
First-Principles Prediction of New 2D p-SiPN: A Wide Bandgap Semiconductor



Abstract
1. Introduction
2. Computational Details
3. Results and Discussions
3.1. Structural Properties
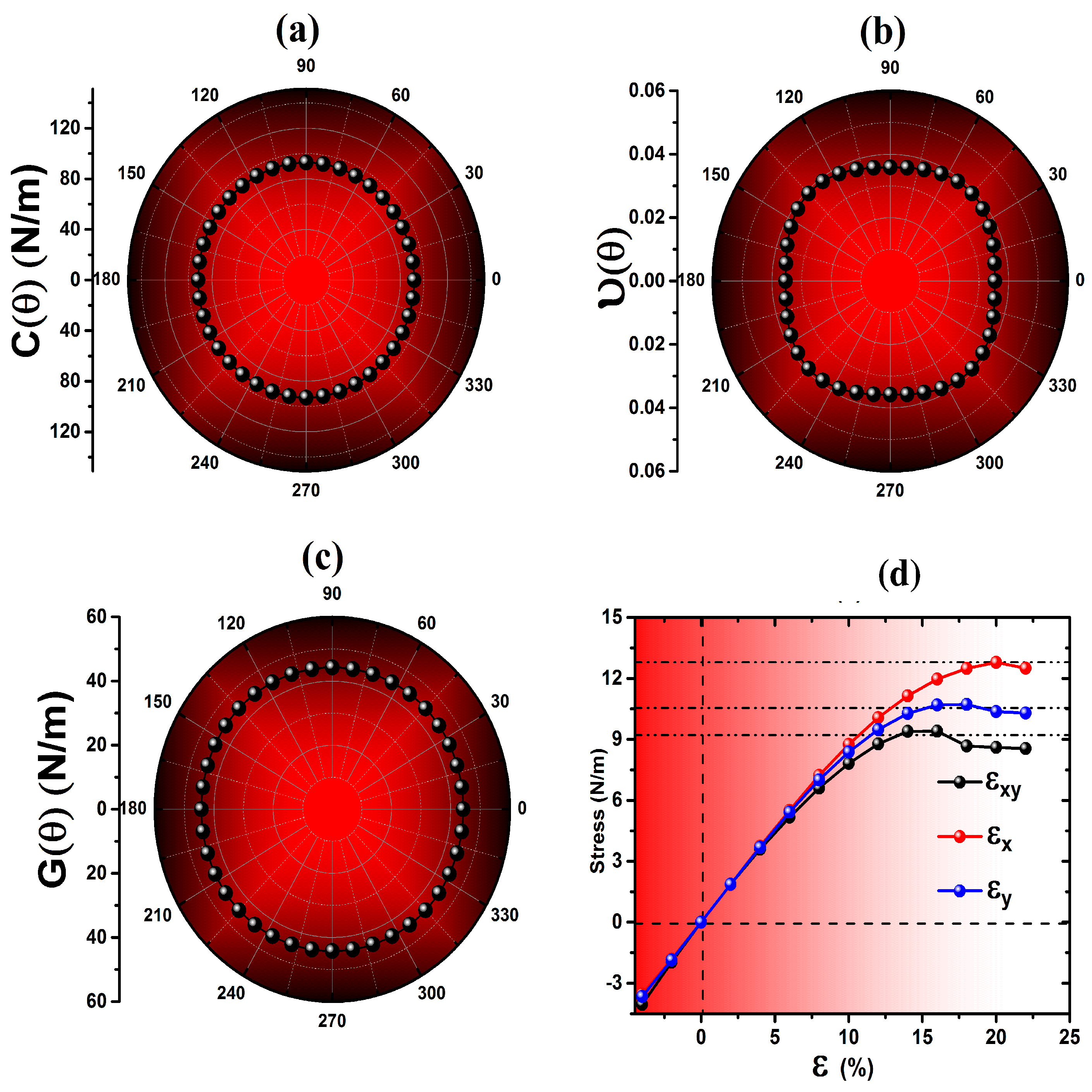
3.2. Mechanical Properties
3.3. Electronic Properties
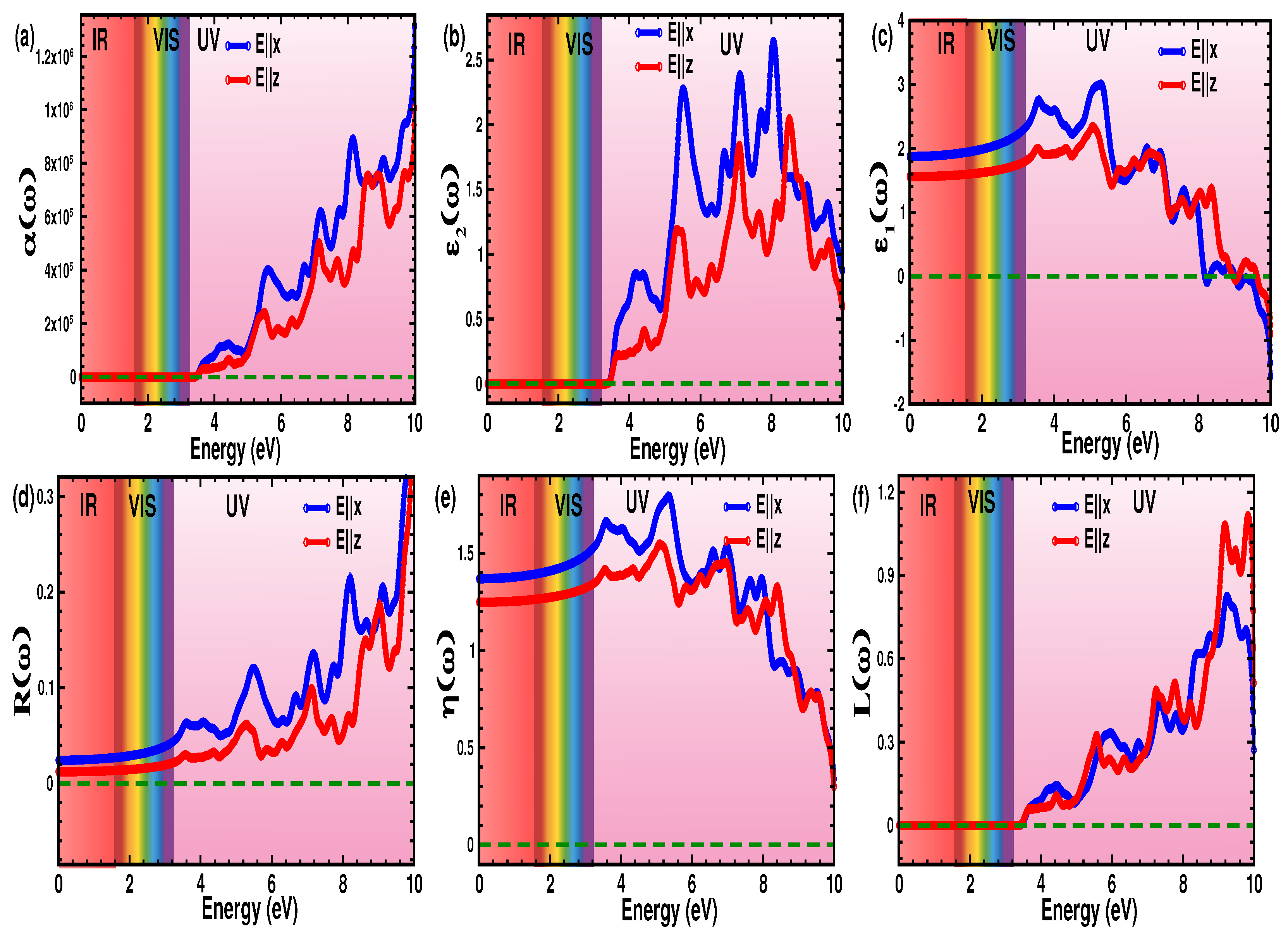
3.4. Optical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Zhou, J.; Wang, Q.; Chen, X.; Kawazoe, Y.; Jena, P. Penta-graphene: A new carbon allotrope. Proc. Natl. Acad. Sci. USA 2015, 112, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Nazir, M.A.; Hassan, A.; Shen, Y.; Wang, Q. Research progress on penta-graphene and its related materials: Properties and applications. Nano Today 2022, 44, 101501. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, Q. Pentagon-based 2D materials: Classification, properties and applications. Phys. Rep. 2022, 964, 1–42. [Google Scholar] [CrossRef]
- Oyedele, A.D.; Yang, S.; Liang, L.; Puretzky, A.A.; Wang, K.; Zhang, J.; Yu, P.; Pudasaini, P.R.; Ghosh, A.W.; Liu, Z.; et al. PdSe2: Pentagonal two-dimensional layers with high air stability for electronics. J. Am. Chem. Soc. 2017, 139, 14090–14097. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Wang, D.; Fan, P.; Zhang, R.; Zhang, Y.Y.; Li, G.; Mao, J.; Wang, Y.; Lin, X.; Du, S.; et al. Construction of bilayer PdSe2 on epitaxial graphene. Nano Res. 2018, 11, 5858–5865. [Google Scholar] [CrossRef]
- Cheng, P.K.; Liu, S.; Ahmed, S.; Qu, J.; Qiao, J.; Wen, Q.; Tsang, Y.H. Ultrafast Yb-doped fiber laser using few layers of PdS2 saturable absorber. Nanomaterials 2020, 10, 2441. [Google Scholar] [CrossRef]
- Zhang, X.; Su, G.; Lu, J.; Yang, W.; Zhuang, W.; Han, K.; Wang, X.; Wan, Y.; Yu, X.; Yang, P. Centimeter-Scale Few-Layer PdS2: Fabrication and Physical Properties. ACS Appl. Mater. Interfaces 2021, 13, 43063–43074. [Google Scholar] [CrossRef]
- Bykov, M.; Bykova, E.; Ponomareva, A.V.; Tasnadi, F.; Chariton, S.; Prakapenka, V.B.; Glazyrin, K.; Smith, J.S.; Mahmood, M.F.; Abrikosov, I.A.; et al. Realization of an ideal cairo tessellation in nickel diazenide NiN2: High-pressure route to pentagonal 2D materials. ACS Nano 2021, 15, 13539–13546. [Google Scholar] [CrossRef]
