progress of Ni(OH)2 Thin Films and Its Subsequent Effect on Electrochemical Supercapacitive Properties. J. Mater. Chem. A 2013, 1, 4793. [Google Scholar] [CrossRef]
- Werner, D.; Griesser, C.; Stock, D.; Griesser, U.J.; Kunze-Liebhäuser, J.; Portenkirchner, E. Substantially Improved Na-Ion Storage Capability by Nanostructured Organic-Inorganic Polyaniline-TiO2 Composite Electrodes. ACS Appl. Energy Mater. 2020, 3, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Muench, S.; Wild, A.; Friebe, C.; Häupler, B.; Janoschka, T.; Schubert, U.S. Polymer-Based Organic Batteries. Chem. Rev. 2016, 116, 9438–9484. [Google Scholar] [CrossRef] [PubMed]
- Liebl, S.; Werner, D.; Apaydin, D.H.; Wielend, D.; Geistlinger, K.; Portenkirchner, E. Perylenetetracarboxylic Diimide as Diffusion-Less Electrode Material for High-Rate Organic Na-Ion Batteries. Chem. A Eur. J. 2020, 26, 17559–17566. [Google Scholar] [CrossRef]
- Werner, D.; Apaydin, D.H.; Wielend, D.; Geistlinger, K.; Saputri, W.D.; Griesser, U.J.; Dražević, E.; Hofer, T.S.; Portenkirchner, E. Analysis of the Ordering Effects in Anthraquinone Thin Films and Its Potential Application for Sodium Ion Batteries. J. Phys. Chem. C 2021, 125, 3745–3757. [Google Scholar] [CrossRef]
- Werner, D.; Apaydin, D.H.; Portenkirchner, E. An Anthraquinone/Carbon Fiber Composite as Cathode Material for Rechargeable Sodium-Ion Batteries. Batter. Supercaps 2018, 1, 160–168. [Google Scholar] [CrossRef]
- Portenkirchner, E.; Werner, D.; Liebl, S.; Stock, D.; Auer, A.; Kunze-Liebhäuser, J. Self-Improving Na Ion Storage in Oxygen Deficient, Carbon Coated Self-Organized TiO2 Nanotubes. ACS Appl. Energy Mater. 2018, 1, 6646–6653. [Google Scholar] [CrossRef]
- Brumbarov, J.; Vivek, J.P.; Leonardi, S.; Valero-Vidal, C.; Portenkirchner, E.; Kunze-Liebhäuser, J. Oxygen Deficient, Carbon Coated Self-Organized TiO2 Nanotubes as Anode Material for Li-Ion Intercalation. J. Mater. Chem. A 2015, 3, 16469–16477. [Google Scholar] [CrossRef]
- Steiner, D.; Auer, A.; Portenkirchner, E.; Kunze-Liebhäuser, J. The Role of Surface Films during Lithiation of Amorphous and Anatase TiO2 Nanotubes. J. Electroanal. Chem. 2018, 812, 166–173. [Google Scholar] [CrossRef]
- Portenkirchner, E.; Rommel, S.; Szabados, L.; Griesser, C.; Werner, D.; Stock, D.; Kunze-Liebhäuser, J. Sodiation Mechanism via Reversible Surface Film Formation on Metal Oxides for Sodium-ion Batteries. Nano Sel. 2021, 2, 1533–1543. [Google Scholar] [CrossRef]
- Lau, S.; Archer, L.A. Batteries: Discharging the Right Product. Nat. Energy 2016, 1, 16019. [Google Scholar] [CrossRef]
- Kim, J.; Park, H.; Lee, B.; Seong, W.M.; Lim, H.-D.; Bae, Y.; Kim, H.; Kim, W.K.; Ryu, K.H.; Kang, K. Dissolution and Ionization of Sodium Superoxide in Sodium–Oxygen Batteries. Nat. Commun. 2016, 7, 10670. [Google Scholar] [CrossRef]
- Yadegari, H.; Sun, Q.; Sun, X. Sodium-Oxygen Batteries: A Comparative Review from Chemical and Electrochemical Fundamentals to Future Perspective. Adv. Mater. 2016, 28, 7065–7093. [Google Scholar] [CrossRef] [PubMed]