- Cerdá, J.I.; Sławińska, J.; Le Lay, G.; Marele, A.C.; Gómez-Rodríguez, J.M.; Dávila, M.E. Unveiling the pentagonal nature of perfectly aligned single-and double-strand Si nano-ribbons on Ag (110). Nat. Commun. 2016, 7, 13076. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, J.; Wang, Q.; Jena, P. Beyond graphitic carbon nitride: Nitrogen-rich penta-CN2 sheet. J. Phys. Chem. C 2016, 120, 3993–3998. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Z.; Liu, Z. Penta-CN2 revisited: Superior stability, synthesis condition exploration, negative Poisson’s ratio and quasi-flat bands. Appl. Surf. Sci. 2022, 585, 152536. [Google Scholar] [CrossRef]
- Wang, H.; Qin, G.; Qin, Z.; Li, G.; Wang, Q.; Hu, M. Lone-pair electrons do not necessarily lead to low lattice thermal conductivity: An exception of two-dimensional penta-CN2. J. Phys. Chem. Lett. 2018, 9, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Bouziani, I.; Haman, Z.; Kibbou, M.; Benhouria, Y.; Essaoudi, I.; Ainane, A.; Ahuja, R. Ab initio study of electronic and optical properties of penta-SiC2 and-SiGeC4 monolayers for solar energy conversion. Superlattices Microstruct. 2020, 142, 106524. [Google Scholar] [CrossRef]
- Xu, Y.; Ning, Z.; Zhang, H.; Ni, G.; Shao, H.; Peng, B.; Zhang, X.; He, X.; Zhu, Y.; Zhu, H. Anisotropic ultrahigh hole mobility in two-dimensional penta-SiC2 by strain-engineering: Electronic structure and chemical bonding analysis. RSC Adv. 2017, 7, 45705–45713. [Google Scholar] [CrossRef]
- Liu, H.; Qin, G.; Lin, Y.; Hu, M. Disparate strain dependent thermal conductivity of two-dimensional penta-structures. Nano Lett. 2016, 16, 3831–3842. [Google Scholar] [CrossRef]
- Naseri, M. Penta-SiC5 monolayer: A novel quasi-planar indirect semiconductor with a tunable wide band gap. Phys. Lett. A 2018, 382, 710–715. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, Y.; Huang, B.; Dai, Y. Two-dimensional penta-BN2 with high specific capacity for Li-ion batteries. ACS Appl. Mater. Interfaces 2019, 11, 6104–6110. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhang, X.; Zhang, H.; Gao, J.; Liu, H.; Yu, X.; Dai, X.; Liu, G.; Chen, G. Pentagonal B2C monolayer with extremely high theoretical capacity for Li-/Na-ion batteries. Phys. Chem. Chem. Phys. 2021, 23, 6278–6285. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, T.; Zhang, D.; Huang, H.; Wang, H.; Wang, H.; Ni, Y. First-principles calculations of phonon transport in two-dimensional penta-X2C family. J. Appl. Phys. 2020, 127, 205106. [Google Scholar] [CrossRef]
- Liu, S.; Liu, B.; Shi, X.; Lv, J.; Niu, S.; Yao, M.; Li, Q.; Liu, R.; Cui, T.; Liu, B. Two-dimensional penta-BP5 sheets: High-stability, strain-tunable electronic structure and excellent mechanical properties. Sci. Rep. 2017, 7, 2404. [Google Scholar] [CrossRef]
- Zhao, K.; Guo, Y.; Shen, Y.; Wang, Q.; Kawazoe, Y.; Jena, P. Penta-BCN: A new ternary pentagonal monolayer with intrinsic piezoelectricity. J. Phys. Chem. Lett. 2020, 11, 3501–3506. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Shen, Y.; Guo, Y.; Chen, Y.; Wang, Q. 1, 2, 4-Azadiphosphole-based piezoelectric penta-CNP sheet with high spontaneous polarization. Appl. Surf. Sci. 2021, 554, 149499. [Google Scholar] [CrossRef]
- Varjovi, M.J.; Kilic, M.; Durgun, E. Ternary pentagonal BNSi monolayer: Two-dimensional structure with potentially high carrier mobility and strong excitonic effects for photocatalytic applications. Phys. Rev. Mater. 2022, 6, 034004. [Google Scholar] [CrossRef]
- Sharma, S.B.; Qattan, I.A.; Jaishi, M.; Paudyal, D. Penta-SiCN: A Highly Auxetic Monolayer. ACS Appl. Electron. Mater. 2022, 4, 2561–2569. [Google Scholar] [CrossRef]
- Hou, C.; Shen, Y.; Sun, W.; Chen, Y.; Ni, D.; Wang, Q. A penta-BCP sheet with strong piezoelectricity and a record high positive Poisson’s ratio. J. Mater. Chem. C 2022, 10, 10302–10309. [Google Scholar] [CrossRef]
- Sharma, S.B.; Qattan, I.A.; KC, S.; Alsaad, A.M. Large Negative Poisson’s Ratio and Anisotropic Mechanics in New Penta-PBN Monolayer. ACS Omega 2022, 7, 36235–36243. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Artacho, E.; Sánchez-Portal, D.; Ordejón, P.; Garcia, A.; Soler, J.M. Linear-scaling ab-initio calculations for large and complex systems. Phys. Status Solidi (b) 1999, 215, 809–817. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter. Mater Phys. 1999, 59, 1758. [Google Scholar] [CrossRef]
- Saha, S.; Sinha, T.; Mookerjee, A. Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3. Phys. Rev. B 2000, 62, 8828. [Google Scholar] [CrossRef]
- Bohm, D.; Pines, D. A collective description of electron interactions. I. Magnetic interactions. Phys. Rev. 1951, 82, 625. [Google Scholar] [CrossRef]
- Bohm, D.; Pines, D. A collective description of electron interactions: III. Coulomb interactions in a degenerate electron gas. Phys. Rev. 1953, 92, 609. [Google Scholar] [CrossRef]