- Portenkirchner, E.; Neri, G.; Lichtinger, J.; Brumbarov, J.; Rüdiger, C.; Gernhäuser, R.; Kunze-Liebhäuser, J. Tracking Areal Lithium Densities from Neutron Activation—Quantitative Li Determination in Self-Organized TiO2 Nanotube Anode Materials for Li-Ion Batteries. Phys. Chem. Chem. Phys. 2017, 19, 8602–8611. [Google Scholar] [CrossRef] [PubMed]
- Solvonic. Catalogue-Electrolytes-Materials; Solvionic: Toulouse, France, 2017. [Google Scholar]
- Young, L. Anodic Oxide Films; Elsevier: London, UK; New York, NY, USA, 1961. [Google Scholar]
- Auer, A.; Kunze-Liebhäuser, J. Recent Progress in Understanding Ion Storage in Self-Organized Anodic TiO2 Nanotubes. Small Methods 2018, 1800385, 1800385. [Google Scholar] [CrossRef]
- Macak, J.M.; Tsuchiya, H.; Ghicov, A.; Yasuda, K.; Hahn, R.; Bauer, S.; Schmuki, P. TiO2 Nanotubes: Self-Organized Electrochemical Formation, Properties and Applications. Curr. Opin. Solid State Mater. Sci. 2007, 11, 3–18. [Google Scholar] [CrossRef]
- Hahn, R.; Schmidt-Stein, F.; Sahnen, J.; Thiemann, S.; Song, Y.; Kunze, J.; Lehto, V.P.; Schmuki, P. Semimetallic TiO2 Nanotubes. Angew. Chem. Int. Ed. 2009, 48, 7236–7239. [Google Scholar] [CrossRef] [PubMed]
- Auer, A.; Steiner, D.; Portenkirchner, E.; Kunze-Liebhäuser, J. Nonequilibrium Phase Transitions in Amorphous and Anatase TiO2 Nanotubes. ACS Appl. Energy Mater. 2018, 1, 1924–1929. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Y.; Xiao, P.; Garcia, B.B.; Zhang, Q.; Zhou, X.; Jeong, Y.-H.; Cao, G. TiO2 Nanotube Arrays Annealed in CO Exhibiting High Performance for Lithium Ion Intercalation. Electrochim. Acta 2009, 54, 6816–6820. [Google Scholar] [CrossRef]
- Fu, L.; Chen, C.C.; Samuelis, D.; Maier, J. Thermodynamics of Lithium Storage at Abrupt Junctions: Modeling and Experimental Evidence. Phys. Rev. Lett. 2014, 112, 208301. [Google Scholar] [CrossRef]
- Shin, J.; Joo, J.H.; Samuelis, D.; Maier, J. Oxygen-Deficient TiO2−δ Nanoparticles via Hydrogen Reduction for High Rate Capability Lithium Batteries. Chem. Mater. 2012, 24, 543–551. [Google Scholar] [CrossRef]
- Borghols, W.J.H.; Lützenkirchen-Hecht, D.; Haake, U.; van Eck, E.R.H.; Mulder, F.M.; Wagemaker, M. The Electronic Structure and Ionic Diffusion of Nanoscale LiTiO2 Anatase. Phys. Chem. Chem. Phys. 2009, 11, 3010. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Bresser, D.; Buchholz, D.; Giffin, G.A.; Castro, C.R.; Ochel, A.; Passerini, S. Unfolding the Mechanism of Sodium Insertion in Anatase TiO2 Nanoparticles. Adv. Energy Mater. 2015, 5, 1401142. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Sun, Q.; Banis, M.N.; Sun, X.; Sham, T.-K. Tracking the Effect of Sodium Insertion/Extraction in Amorphous and Anatase TiO2 Nanotubes. J. Phys. Chem. C 2017, 121, 11773–11782. [Google Scholar] [CrossRef]
- Conway, B.E.; Birss, V.; Wojtowicz, J. The Role and Utilization of Pseudocapacitance for Energy Storage by Supercapacitors. J. Power Sources 1997, 66, 1–14. [Google Scholar] [CrossRef]
- Wang, J.; Polleux, J.; Lim, J.; Dunn, B. Pseudocapacitive Contributions to Electrochemical Energy Storage in TiO2 (Anatase) Nanoparticles. J. Phys. Chem. C 2007, 111, 14925–14931. [Google Scholar] [CrossRef]