- Pines, D.; Bohm, D. A collective description of electron interactions: II. Collective vs individual particle aspects of the interactions. Phys. Rev. 1952, 85, 338. [Google Scholar] [CrossRef]
- Yu, P.Y.; Cardona, M. Optical properties I. In Fundamentals of Semiconductors; Springer: Berlin, Germnay, 2010; pp. 243–344. [Google Scholar]
- Fadaie, M.; Shahtahmassebi, N.; Roknabad, M.; Gulseren, O. Investigation of new two-dimensional materials derived from stanene. Comput. Mater. Sci. 2017, 137, 208–214. [Google Scholar] [CrossRef]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Li, R.; Shao, Q.; Gao, E.; Liu, Z. Elastic anisotropy measure for two-dimensional crystals. Extrem. Mech. Lett. 2020, 34, 100615. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Kube, C.M.; De Jong, M. Elastic constants of polycrystals with generally anisotropic crystals. J. Appl. Phys. 2016, 120, 165105. [Google Scholar] [CrossRef]
- Born, M.; Misra, R.D. On the stability of crystal lattices. IV. In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: Cambridge, UK, 1940; Volume 36, pp. 466–478. [Google Scholar]
- Sharma, S.B.; Bhatta, R.; Adhikari, R.; Paudyal, D. Strain dependent electronic and optical responses of penta-BCN monolayer. Carbon Trends 2022, 7, 100162. [Google Scholar] [CrossRef]
- Sharma, S.B.; Qattan, I. Enhanced mechanical, electronic, magnetic, and optical properties of penta-BCN by Ni-doping. Appl. Surf. Sci. 2022, 599, 153997. [Google Scholar] [CrossRef]
- Sharma, S.B.; Santosh, K.; Paudyal, D. Enhanced optoelectronic and elastic responses in fluorinated penta-BCN. Appl. Surf. Sci. 2022, 593, 153239. [Google Scholar] [CrossRef]
- Jappor, H.R.; Habeeb, M.A. Optical properties of two-dimensional GaS and GaSe monolayers. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 101, 251–255. [Google Scholar] [CrossRef]
- Tornatzky, H.; Gillen, R.; Uchiyama, H.; Maultzsch, J. Phonon dispersion in MoS2. Phys. Rev. B 2019, 99, 144309. [Google Scholar] [CrossRef]
- Dai, X.; Shen, T.; Feng, Y.; Liu, H. Structure, electronic and optical properties of Al, Si, P doped penta-graphene: A first-principles study. Phys. B Condens. Matter 2019, 574, 411660. [Google Scholar] [CrossRef]
- John, R.; Merlin, B. Optical properties of graphene, silicene, germanene, and stanene from IR to far UV—A first principles study. J. Phys. Chem. Solids 2017, 110, 307–315. [Google Scholar] [CrossRef]
- Laturia, A.; Van de Put, M.L.; Vandenberghe, W.G. Dielectric properties of hexagonal boron nitride and transition metal dichalcogenides: From monolayer to bulk. NPJ 2D Mater. Appl. 2018, 2, 6. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, F.; Shen, B.; Zhang, R.; Zheng, Y.; Chen, L.; Wang, S.; Wang, C.; Ho, K.; Fan, Y.J.; et al. Electronic and optical properties of novel carbon allotropes. Carbon 2016, 101, 77–85. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Ref | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| p-SiPN | This work | 91.76 | 92.98 | 3.29 | 44.23 | 91.64 | 92.86 | 0.035 | 0.036 |
| p-SiCN | Ref. [24] | 132.15 | 133.59 | −17.44 | 74.80 | 129.88 | 131.29 | −0.131 | −0.132 |
| p-BNSi | Ref. [23] | 114.46 | 112.21 | 12.76 | 48.97 | 113 | 109 | 0.11 | 0.11 |
| p-BCN | Ref. [45] | 210.15 | 170.77 | 4.27 | 102.93 | 210.05 | 170.66 | 0.020 | 0.025 |
| Ref. [21] | 223.56 | 189.16 | 4.90 | 104.80 | 223.45 | 189.03 | 0.022 | 0.026 | |
| p-CNP | Ref. [22] | 173.32 | 183.57 | 4.52 | 99.01 | 172 | 190 | - | - |
| Materials | Ref | K | ||||||
|---|---|---|---|---|---|---|---|---|
| p-SiPN | This work | 44.38 | 44.38 | 47.83 | 47.82 | 0.0000 | 0.0001 | 0.0000 |
| p-BCN | Refs. [45,46] | 98.01 | 97.21 | 97.36 | 96.32 | 0.0116 | 0.0273 | 0.0069 |
| MLs | Methods | (eV) | |||||
|---|---|---|---|---|---|---|---|
| Ex | Ez | Ex | Ez | Ex | Ez | ||
| p-SiPN | GGA | 1.37 | 1.25 | 1.88 | 1.55 | 3.43 | 3.44 |
| p-SiCN [24] | GGA | 3.17 | 1.30 | 10.01 | 1.98 | - | - |
| p-BCN [45] | GGA | 1.43 | - | 2.04 | - | 1.69 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhandari Sharma, S.; Qattan, I.; KC, S.; Abedrabbo, S. First-Principles Prediction of New 2D p-SiPN: A Wide Bandgap Semiconductor. Nanomaterials 2022, 12, 4068. https://doi.org/10.3390/nano12224068
Bhandari Sharma S, Qattan I, KC S, Abedrabbo S. First-Principles Prediction of New 2D p-SiPN: A Wide Bandgap Semiconductor. Nanomaterials. 2022; 12(22):4068. https://doi.org/10.3390/nano12224068
Chicago/Turabian StyleBhandari Sharma, Shambhu, Issam Qattan, Santosh KC, and Sufian Abedrabbo. 2022. "First-Principles Prediction of New 2D p-SiPN: A Wide Bandgap Semiconductor" Nanomaterials 12, no. 22: 4068. https://doi.org/10.3390/nano12224068
APA StyleBhandari Sharma, S., Qattan, I., KC, S., & Abedrabbo, S. (2022). First-Principles Prediction of New 2D p-SiPN: A Wide Bandgap Semiconductor. Nanomaterials, 12(22), 4068. https://doi.org/10.3390/nano12224068