- Lindström, H.; Södergren, S.; Solbrand, A.; Rensmo, H.; Hjelm, J.; Hagfeldt, A.; Lindquist, S.-E. Li+ Ion Insertion in TiO2 (Anatase). 2. Voltammetry on Nanoporous Films. J. Phys. Chem. B 1997, 101, 7717–7722. [Google Scholar] [CrossRef]
- Chen, C.; Wen, Y.; Hu, X.; Ji, X.; Yan, M.; Mai, L.; Hu, P.; Shan, B.; Huang, Y. Na+ Intercalation Pseudocapacitance in Graphene-Coupled Titanium Oxide Enabling Ultra-Fast Sodium Storage and Long-Term Cycling. Nat. Commun. 2015, 6, 6929. [Google Scholar] [CrossRef]
- Marmitt, G.G.; Nandi, S.K.; Venkatachalam, D.K.; Elliman, R.G.; Vos, M.; Grande, P.L. Oxygen Diffusion in TiO2 Films Studied by Electron and Ion Rutherford Backscattering. Thin Solid Films 2017, 629, 97–102. [Google Scholar] [CrossRef]
- Bella, F.; Muñoz-García, A.B.; Meligrana, G.; Lamberti, A.; Destro, M.; Pavone, M.; Gerbaldi, C. Unveiling the Controversial Mechanism of Reversible Na Storage in TiO2 Nanotube Arrays: Amorphous versus Anatase TiO2. Nano Res. 2017, 10, 2891–2903. [Google Scholar] [CrossRef]
- Pfeifer, K.; Arnold, S.; Becherer, J.; Das, C.; Maibach, J.; Ehrenberg, H.; Dsoke, S. Can Metallic Sodium Electrodes Affect the Electrochemistry of Sodium-Ion Batteries? Reactivity Issues and Perspectives. ChemSusChem 2019, 12, 3312–3319. [Google Scholar] [CrossRef]
- Zwilling, V.; Aucouturier, M.; Darque-Ceretti, E. Anodic Oxidation of Titanium and TA6V Alloy in Chromic Media. An Electrochemical Approach. Electrochim. Acta 1999, 45, 921–929. [Google Scholar] [CrossRef]
- LeClere, D.J.; Velota, A.; Skeldon, P.; Thompson, G.E.; Berger, S.; Kunze, J.; Schmuki, P.; Habazaki, H.; Nagata, S. Tracer Investigation of Pore Formation in Anodic Titania. J. Electrochem. Soc. 2008, 155, C487. [Google Scholar] [CrossRef]
- Leonardi, S.; Li Bassi, A.; Russo, V.; Di Fonzo, F.; Paschos, O.; Murray, T.M.; Efstathiadis, H.; Kunze, J. TiO2 Nanotubes: Interdependence of Substrate Grain Orientation and Growth Characteristics. J. Phys. Chem. C 2012, 116, 384–392. [Google Scholar] [CrossRef]
- Werner, D.; Alexander, T.; Winkler, D.; Apaydin, D.H.; Loerting, T.; Portenkirchner, E. Substrate Dependent Charge Transfer Kinetics at the Solid/Liquid Interface of Carbon-Based Electrodes with Potential Application for Organic Na-Ion Batteries. Isr. J. Chem. 2022, 62, e202100082. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portenkirchner, E. Substantial Na-Ion Storage at High Current Rates: Redox-Pseudocapacitance through Sodium Oxide Formation. Nanomaterials 2022, 12, 4264. https://doi.org/10.3390/nano12234264
Portenkirchner E. Substantial Na-Ion Storage at High Current Rates: Redox-Pseudocapacitance through Sodium Oxide Formation. Nanomaterials. 2022; 12(23):4264. https://doi.org/10.3390/nano12234264
Chicago/Turabian StylePortenkirchner, Engelbert. 2022. "Substantial Na-Ion Storage at High Current Rates: Redox-Pseudocapacitance through Sodium Oxide Formation" Nanomaterials 12, no. 23: 4264. https://doi.org/10.3390/nano12234264
APA StylePortenkirchner, E. (2022). Substantial Na-Ion Storage at High Current Rates: Redox-Pseudocapacitance through Sodium Oxide Formation. Nanomaterials, 12(23), 4264. https://doi.org/10.3390/nano12234264